Characterization of Structure and Dynamics of Membrane Proteins from Solid-State NMR
by Byungsu Kwon B. S. Chemistry
Seoul National University of Science and Technology, 2008 Seoul, South Korea
Submitted to the Department of Chemistry
in Partial Fulfilment of the Requirements for the Degree of Doctor of Philosophy
in Physical Chemistry at the
MASSACHUSETTS INSTITUTE OF TECHNOLOGY
June 2018
2018 Massachusetts Institute of Technology. All rights reserved
Signature redacted
Signature of Author:Department of Chemistry Fabruary 9, 2018
Certified by:
Signature redacted
Mei Hong Professor of Chemistry Thesis Supervisor
Accepted by:
Signature redacted
Robert W. Field Haslam and Dewey Professor of Chemistry Chairman, Departmental Committee on Graduate Students MASSACHUSETTS INSTITUTE
OF TECHNOLOGY
This doctoral thesis has been examined by a committee of professors from the Department of Chemistry as follows:
Professor Robert W. Field
Professor Mei Hong
Signature redacted
L Thesis Committee Chair
Signature redacted
Thesis Supervisor
Signature redacted
Professor Alex K. ShalekThesis Committee Member
I' ,.
Characterization of Structure and Dynamics of Membrane Proteins from Solid-State NMR
by Byungsu Kwon
Submitted to the Department of Chemistry on Fabruary 9, 2018 in Partial Fulfilment of the Requirements for the Degree of Doctor of Philosophy in Physical Chemistry Abstract
Solid-state nuclear magnetic resonance (ssNMR) spectroscopy is an essential tool for elucidating the structure, dynamics, and function of biomolecules. ssNMR is capable of studying membrane proteins in near-native lipid bilayers and is thus preferred over other biophysical techniques for characterizing the structure and dynamics of membrane proteins. This thesis primarily focuses on the study of the following membrane proteins: 1) the N-terminal ectodomain and C-terminal cytoplasmic domain of influenza A virus M2 and 2) HIV-1 glycoprotein gp4l membrane-proximal external region and transmembrane domain (MPER-TMD) in a near native membrane environment.
The cytoplasmic domain of M2 is necessary for membrane scission and virus shedding. The M2(22-71) construct shows random-coil chemical shifts, large motional amplitudes, and a membrane surface-bound location with close proximity to water, indicating the post-amphipathic helix (AH) cytoplasmic domain is a dynamic random coil near the membrane surface. The influenza M2 ectodomain contains highly conserved epitopes but its structure is largely unknown. The M2(1-49) construct containing both the ectodomain and transmembrane domain exhibits an entirely unstructured ectodomain with a motional gradient in which the motion is slower for residues near the TM domain, which attributed to the formation of a tighter helical bundle in the presence of drug that should cause the more tightened C-terminal ectodomain, thereby slowing its local motions.
HIV-1 virus gp4l is directly involved in virus-cell membrane fusion. However, the structural topologies of the gp4l MPER-TMD are still controversial and the biologically-relevant intrinsic conformational state of MPER has not yet been determined. In order to obtain near native structural information of gp4l, we have studied gp41 (665-704) and found a primarily a-helical conformation, membrane-anchored trimeric TMD and water-exposed membrane surface-bound MPER. Intra- and intermolecular distances measured using "C-19F REDOR and 19F-19F CODEX revealed that MPER-TMD has a significant kink between MPER and TMD, which has aided a deeper understanding of the HIV virus entry mechanism and the design of vaccines.
Thesis Supervisor: Mei Hong Title: Professor of Chemistry
Acknowledgments
I had never thought about studying abroad and completing a doctoral program in MIT when
I was in Korea. I thank God more than I can say and more than I can write. It is not because I got
a good university degree but He has intervened in my life and come with me so far and I believe
that He will be with me for the rest of my life.
I sincerely appreciate my advisor, Mei, who has shaped me as a scientist during my Ph.D.
journey. I could not have been able to conduct this research without her consistent support. Her
solid knowledge about solid-state NMR and an insightful point of view on my research has thereby
aided me accomplish each of my project smoothly. I especially thank her for her understanding
and patience when I was faced with trouble during the research. She always provides me with
waits me with words of warm encouragement and helpful guidance until I complete each project.
