THE EFFECT OF ALCOHOL AND CARBONYL FUNCTIONAL
GROUPS ON THE COMPETITION BETWEEN
UNIMOLECULAR DECOMPOSITION AND ISOMERIZATION
IN C
4and C
5ALKOXY RADICALS
Enoch E. Dames, William H. Green*
Massachusetts Institute of Technology, Department of Chemical Engineering Cambridge, MA 02139
*Author to whom correspondence should be addressed: William H. Green – whgreen@mit.edu
Department of Chemical Engineering Massachusetts Institute of Technology Cambridge, MA 02139
Abstract
Presented here are computed rates for the thermal unimolecular decomposition of a variety of alkoxy radicals with 4- and 5-carbon length backbones. Three classes of molecules are examined: alkoxy radicals with saturated hydrocarbon backbones, those with alcohol functional groups, and those with carbonyl functional groups. The chosen species represent many of those found during the combustion of fossil fuels as well as bio-derived alternatives. Density functional theory calculations were benchmarked against higher level coupled cluster calculations and used to explore the potential energy surfaces of these systems. Transition state theory was used to calculate high-pressure limit rate coefficients of all radical intermediates in the regimes relevant to atmospheric chemistry and combustion. We show that the assumption that alkoxy radicals quickly decompose via -scission to aldehydes and other radicals is not valid for some of the alkoxy radicals investigated in this work. We further illustrate how intra-H migrations in larger alkoxy radicals with carbonyl and alcohol functional groups can dominate unimolecular decomposition under combustion and atmospheric relevant conditions. Finally, we discuss why carbonyl groups can increase or decrease intra-H migration barriers depending on their location relative to the transferring H-atom.
Keywords
Introduction
Alkoxy radicals are formed during the combustion of nearly all oxygenated and hydrocarbon fuels. Under intermediate- to high-temperature combustion conditions, these radicals quickly undergo unimolecular decomposition to an aldehyde/ketone and another radical species. Indeed, these decomposition reactions are so fast that most combustion kinetic models containing alkoxy radicals only include -scission pathways. However, this is not the case under low- to intermediate-temperature combustion conditions and when alkoxy radicals are large enough to undergo facile internal H-migrations1,2. Such considerations are more commonly recognized in the field of atmospheric
chemistry3,4, but have been largely neglected in the combustion community.
Much of our knowledge on gas-phase alkoxy radical decomposition stems from the extensive work of Hippler and coworkers, who have systematically investigated the temperature and pressure dependent kinetics of alkoxy decomposition for many systems.5–8 The decomposition of alkoxy radicals is also
important in atmospheric chemistry. To this end, Somnitz and Zellner have computationally investigated the decomposition of C2-C5 alkoxy radicals.2,9 Vereecken and Peeters have formulated
structure additivity relationships for a large body of alkoxy decompositions10. In a subsequent paper,
those authors performed a similar study, illustrating how H-migration reactions can compete with decomposition reactions for some alkoxy radicals1. In their work, Vereeken and Peeters conducted a
comprehensive computational study on H-migrations for a wide variety of substituted alkoxy radicals for the conditions of 1 atm and 250-350 K, noting that H-shift reactions for some larger carbonyl substituted alkoxy radicals dominate all other possibilities. The authors also note the expected dominance of 1,5-hydrogen shifts in comparison with other H-shift possibilities. Although the authors performed multi-structural TST calculations, they did not provide parameterized rate constants
desirable for detailed kinetic modeling. The importance of 1,5 H-migrations in alkoxy radicals has also been demonstrated by Davis and Francisco11 in a semi-empirical study of reactivity trends for methoxy
through heptoxy radicals, with emphasis on the increased overall complexity in intermediate and product species.11 However, Davis and Francisco did not include parameterized rate constants
desirable for detailed kinetic modeling, nor did they explicitly account for the effects of hindered internal rotations on resulting rate computations. As discussed below, accounting for internal hindered rotors can have over an order of magnitude influence on computed intra-H migration rate constants. The intra-H migrations that are the focus of this work are largely missing from detailed combustion kinetic models relevant to both hydrocarbon and oxygenated fuels, where the formation of alkoxy radicals containing carbonyl and alcohol function groups is expected. Thus, the objective here is to identify alkoxy radicals with H-migration pathways that compete with simple -scission, while providing reliable corresponding high-pressure limit rate coefficients for combustion relevant temperatures.
Of specific interest are substituted alkoxys that can be expected to form as radical intermediates during the oxidation of hydrocarbons, as well as during the combustion of oxygenated fuels. For example, alkoxy radicals with carbonyl groups (ketoalkoxys) readily form during the low temperature oxidation of most larger fuel compounds (RH) through the following sequence of reactions:
R + O2 = RO2
RO2 = QOOH
O2 + QOOH = OOQOOH
OOQOOH = OQ´OOH (ketohydroperoxide) + OH OQ´OOH = OQ´O (ketoalkoxy) + OH
The role of ketohydroperoxides and their decomposition to OH and a ketoalkoxy radical in low temperature autoignition is well known and is described in great detail elsewhere12,13. In this work, the
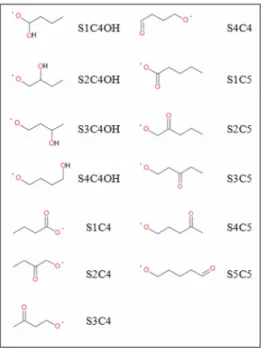
high-pressure kinetics of the alkoxy radicals with hydroxy and carbonyl functional groups were determined using transition state theory (TST) for alkoxy radicals with 4 and 5 carbon backbones. These species are illustrated in Figure 1. Although the isomerization and decomposition of the alkoxys in this work are expected to be pressure-dependent (e.g., see the work of Somnitz and Zellner14), only
the high-pressure limit values were computed since in most cases an accurate pressure dependent calculation would require computation of many secondary reaction steps in the multiwell system, not just the primary -scission and H-migration steps studied here. The supplemental information provides parameters necessary for future pressure-dependent calculations. For tools like the Reaction Mechanism Generator (RMG), pressure dependence can be computed approximately on-the-fly through the high-pressure limit rate and thermodynamic parameters of the reacting system, estimating rates for the secondary reactions15. Such tools can also be useful for estimating rates of other potentially
competing pathways available to the species studied in this work but not explicitly computed here, like internal H-transfers between the hydrogen on the hydroxyl groups where the ring strain of the transition state is low. Additionally, -scission of some species (e.g., S1C5 and S1C4OH) is expected to be competitive with intra-H migrations. Such pathways should be explored and/or included in full detailed kinetic models for these and similar compounds
Methodology
Molecular geometries, force constants, and energies for all species and saddle points were determined at the M08SO/MG3S16,17 level of theory, utilizing QChem 4.1.18 A computational grid with 75 radial
were scaled by the recommended value of 0.985. Loose internal degrees of freedom for relevant adducts and transition states (i.e., hindered rotors (HRs)) were treated separately by performing relaxed potential energy scans about the bond defining the internal rotor; these calculations were performed in Gaussian 0319 at the BMK /6-311+G(d,p) level of theory.20 Reduced internal moments of inertia for all
internal rotors were estimated at the I(2,3) level as defined be East and Radom.21
CCSD(T)-F12a/cc-pVTZ-F1222–25 calculations used to benchmark the M08SO/MG3S relative energies were computed
using Molpro26. Cantherm (a utility of the MIT RMG, found at
http://reactionmechanismgenerator.github.io)15 was used for all transition state theory calculations,
which were performed in the range of 300-2000 K. Eckart tunneling corrections were applied to all H-migration reactions.27 Intramolecular H-migration reactions studied in this work are given a reaction
type notation corresponding to the nature of the transition state:
ixy where i = 5, 6, 7; x,y = [p]rimary, [s]econdary, [t]ertiary, keto-endo, keto-exo, acyl, OH
i in the above expression denotes whether or not the transition state is a 5-, 6-, or 7-membered ring structure, while the remaining prefixes correspond to the carbon type from which a H-atom is being abstracted. The specific alcohol and aldehyde alkoxy radicals studied in this work are illustrated below in Figure 1. All of the mono-substituted alkoxy radicals and corresponding reactions studied in this work have also been investigated by Vereeken and Peeters1, who performed conformational analyses
to determine the lowest energy pathways (in addition to multi-conformer TST for the temperature range of 250 – 350 K). Only the lowest energy conformers were explicitly considered in this work, with conformers being treated by the independent hindered rotors approximation. Geometries, energies, force constants, and hindered rotor scans for all species studied in this work can be found in the supplemental information.
Figure 1. Nomenclature and structure of alkoxy-aldehyde and alkoxy-alcohol radical species studied in this work.
Results and Discussion
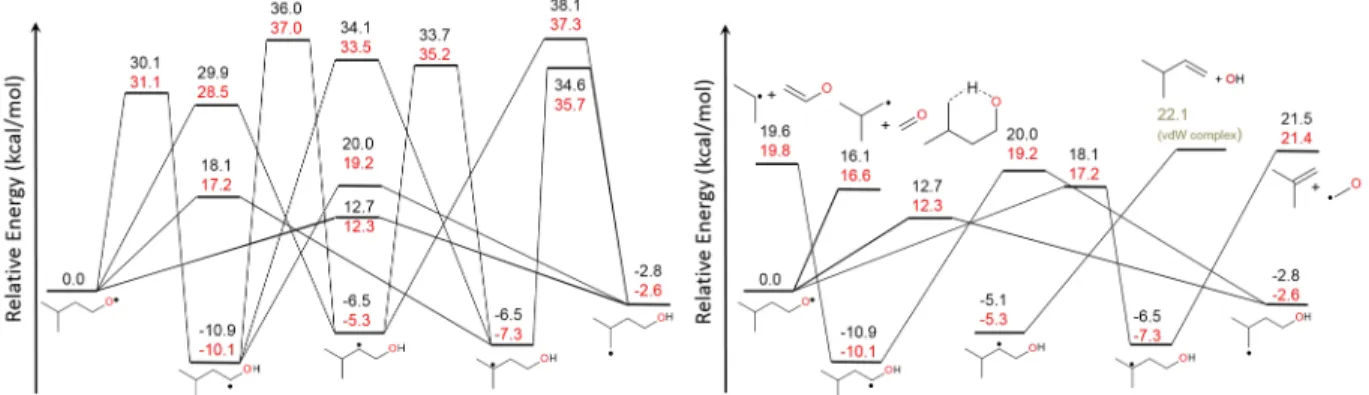
Relative energies computed at the M08SO/MG3S level of theory are compared to higher level calculations for the intra-H migrations in iso-pentoxy, as illustrated in Figure 2. M08SO/MG3S relative energies are all within 1.5 kcal/mol of the CCSD(T)-F12a/cc-pVTZ-F12 values, with an average deviation in absolute values of the energy differences of 0.9 kcal/mol. Thus, the M08SO/MG3S model chemistry was used to compute the relative energies of all species in this work, being a good compromise between computational speed and relative energy accuracy.
Figure 2. Zero Kelvin (zpe not included) relative energy comparisons for select isomerization (left) and elimination reactions (right) in iso-pentoxy between two levels of quantum chemistry theory. Red: CCSD(T)-F12/VTZ-F12//M08SO/MG3S; Black: M08SO/MG3S. PES split into two panels for increased clarity of presentation.