I would also like to express my gratitude to Prof. Klaus who gave insightful NMR lectures and
valuable comments on my research during our weekly group meeting in Ames.
I would like to thank Prof. Robert W. Field and Prof. Alex K. Shalek for being my thesis
committee members. I thank Bob for the helpful discussions about my M2 research and
encouragement during the annual meeting. It was also great to meet Alex before my thesis defense
to talk about my research and future plan after graduation. I'd like to also thank Prof. Alan Waring
at the University of California, Irvine, who is our great collaborator for the gp4l project, for
helping prepare samples and advising on MD simulations.
I would also like to thank my fellow group members who have been awesome collaborators
and friends during my graduate studies in Ames and Cambridge. Sarah, Yongchao, and Yuan were
great senior lab members and mentors in my first years at Ames. A virtuous Tuo, a pleasant Aaron,
a cheerful Keith, and a reticent but kind Jon, who have all worked with me and have been friends
and teachers over the past 6 years. Thanks also to Daniel and Paul who introduced me to organic
chemistry, thus allowing me to finally synthesize membrane proteins and understand the
mechanism of every step of the synthesis. I would especially like to thank Hongwei and his wife
Yu and Tuo's better half, Weiwei, who have been my sincere friends through the tough times.
Many thanks to Myungwoon who has been my friend, teacher, and mentor for helping me go
through real life. I am also very grateful for the time spent with and help from our EHS instructor
Matt, Shiva who is the only the guy to call me "handsome man", always kind and ever-positiveMarty, sensitive but with pure heart Pyae, and reticent and good-natured Alex. It was also enjoyable to have Chandan, Matthias, Chloe, and Aurelio in 150 Albany Street.
My time in Ames would not have been enjoyable without the companionship of my many friends from school and Korean Grace Church. The endless support and prayer from KGC members allowed me to stand right next to Jesus. The Wednesday Prayer meeting was a crucial stimulus for my weary life. Many thanks to all of them. I would also thank to my English conversation and academic life mentor, Prof. Bob, who was a sincere friend for international students to listen to their distress and encourage them to overcome the long and difficult journey to receiving a Ph.D.
My second journey in Cambridge was really exciting due not only to MIT but also to First Korean Church in Cambridge where I learned more about Jesus and met honorable disciples of Christ. Many thanks to Supernova Club, Bible Couple, many group leaders and members, and especially Hong & Jung. I learned from the Church how to obey and live as a Christian and I realized that it is really difficult to lay me down and follow the will of God but I know this is the way to breathe and love Him and people.
Finally, special thanks to my Korean advisor, Lee who introduced me to solid-state NMR and allowed me to taste college life in the U.S. with full financial and moral support. Dr. Kim and Kang who were my spiritual mentors during my college life, I really appreciate their wholehearted counseling along with material and spiritual support. I could never have imagined to be where I am at today without encouragement from my friends and family. Unconditional and endless love from my family makes me a better person through their sacrifice and dedication. I am sure I would not be able to understand how parents can give everything to their kids before being a dad.