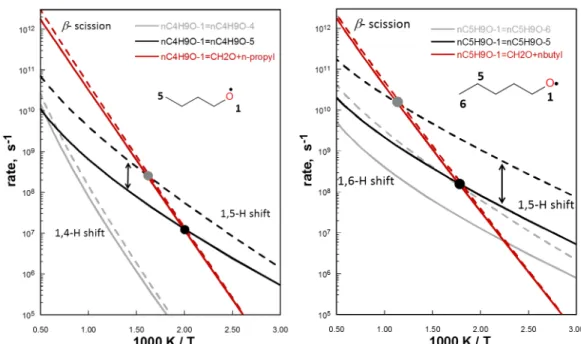
The influence of accounting for loose internal degrees of freedom as 1D hindered rotors (1D HRs) compared to the rigid rotor harmonic oscillator treatment (RRHO) is illustrated in Figure 3 for n-butoxy and n-pentoxy radicals. The difference in computed rates (RRHO vs 1D HR) can exceed one order of magnitude, illustrating the importance of properly including loose internal degrees of freedom. In n-pentoxy for instance, the reactant species has 4 internal modes corresponding to internal rotations that were described as 1D HRs in the final TST-derived rate coefficient. The influence of HRs on rates is more evident for intra-H migrations than for -scissions because many of the internal rotors are 'tied up' in the transition state (rates computed using the RRHO and HR methods for all substituted alkoxys are illustrated in the supplemental information). In contrast, inclusion of 1D HRs does not have a large effect on computed -scission reactions since internal rotors within the reactant and transition state do not noticeably change. We note that two species in this work (S4C5 and S3C4OH) have internal rotors with internal relaxed scan energy profiles that do not end at the same place they began. For such cases, it is more appropriate to use a multistructural TST, but this is outside the scope of the current work. However, we further note that resulting rates of H-migrations for S4C5 and S3C4OH have additional uncertainties for this reason. Figure 3 illustrates an additional important point: the ‘crossover
temperature’– the temperature at which each channels’ branching fraction for the -scission and H-migration channels are equal (50%) – can vary widely depending on how the internal hindered rotors are treated in rate calculations. Thus, proper treatment of internal hindered rotors is critical to more accurately compute rates for reactions that are the focus of this work. With this in mind, we recognize more advanced methods of treating contributions to H-transfer rates from internal rotors, but such treatments were not attempted here28,29,30.
Figure 3. M08SO/MG3S computed high-pressure limit rate coefficients for the dominant unimolecular fates of n-butoxy (left) and n-pentoxy (right). Dashed lines: TST computed rates using the RRHO assumption for all internal degrees of freedom; solid lines: TST computed rates using the 1-D HR approximation for all loose internal rotors, as described in the text; black and grey circles: intra-H migration/-scission competitive crossover temperature with and without the 1-D HR approximation, respectively. Arrows used to highlight difference between 1-D HR and RRHO computed rate results. Red lines: -scission reactions; black lines 1,5 H-transfer rates; grey lines: 1,4 H-transfer rates (n-butoxy) and 1,6 H-transfer rates (n-pentoxy).
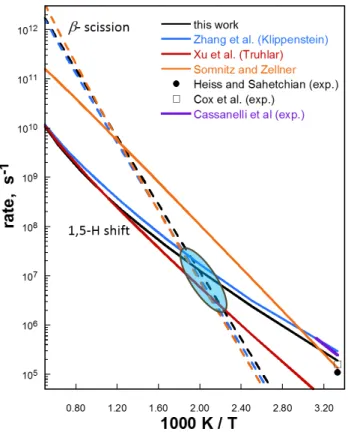
Figure 4 compares intra-H migration rates computed here with high-level results computed by Zhang et al.31, Somnitz and Zellner2, and Xu et al.32, along with available experimental values33,34,35. All
computed -scission rates are in good agreement, within 30% at 1400 K. The -scission to CH2O +
n-propyl and intra-H migration rates computed here are in fair agreement with those computed by Zhang et al.31 (lower by 40% at 300 K, decreasing to 20 % at 1500 K). However, the intra-H migration rates
computed by Xu and coworkers deviates from other predictions at lower temperatures (over an order of magnitude lower than rate computed in this work at 300 K); the authors used multi-structural variational TST, and attribute the lower computed rates to the use of a more stable energy reactant structure [compared to their previous calculations] and lower tunneling contributions than when computed with conventional TST. Regarding the intra-H migration rates computed by Somnitz and Zeller, we note that their work focused on the computation of rates relevant to atmospheric conditions. Thus, agreement between the 1,5 H-shift rates computed here and those of Somnitz and Zeller are in fact within 30% at 300 K, and both are in good agreement with the available experimental values. At higher temperatures, all computed 1,5 H-shift rates agree well, although this is somewhat meaningless in the context of the n-butoxy radical in any high-pressure system at this temperature, since the -scission branching fraction begins to exceed 95% at temperatures at and above 1000 K. Regardless, the varying computed rates result in different locations of the crossover temperature. Depending on the combination of model chemistry and rate theory used, this crossover temperature can vary greatly. Additionally uncertainties arise from the treatment of hindered rotors, as explained above. For these two aforementioned reasons, this crossover temperature is only qualitatively addressed in this work, but is nonetheless useful for comparing and inspecting the relative influence of substitution type (i.e., alcohol and carbonyl groups) and position along the alkoxy carbon backbone.
Figure 4. Comparison of n-butoxy intra-H migration and -scission rate coefficients computed in this work and compared with previously computed and measured values. Solid lines: 1,5 intra-H transfer; dashed lines: -scission to CH2O + n-propyl Blue: Klippenstein et al.31; Red: Truhlar et al.32; Orange:
Somnitz and Zellner9; black: this work. Experimental measurements of isomerization channels: open
square: Cox et al.34; filled circle: Heiss and Sahetchian 35; purple line: experimentally fitted data of
Cassanelli et al.33 Blue transparent circle included to highlight variations in computed crossover
temperatures.
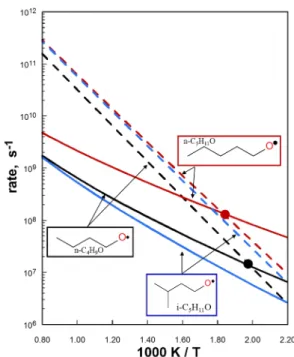
For the alkoxy radicals studied in this work, the 1,5 and 1,6 H-migration rates are generally faster than -scission pathways below 550 K (in the high pressure limit). However, -scission pathways dominate above the crossover temperature, which is typically between 350 and 750 K (e.g., see Figures 5-7). As illustrated in Figure 6, the 1,5 H-shift of S4C4 is faster than the -scission of n-butoxy producing formaldehyde and n-propyl up to 750 K (this reaction is used as a prototypical -scission for alkoxy radicals, and accurately describes the -scission for alkoxys with functional groups 2 or more carbons from the oxygen radical). In general, alcohol functional groups do not offer greatly enhanced
preference for intra-H migration; as shown in Figure 6, none of the selected species exhibit crossover temperatures above 600 K, primarily because the OH group only reduces H-shift barrier heights by up to 4 kcal/mol in S4C4OH, compared to 5 kcal/mol for some of the carbonyl alkoxy radicals discussed below. However, unlike in the carbonyl substituted alkoxy radicals, hydroxyl groups do not increase the H-transfer barrier height relative to in the n-butoxy H-transfer. In general, the presence of hydroxyl groups does not greatly affect the transition state for the H-shift reaction. We also note the additional complicating factor of internal hydrogen bonding for the reactions involving alkoxys with hydroxyl functional groups, which can further stabilize the molecule by up to 5 kcal/mol. In this work, only the lowest energy conformer for reactants and corresponding transition states were used in TST calculations, and taken from the supporting information of Vereecken and Peeters1.