Table of Contents
Chapter 1. Solid-State Nuclear Magnetic Resonance Spectroscopy and Membrane Proteins
1.1 Applications and Advantages of Solid-State NMR 10
1.2 Basic Concepts of NMR 10
1.3 Fundamental NMR Interactions and Their Hamiltonians 11
1.4 Anisotropic Nuclear Interactions and Magic Angle Spinning (MAS) 13
1.5 One-Dimensional MAS NMR Spectra 15
1.6 Motional Amplitude of Lipids and Membrane Proteins 17
1.7 Conformation of Membrane-Bound Proteins 19
1.8 Depth of Insertion of Membrane-Bound Proteins in Lipid Bilayers 22
1.9 Heteronuclear and Homonuclear Distance Measurements 24
1.10 References 29
Chapter 2. Solid-Phase Peptide Synthesis and Native Chemical Ligation for ssNMR
2.1 Introduction 32
2.2 Solid-Phase Peptide Synthesis for Site-Specifically Isotope Labeled Proteins 33 2.3 Native Chemical Ligation for Membrane Proteins 37
2.4 Thesis Organization 41
2.5 Copyright Permissions 42
2.6 References 43
Chapter 3. A 2H Solid-State NMR Study of Lipid Clustering by Cationic Antimicrobial and Cell-Penetrating Peptides in Model Bacterial Membranes
3.1 Abstract 44
3.2 Introduction 45
3.3 Material and Methods 47
3.4 Results 49
3.5 Discussion 57
3.6 Acknowledgment 63
3.7 References 64
Chapter 4. Chemical Ligation of the Influenza M2 Protein for Solid-State NMR Characterization of the Cytoplasmic Domain
4.1 Abstract 69
4.2 Introduction 70
4.3 Results 72
4.4 Discussion 83
4.5 Material and Methods 85
4.6 Acknowledgment 88
4.7 References 89
Chapter 5. The Influenza M2 Ectodomain Regulates the Conformational Equilibria of the Transmembrane Proton Channel: Insights from Solid-State NMR
5.1 Abstract 91
5.2 Introduction 92
5.3 Material and Methods 93
5.4 Results 97
5.5 Discussion 105
5.6 Acknowledgment 109
5.7 References 110
Chapter 6. Membrane-Bound Structural Topology and Oligomeric Assembly of the HIV gp41 MPER and Transmembrane Domain in Lipid Bilayers from Solid-State NMR
6.1 Abstract 113
6.2 Introduction 114
6.3 Material and Methods 116
6.4 Results 121
6.5 Discussion 133
6.6 Acknowledgement 135
6.7 References 136
Chapter 1
Solid-State Nuclear Magnetic Resonance Spectroscopy and Membrane Proteins
1.1 Applications and Advantages of Solid-State NMR
Nuclear Magnetic Resonance (NMR) spectroscopy is a robust technique for characterizing the structure, dynamics and components of chemical species including disease-related peptides or proteins, small molecules for drug discovery, nutritional and genetically modified foods and cosmetic materials, semicrystalline polymers and nanocomposites for energy materials at atomic resolution. Its application to disease-related membrane protein structure elucidation has many advantages over X-ray crystallography because it is difficult to obtain well-ordered crystals, which are prerequisite for X-ray structure determination. Structural analysis of membrane proteins by solution NMR bypasses the crystal sample preparation, however, it requires membrane proteins that experience the fast-tumbling in solution to achieve the narrow linewidths necessary for assignment in highly labelled proteins. Sample preparation for solution NMR studies of membrane proteins can be achieved by dispersing them in lipid micelles, mixing with bicelles, or encompassing them within amphipathic lipoprotein belts to form nanodiscs. However, all of these solution NMR sample preparations do not result in near native environments. The rise of cryogenic electron microscopy (cryo-EM), shows that cryo-EM is capable of determining membrane-embedded protein structures below 3.4 A atomic resolution (1) in near native membrane systems, but protein size limitations of current cryo-EM techniques hamper structure and dynamics elucidation of membrane proteins in the range of 10 to 40 kDa (2). In addition, the cryogenic temperature required for cryo-EM may perturb the protein structure (3, 4). Thus, solid-state NMR (ssNMR) is an incomparable technique to gain insights into the conformation, mobility, membrane insertion depth, oligomerization, and intermolecular distances of membrane proteins in near native membranes at an atomic level.
1.2 Basic Concepts of NMR
Atomic nuclei have a unique feature called spin, which is characterized by a spin quantum number, I (I =
o,!,
1,..., 6) and associated with magnetic moment, yI = yhI. When nucleiPlank constant (h/2 ), y is the gyromagnetic ratio of a nucleus, and I is the angular momentum
operator. The vector sum of the individual magnetic moments is called bulk magnetization and can be expressed as:
This bulk magnetization depends on both temperature and an external magnetic field Bo, and in the high-temperature limit can be expressed by:
_.oBo
M oc yh
-where, k is the Boltzmann constant and T is the absolute temperature. In quantum mechanical aspects, magnetic moments are randomly oriented in the absence of a magnetic field (M = 0), but tiny excess number of magnetic moments in the lower energy state 1a) will align parallel with respect to Bo in the presence of a magnetic field. This net magnetization experiences a torque (r) in a magnetic field aligned along the z-axis, B = (0,0, BO), and precesses around B at the Larmor
frequency:
coo = -y- Bo
At equilibrium, the magnetization precesses parallel to the external magnetic field. Radiofrequency (rf) pulses (BI field), when applied to a direction perpendicular to Bo and close to the Larmor frequency (wrf = wo) of nuclear spins of interest, move the bulk magnetization away from Bo as
it precesses around B1. Once the magnetization is along the x-y plane, we can detect resonance
frequencies containing valuable structural information of nuclei in molecules.
1.3 Fundamental NMR Interactions and Their Hamiltonians
Zeeman interaction
When nuclei that possess non-zero spins are placed in an external magnetic field (Bo), they split into different energy states, which is called the Zeeman Effect. The Zeeman Effect is the dominant interaction in NMR, and can be expressed as the scalar product between the nuclear magnetic moment (M = yhl) and Bo. The corresponding Hamiltonian is:
where
I,
is the angular momentum operator representing the z component of the angular momentum.Chemical shift interaction
Electron currents in orbitals surrounding the nuclei of interest produce additional local fields (Bcs), which affect the total field that felt by nuclei. The chemical shift interaction arises from perturbations to the external magnetic fields (BO) from local fields resulting from electron currents (Bcs) experienced by the nuclei, which results in different NMR frequencies. For non-spherical electron density distributions around the nuclei, the chemical shift interaction is anisotropic (orientation dependent). A molecule in solution experiences fast isotropic tumbling; the anisotropic chemical shift of the molecule is thus averaged to an isotropic chemical shift (ais).
The chemical shift Hamiltonian can be expressed as (5):
Hcs = yhI - Bcs = yhI - aBO = yhBo(Yxcrzab + I b + jzczzb)
Where a is the second rank chemical shift tensor. In most cases, chemical shift interactions are much smaller than the Zeeman interaction, thereby Rcs is truncated by the Zeeman interaction, leaving only spin operators that commute with Tz. The truncated chemical shift Hamiltonian is:
R (0) La - .ULab= WS
s
yh
Bolz
- = hwhz
0Iz -ZzZ
CSZ thus, the combined Zeeman and chemical shift interactions can be expressed as:+ )= -hyB 0(1 - -Lab) -hwo(1 - ZLZab)
where w = wo (1 - Ua b) is the observed precession frequency.
Dipolar interaction
The field independent dipolar interaction between spins I and Ik is represented by (5):
where yo is magnetic constant, yj and Yk are the gyromagnetic ratios of the two spins (i and k), rk is the internuclear distance betweenj and k, and ejk is the unit vector parallel to the vector between
j
and k. The dipolar interactions can be separated as truncated Heteronuclear and Homonuclear
dipolar interactions.
The truncated heteronuclear Hamiltonian is:
o ho
1
(3
cos2 0 - 1)H0s = is
- h
47r1!jk r y k3 2 2 -7* IZ SZzand the truncated homonuclear Hamiltonian is (6):
4r rik3
2
The spin part of 2izgz and 31Jz - P1 -[k terms from the heteronuclear and homonuclear
Hamiltonians, respectively, is affected by rf pulses, while the spatial parts are governed by magic
angle spinning and sample alignments.
Quadrupolar interaction
Nuclei with spin quantum number I > 1 possess electric quadrupole moments,
Q,
and
couple with surrounding electric field gradients. The quadrupolar interaction can be described as:
eQ
,
.
H =I - V - I
Q 21(21 - 1)h
where e is the elementary charge, V is electric field gradient tensor. The first-order truncated
Hamiltonian of this interaction is (5):
f =1
-1
e Q
-~a VZZ(31z-I (I + 1) i= (31z
- I(I -+ 1)Q 2 2I(2 - 1)h 6
where
Vja -vPAs(3co
- 1- 77 sin
20 cos 2p) and o>( 1).