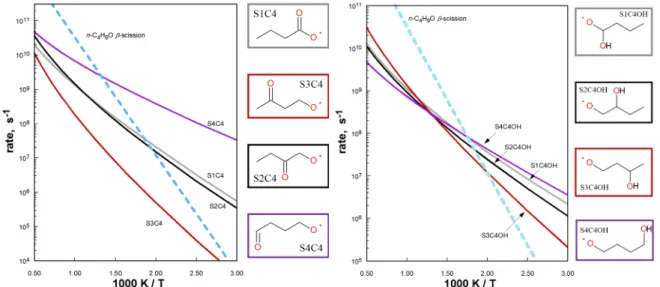
Figure 5. High-pressure limit rate coefficients of select alkoxy -scission (dashed lines) compared with select intra-H migration channels (solid lines). Circles: 1,5 intra-H migration/-scission competitive crossover temperature.
Figure 6. High-pressure limit rate coefficients of 1,5-H shifts for various C4 alcohol and aldehyde radical intermediates. Thick light blue dashed line: rate coefficient for -scission of n-C4H9O to CH2O
+ n-C3H7, shown for reference.
Figure 7. Rate coefficients of 1,5 (solid lines) and 1,6 (dashed lines) H-shifts for various C5 aldehyde radical intermediates. Thick light blue dashed line: rate coefficient for n-C4H9O = CH2O + n-C3H7,
Figure 7 illustrates several reactions of mono-substituted alkoxys where the intra-H migration competes with -scission above 500 K. In one case – S1C5 - the 1,5 H-shift is favored over -scission up to 700 K. The 0 K ZPE-corrected barrier height for this reaction is predicted to be only 5.4 kcal/mol due to the location of the carbonyl group and the resulting weak CH bond. For the same reason, the S5C5 1,6 H-shift is also competitive with -scission, but only up to 650 K. Barrier heights for the reactions examined here are shown inTable 1, along with those computed at the CBS-QB3 level of theory by Vereecken and Peeters1. The use of the CBS-QB336 model chemistry was considered in this work as
well. However, as noted by Vandeputte and coworkers37, CBS-QB3 tends to underestimate reaction
barriers for species with spin contamination in the transition state. The CBS-Q38 model chemistry,
from which the CBS-QB3 model chemistry is derived, is known to present difficulties for species with spin contamination molecules with multiple lone pairs on the same atom (e.g., many of the oxygenated species studied in this work). Thus, the M08SO/MG3S model chemistry was used to compute the relative energies of all species in this work, being a good compromise between computational speed and relative energy accuracy.
As illustrated by the barriers listed in Table 1, the position of the carbonyl groups relative to the oxygen radical can increase or decrease the barrier height to H-shift. In general, the sp2 hybridized C=O bond increases the overall ring strain energy in transition state for reactions that contain a more centrally located carbonyl group. However, for most carbonyl group locations close to the oxygen radical (e.g., S1C5 S1C4, S4C4), H-migration reactions are more exothermic with barriers lower than that for n-butoxy by up to 7 kcal/mol.
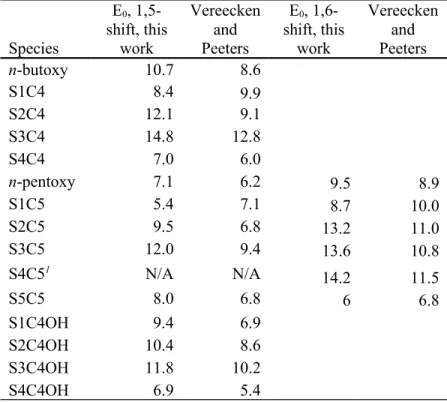
Table 1. Barrier heights (0K, ZPE included, units of kcal/mol) for intra-H migrations in the aldehyde radical intermediates of this work, computed at the M08SO/MG3S level of theory. CBS-QB3 values of Vereecken and Peeters1
also shown. n-butoxy and n-pentoxy included as reference molecules. Species E0, 1,5-shift, this work Vereecken and Peeters E0, 1,6-shift, this work Vereecken and Peeters n-butoxy 10.7 8.6 S1C4 8.4 9.9 S2C4 12.1 9.1 S3C4 14.8 12.8 S4C4 7.0 6.0 n-pentoxy 7.1 6.2 9.5 8.9 S1C5 5.4 7.1 8.7 10.0 S2C5 9.5 6.8 13.2 11.0 S3C5 12.0 9.4 13.6 10.8 S4C51 N/A N/A 14.2 11.5 S5C5 8.0 6.8 6 6.8 S1C4OH 9.4 6.9 S2C4OH 10.4 8.6 S3C4OH 11.8 10.2 S4C4OH 6.9 5.4
1does not have a hydrogen atom available for 1,5 H-shift
Figure 8. 1,5 H-transfer transition state geometry illustrating varying properties of interest for the compounds studied in this work. c: bond angle corresponding to carbonyl location within 6-membered
ring transition state; mid: bond angle corresponding to location directly across from transferring H-atom
in 6-membered ring transition state; : dihedral angle for O-C-C-C bond. Bonds and symbol indicated in blue are meant to aid the eye for defining .
Table 2 displays several properties of interest corresponding to n-butoxy, n-pentoxy, as well as the C4 and C5 carbonyl alkoxy radicals studied in this work. n-Butoxy radical is given as a reference molecule to a normal alkoxy radical with no functional groups. The barrier for this slightly exothermic 1,5-H transfer is 10.7 kcal/mol. c, mid and are all defined in Figure 8. c is chosen because deviations from
the unstrained sp2 hybridized preference of 118 give an indication of how the 6-membered ring transition state is strained. Similarly, mid illustrates how deviations from the unstrained sp3
hybridization preference angle of 109 correspond to deviations from n-butoxy’s reference barrier height of 10.7 kcal/mol. Finally, the dihedral angle, , is also shown since large deviations from that found in n-butoxy were observed in some cases. From the results shown in Table 2, it is clear that H-shift barriers are lower than in n-butoxy when the carbonyl group is located at a terminal position in the parent alkoxy (e.g., S1C4, S4C4, S1C5, S5C5). With reference to S1C4 and S1C5, we note how the O=CO (carboxyl) groups in the reactants are resonantly stabilized. This resonance leads to increased electron donation in the transition state, thereby resulting in a lower energy barrier. For S4C4 and S5C5 however, the carbonyl CH bond energy is lower than compared to both primary and secondary alkyl CH bonds. For example, the CH bond strength in acetaldehyde (CH3C(=O)H) is 88.9 ±1.0
kcal/mol39. Compared to the primary and secondary CH bonds (4- and 3- carbon positions,
respectively) in 1-butanol (n-butanol), the acetyl bond strength is lower by 10 kcal/mol40,41. Hence,
the reactions abstracting aldehydic hydrogens are therefore faster, and explain why the 1,6 H-shift in S5C5 is faster than the respective 1,5 H-shift.
Table 2. 1,5 H-shift reaction and transition state properties (M08-SO) for select molecules studied in this work. Energies in kcal/mol and include contributions from ZPE. Angles in degrees. See Figure 1 for species identification. E0: Zero Kelvin barrier height with
exothermicity for 1,5 H-shift (kcal/mol). See Figure 8 for definitions of geometric properties.
Species E0 rxn c mid n-butoxy 10.7 -4 N/A 108 -59 S1C4 8.4 -13 116 112 -35 S2C4 12.1 -3 116 c -3 S3C4 14.8 -12 113 106 -59 S4C4 7.0 -12 109 111 -63 n-pentoxy 7.1 -7 N/A 108 -59 S1C5 5.4 -13 117 112 -36 S2C5 9.5 -7 115 c 2 S3C5 12.0 -18 114 106 -59 S4C51 N/A S5C5 8.0 -15 116 108 60
1does not have a hydrogen atom available for 1,5 H-shift
The largest H-transfer barrier height among the reactions in this work corresponds to S3C4, at 14.8 kcal/mol, 4 kcal/mol higher than in the reference molecule n-butoxy. The c values for S3C4 and S3C5
are lower than most other corresponding values in Table 2, and are different from the 118-120 bond orientation preferred by sp2 hybridized orbitals. This deviation likely increases the overall ring strain. In addition, the resulting product species for the 1,5 H-shifts in S3C4 and S3C5 are comparatively exothermic due to their resonance stabilization. A few of the computed reaction exothermicities are a few kcal/mole different than expected, possibly indicating errors of magnitude in the M08SO model chemistry. We note that empirical bond-additivity corrections (BAC) have been developed for use with several methods, including CBS-QB3 [ref], to improve the accuracy of computed enthalpies of formation, but to our knowledge no BAC have been developed yet for MO8SO.Of course, the precise thermochemistry of most of the radicals considered in this work is unknown.
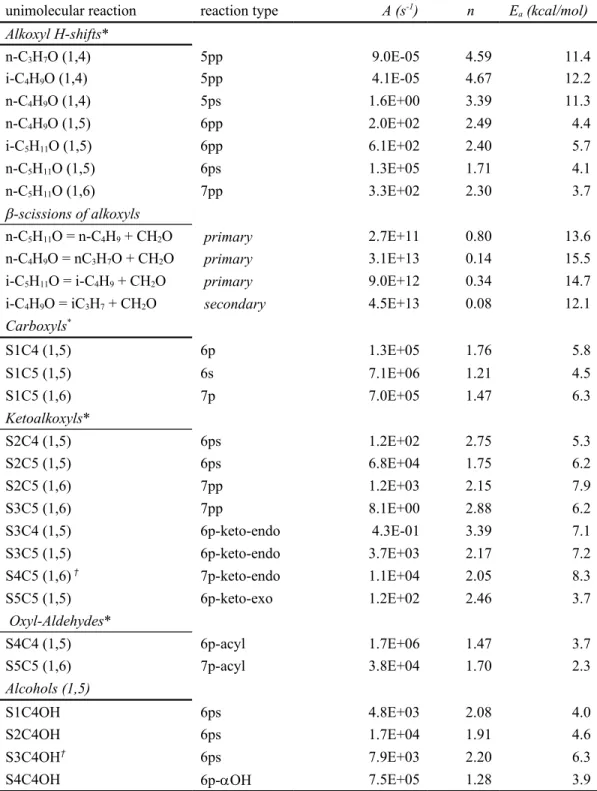
shows the reactions and corresponding high-pressure limit rates computed in this work, which are suitable for use in detailed kinetic models. Systems like those studied here are expected to be common
in the combustion of both oxygenated and pure hydrocarbon compounds, especially under low-temperature oxidative conditions. The results of this work clearly illustrate that scissions in larger alkoxy radicals (especially those with carbonyl substitutions) cannot be the assumed exclusive pathway since intra-H migrations can compete up to 750 K, well into the first stage autoignition regime of many fuel compounds and future alternative fuel candidates.
Table 3. High pressure limit rate constants for the reactions studied in this work*.
unimolecular reaction reaction type A (s-1) n E
a (kcal/mol) Alkoxyl H-shifts* n-C3H7O (1,4) 5pp 9.0E-05 4.59 11.4 i-C4H9O (1,4) 5pp 4.1E-05 4.67 12.2 n-C4H9O (1,4) 5ps 1.6E+00 3.39 11.3 n-C4H9O (1,5) 6pp 2.0E+02 2.49 4.4 i-C5H11O (1,5) 6pp 6.1E+02 2.40 5.7 n-C5H11O (1,5) 6ps 1.3E+05 1.71 4.1 n-C5H11O (1,6) 7pp 3.3E+02 2.30 3.7 -scissions of alkoxyls n-C5H11O = n-C4H9 + CH2O primary 2.7E+11 0.80 13.6 n-C4H9O = nC3H7O + CH2O primary 3.1E+13 0.14 15.5
i-C5H11O = i-C4H9 + CH2O primary 9.0E+12 0.34 14.7
i-C4H9O = iC3H7 + CH2O secondary 4.5E+13 0.08 12.1 Carboxyls* S1C4 (1,5) 6p 1.3E+05 1.76 5.8 S1C5 (1,5) 6s 7.1E+06 1.21 4.5 S1C5 (1,6) 7p 7.0E+05 1.47 6.3 Ketoalkoxyls* S2C4 (1,5) 6ps 1.2E+02 2.75 5.3 S2C5 (1,5) 6ps 6.8E+04 1.75 6.2 S2C5 (1,6) 7pp 1.2E+03 2.15 7.9 S3C5 (1,6) 7pp 8.1E+00 2.88 6.2 S3C4 (1,5) 6p-keto-endo 4.3E-01 3.39 7.1 S3C5 (1,5) 6p-keto-endo 3.7E+03 2.17 7.2 S4C5 (1,6) † 7p-keto-endo 1.1E+04 2.05 8.3 S5C5 (1,5) 6p-keto-exo 1.2E+02 2.46 3.7 Oxyl-Aldehydes* S4C4 (1,5) 6p-acyl 1.7E+06 1.47 3.7 S5C5 (1,6) 7p-acyl 3.8E+04 1.70 2.3 Alcohols (1,5) S1C4OH 6ps 4.8E+03 2.08 4.0 S2C4OH 6ps 1.7E+04 1.91 4.6 S3C4OH† 6ps 7.9E+03 2.20 6.3
S4C4OH 6p-OH 7.5E+05 1.28 3.9
*type of H-transfer reaction indicated in parentheses. †Higher overall computed rate uncertainty due to
Uncertainties
Because TST-derived rates are sensitive to the low energy internal harmonic modes, frequencies for the transition state were computed at the CCSD(T)/cc-pVDZ level of theory using Molpro26 (we note that
performing frequency calculations using this model chemistry for all species in this work is computationally prohibitive at this time). Select results are given in Table 4 and give a sense of the uncertainty in computed frequencies using the M08SO functional. As seen in Table 4, the computed vibrational frequencies for the imaginary and low modes differ by up to a substantial 16%. Although a fortuitous cancellation of errors in frequencies of the transition state and reactant likely reduces the overall uncertainty in computed rate constants, there is nevertheless uncertainty in computed rates associated with the frequencies alone, discussed below (the influence of anharmonic corrections are acknowledged, but neglected here [and in most rate calculations]).
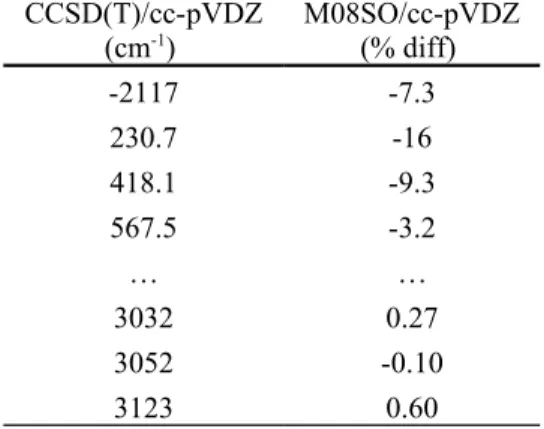
Table 4. Comparison of select vibrational frequencies (cm-1) for the transition state in the 1,4-intra-H migration of 1-propoxy computed using two different model chemistries and the same basis sets. CCSD(T)/cc-pVDZ results scaled by 0.979; M08SO/cc-pVDZ results scaled by 0.983. Note: only lowest and highest frequencies shown; only percent relative difference shown for M08SO results. CCSD(T)/cc-pVDZ (cm-1) M08SO/cc-pVDZ(% diff) -2117 -7.3 230.7 -16 418.1 -9.3 567.5 -3.2 … … 3032 0.27 3052 -0.10 3123 0.60
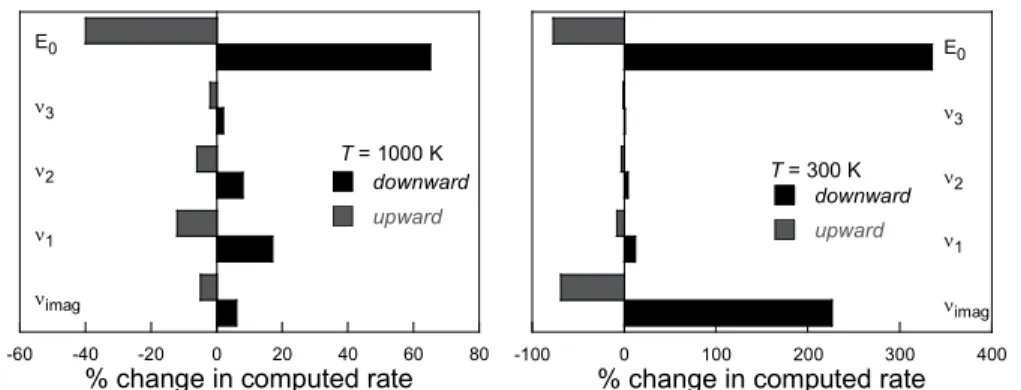
A straightforward uncertainty analysis was performed on select input parameters for the 1,4- intra-H migration in 1-propoxy. Percent changes in high-pressure limit rate constants at both 300 K and 1000 K were computed for the first three vibrational frequencies of the transition state (including the imaginary frequency) and the reaction barrier height. For these calculations, the vibrational frequencies of the transition state were perturbed upward and downwards by the computed percent difference between CCSD(T)/cc-pVDZ and M08SO frequencies listed for each corresponding vibrational mode in Table 4. This represents an estimate of the upper bound in computed uncertainties based on comparisons with higher level CCSD(T)/cc-pVDZ frequency calculations. This is designated as a upper bound because only the frequencies in the transition state were perturbed, and there is therefore no [fortuitous] cancellation of errors in the resulting rate constants. The 0 K reaction barrier height was perturbed by ± 1 kcal. Figure 9 shows histograms for perturbations in upward and downward directions from nominally computed values, which are available in the supporting information.
Figure 9 illustrates that the contributions to global kinetic uncertainty for 1,4 H-shift in n-propoxy from uncertainties in the barrier height are dominant at all temperatures. However, the relative contribution to rate uncertainty due to uncertainties in the normal mode frequencies is slightly higher at 1000 K, with a maximum computed rate increase of 17% due to a 7.3% reduction of the lowest vibrational mode. As discussed above, properly accounting for hindered rotors in H-shift reactions where the loose internal modes are tied up in the transition state is of critical importance. In the 1,4 H-shift in n-propoxy, there exist two such modes in the reactant (the methyl rotor, and the CH3CH2CH2O rotor).
By not accounting for these through the independent rotor treatment used for all reactions of this work and simply using the RRHO treatment instead, the computed rates at 1000 K and 300 K are higher by 66% and 34% respectively. This difference of course increases with increase transition state ring size. Thus, uncertainties associated with the treatment of low vibrational modes can be comparable to the uncertainties resulting from the barrier height uncertainty.
-60 -40 -20 0 20 40 60 80 imag 1 2 3 E0 downward upward
% change in computed rate T = 1000 K -100 0 100 200 300 400 imag 1 2 3 E0 downward upward
% change in computed rate T = 300 K
Figure 9. Influence of selected dependent parameters on computed rates for the 1-, 4- intra-H migration in 1-propoxy at 1000 K (left panel) and 500 K (right panel) and in the high-pressure limit. downward: percent change in rate due to decreases in parameters of the cyclic transition state percent decrease in force constants from corresponding values in Table 4, i (i = frequency of internal mode beginning at the
lowest energy, i = 1), and 1 kcal decrease for the electronic energy, E0; upward:
It is well known that computed rate uncertainties due to uncertainties in reaction barrier heights tend to dominate contributions to the global kinetic uncertainty at the high pressure limit. In the above exercise, the barrier height was perturbed by 1 kcal, which is lower than the estimated 1.5 kcal/mol uncertainty estimated for the M08SO model chemistry used through this work. In the above example, the greatest influence on computed rate stems form from a 1 kcal decrease in barrier height. At 300 K, the resulting rate is a factor of 3.4 higher than the nominal value. At 1000 K, the resulting rates differ by between 40-65 %. The resulting uncertainties due to barrier height alone are thus highly variable, depending not just on temperature, but also on overall reaction barrier height.
As illustrated in Figure 9 and extensively discussed in previous work42, the uncertainty in the computed
imaginary frequency influences the Eckart quantum transmission coefficient, especially at lower temperatures relevant to atmospheric and aqueous chemistry, underscoring the value of low-temperature experimental data which may be used to benchmark rate theory calculations. Although uncertainties due to treatment of H-atom tunneling fall below 6% at ~1000 K and continue to diminish at higher temperatures, they exceed 220% at and 300 K. Thus, uncertainties in global kinetic rates for H-transfer reactions at low temperatures are dominated by uncertainties in both barrier height and quantum tunneling. It is clear from the discussion above that the global parametric high-pressure rate uncertainties are highly dependent on temperature. Based on the uncertainty analysis above and preceding discussion on hindered rotor uncertainty, it is not possibly to report reliable overall uncertainties for all of the reactions of this work. However, we estimate the uncertainties in total rates range from a factor of 5-20 at lower temperatures (298 K – 500 K) and decrease to a factor of 2-4 at temperatures above 500 K.
Conclusions
Three classes of molecules were examined in this work: alkoxy radicals with hydrocarbon backbones, those with alcohol functional groups, and those with carbonyl functional groups. TST was used to compute rates for all species in this work based on electronic structure calculations at the M08SO/MG3S level of theory. Although alkoxy radicals with pure hydrocarbon backbones can take part in intra-H migration reactions, their -scission to aldehydes/ketones and other radicals dominate above ~500 K. Likewise, alkoxy radicals with single alcohol functional groups do not significantly increase H-transfer crossover temperatures. On the other hand, 1,5 and 1,6 intra-H migrations for alkoxy radials with C=O groups at terminal locations on the carbon backbone (i.e., abstraction by carboxyl radicals, or from formyl CH bonds) are shown to exhibit higher exothermicity and low barrier heights with corresponding rates competitive with unimolecular decompositions up to ~750 K. The reactions studied in this work have been parameterized and are suitable for use in detailed kinetic models of atmospheric and combustion chemistry.
Acknowledgments
This work is supported by the U.S. Department of Energy, Office of Basic Energy Sciences under the Energy Frontier Research Center for Combustion Science (Grant No. DE-SC0001198). This research used resources of the National Energy Research Scientific Computing Center, a DOE Office of Science User Facility supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. We greatly acknowledge additional financial support from the MIT Energy Initiative Seed Fund.
References
(1) Vereecken, L.; Peeters, J. Phys. Chem. Chem. Phys. 2010, 12 (39), 12608–12620. (2) Somnitz, H.; Zellner, R. Phys. Chem. Chem. Phys. 2000, 2 (9), 1899–1905. (3) Atkinson, R. Atmos. Environ. 2007, 41 (38), 8468–8485.
(4) Peeters, J.; Fantechi, G.; Vereecken, L. J. Atmospheric Chem. 2004, 48 (1), 59–80.
(5) Hippler, H.; Striebel, F.; Viskolcz, B. Phys. Chem. Chem. Phys. 2001, 3 (12), 2450–2458. (6) Caralp, F.; Devolder, P.; Fittschen, C.; Gomez, N.; Hippler, H.; Mereau, R.; T. Rayez, M.;
Striebel, F.; Viskolcz, B. Phys. Chem. Chem. Phys. 1999, 1 (12), 2935–2944.
(7) Fittschen, C.; Hippler, H.; Viskolcz, B. Phys. Chem. Chem. Phys. 2000, 2 (8), 1677–1683. (8) Fittschen, C.; Frenzel, A.; Hippler, H.; Poskrebyshev, G.; Striebel, F. Phys. Chem. Chem. Phys.
1999, 1 (4), 675–681.
(9) Somnitz, H.; Zellner, R. Phys. Chem. Chem. Phys. 2000, 2 (9), 1907–1918. (10) Vereecken, L.; Peeters, J. Phys. Chem. Chem. Phys. 2009, 11 (40), 9062–9074. (11) Davis, A. C.; Francisco, J. S. J. Am. Chem. Soc. 2011, 133 (45), 18208–18219.
(12) Merchant, S. S.; Goldsmith, C. F.; Vandeputte, A. G.; Burke, M. P.; Klippenstein, S. J.; Green, W. H. Combust. Flame 2015, 162 (10), 3658–3673.
(13) Zádor, J.; Taatjes, C. A.; Fernandes, R. X. Prog. Energy Combust. Sci. 2011, 37 (4), 371–421. (14) Somnitz, H.; Zellner, R. Z. Für Phys. Chem. 2006, 220 (8), 1029–1048.
(15) Cantherm/RMGPy http://greengroup.github.io/RMG-Py.
(16) Zhao, Y.; Truhlar, D. G. J. Chem. Theory Comput. 2008, 4 (11), 1849–1868. (17) Lynch, B. J.; Zhao, Y.; Truhlar, D. G. J. Phys. Chem. A 2003, 107 (9), 1384–1388.
(18) Shao, Y.; Molnar, L. F.; Jung, Y.; Kussmann, J.; Ochsenfeld, C.; Brown, S. T.; Gilbert, A. T. B.; Slipchenko, L. V.; Levchenko, S. V.; O’Neill, D. P.; DiStasio, R. A.; Lochan, R. C.; Wang, T.; Beran, G. J. O.; Besley, N. A.; Herbert, J. M.; Lin, C. Y.; Van Voorhis, T.; Chien, S. H.; Sodt, A.; Steele, R. P.; Rassolov, V. A.; Maslen, P. E.; Korambath, P. P.; Adamson, R. D.; Austin, B.; Baker, J.; Byrd, E. F. C.; Dachsel, H.; Doerksen, R. J.; Dreuw, A.; Dunietz, B. D.; Dutoi, A. D.; Furlani, T. R.; Gwaltney, S. R.; Heyden, A.; Hirata, S.; Hsu, C. P.; Kedziora, G.; Khalliulin, R. Z.; Klunzinger, P.; Lee, A. M.; Lee, M. S.; Liang, W.; Lotan, I.; Nair, N.; Peters, B.; Proynov, E. I.; Pieniazek, P. A.; Rhee, Y. M.; Ritchie, J.; Rosta, E.; Sherrill, C. D.; Simmonett, A. C.; Subotnik, J. E.; Woodcock, H. L.; Zhang, W.; Bell, A. T.; Chakraborty, A. K.; Chipman, D. M.; Keil, F. J.; Warshel, A.; Hehre, W. J.; Schaefer, H. F.; Kong, J.; Krylov, A. I.; Gill, P. M. W.; Head-Gordon, M. Phys. Chem. Chem. Phys. 2006, 8 (27), 3172–3191.
(19) Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Montgomery, J. A.; Vreven, T.; Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G. A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Laham, A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Gonzalez, C.; Pople, J. A. Gaussian 03, Revision B.03; 2004.
(20) Boese, A. D.; Martin, J. M. J. Chem. Phys. 2004, 121 (8), 3405–3416. (21) East, A. L. L.; Radom, L. J. Chem. Phys. 1997, 106 (16), 6655–6674.
(22) Adler, T. B.; Knizia, G.; Werner, H.-J. J. Chem. Phys. 2007, 127 (22), 221106–224100. (23) Adler, T. B.; Werner, H.-J. J. Chem. Phys. 2009, 130 (24), 241101.
(24) Adler, T. B.; Werner, H.-J.; Manby, F. R. J. Chem. Phys. 2009, 130 (5), 054106. (25) Knizia, G.; Adler, T. B.; Werner, H.-J. J. Chem. Phys. 2009, 130 (5), 054104.
(26) Werner, H.; Knowles, P. J.; Knizia, G.; Manby, F. R.; Schütz, M. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2 (2), 242–253.
(27) Eckart, C. Phys. Rev. 1930, 35 (11), 1303–1309.
(28) Sharma, S.; Raman, S.; Green, W. H. J. Phys. Chem. A 2010, 114 (18), 5689–5701.
(29) Zheng, J.; Mielke, S. L.; Clarkson, K. L.; Truhlar, D. G. Comput. Phys. Commun. 2012, 183 (8), 1803–1812.
(30) Reinisch, G.; Miki, K.; Vignoles, G. L.; Wong, B. M.; Simmons, C. S. J. Chem. Theory Comput.
2012, 8 (8), 2713–2724.
(31) Zhang, P.; Klippenstein, S. J.; Law, C. K. J. Phys. Chem. A 2013, 117 (9), 1890–1906. (32) Xu, X.; Papajak, E.; Zheng, J.; Truhlar, D. G. Phys. Chem. Chem. Phys. 2012, 14 (12), 4204–
4216.
(33) Cassanelli, P.; Johnson, D.; Cox, R. A. Phys. Chem. Chem. Phys. 2005, 7 (21), 3702–3710. (34) Cox, R. A.; Patrick, K. F.; Chant, S. A. Environ. Sci. Technol. 1981, 15 (5), 587–592. (35) Heiss, A.; Sahetchian, K. Int. J. Chem. Kinet. 1996, 28 (7), 531–544.
(36) Montgomery, J. A.; Frisch, M. J.; Ochterski, J. W.; Petersson, G. A. J. Chem. Phys. 1999, 110 (6), 2822–2827.
(37) Vandeputte, A. G.; Sabbe, M. K.; Reyniers, M.-F.; Van Speybroeck, V.; Waroquier, M.; Marin, G. B. J. Phys. Chem. A 2007, 111 (46), 11771–11786.
(38) Ochterski, J. W.; Petersson, G. A.; Montgomery Jr, J. A. J. Chem. Phys. 1996, 104 (7), 2598– 2619.
(39) Tsang, W. In Energetics of organic free radicals; Springer, 1996; pp 22–58.
(40) Sarathy, S. M.; Vranckx, S.; Yasunaga, K.; Mehl, M.; Oßwald, P.; Metcalfe, W. K.; Westbrook, C. K.; Pitz, W. J.; Kohse-Höinghaus, K.; Fernandes, R. X. Combust. Flame 2012, 159 (6), 2028– 2055.
(41) Yasunaga, K.; Mikajiri, T.; Sarathy, S. M.; Koike, T.; Gillespie, F.; Nagy, T.; Simmie, J. M.; Curran, H. J. Combust. Flame 2012, 159 (6), 2009–2027.