Hetprincipal
ZZ 2 Q 21(2I-l)h
Hr h
value
VZZscan also be expressed as eq which is related to the electric field gradient tensor.
1.4 Anisotropic Nuclear Interactions and Magic Angle Spinning
Orientation-dependent nuclear interactions, such as chemical shift anisotropy (CSA) and
dipolar interactions, lead to ssNMR spectra that are more complicated and less well-resolved
compared to solution NMR spectra. Anisotropic interactions broaden linewidths of the NMR
signals, resulting in ambiguous chemical shift assignment and spectral crowding. These
anisotropic nuclear interactions are averaged to zero from isotropic tumbling in solution NMR.
Techniques to overcome the drawbacks associated with broadened linewidths from dipolar
interactions are the basis for ssNMR; magic angle spinning (MAS) is the fundamental method used
to obtain the high resolution ssNMR spectra by averaging the anisotropic interactions by sample
rotation at the magic angle, thus averaging out many anisotropic interactions.
As mentioned before, the chemical shift frequency is proportional to the zz-element of the
chemical shift tensor:
ocs = -yBcs,z = -azab W
and is defined as:
OLAb WLAb LAb
B
LAS(xx xy Wxz
0=(b7b)A=(Tij)
t0C= Lab
=
01
LAbo4~" wJLbTT--
PS=(T-&LAb WLAb LAb (1)
where bo = B0 is a unit vector along B0 and bSo
Since only the symmetric part of the CS tensor (diagonal) contributes to the frequency, we
obtain chemical shift frequency in the principal axis system (PAS):
I$4b 0 0
4cosP
sin 6OCS
=(cos q sin 0, sin q sin 0, cos 0)
0
Oy^0)
sin 0 sin )
0 0 (J-zZb COS 0
= )PAS COS2
p
sin2 A+ yPAs sin2
p
sin
20
+ WS COS2 0This equation can be modified in terms of the isotropic shift (aso), anisotropy parameter
(S),
and
asymmetry parameter (qj) as (5):S
a(0, ;6,n)cs =-(3 cos
2 0-1 -ijsin
26 cos 2p)+ ot
0s
2
S
U(0,
f;
6, T)csA = -(3 cos28 - 1 -
17
sin
2 0 cos 2q)2
1
= g (PAS + P + PAS)
PAS - PAS
1=P AS
where 0 is the angle between principal z-axis and B0 and 4 is the azimuthal angle in the xy-plane (Fig. 1.1). The chemical shift anisotropy can be averaged out by spinning the sample at the magic angle (7), 6
MAS = 54.74', where the second-order Legendre polynomial is zero (3 cos 2 6 MAS
-1
=
0) and the asymmetry parameter
(q)
is averaged out to zero due to the uniaxial rotation of the
molecules around the rotor axis (unique principal axis), resulting in the isotropic chemical shift being the dominant interaction (wi,) (Fig. 1.1c).(a) molecule-fixed PAS
(b) rotor-fixed frame
(c) MAS
Z PAS Z Roto O MAS
B
0
B
Z Rotor54.7'
_______IMAS
i
)YPAS y Rotor X PAS X Rotor r tFigure 1.1 (a) Molecule-fixed principal axis system (PAS). (b) Rotor-fixed frame, MAS and molecular
motions render the orientation of the molecule relative to the B0 time-dependent. (c) Rotating molecules in a rotor at 54.74' (6MAS =
cos-1
J1/3
= 54.740) with respect to Bo where the molecules experience uniaxial rotation (-q = 0).1.5 One-Dimensional Magic Angle Spinning NMR Spectra
The fundamental structural and dynamic information of membrane proteins can be
investigated by simple one-dimensional (ID) NMR experiments. Naturally abundant nuclei with
high gyromagnetic ratio, y, such as 'H and
9F can be directly polarized by applying
900
pulses
thus generating high resolution spectra with fast MAS. Nuclei with low-y, such as 1
3C and "N,
possess long relaxation times and relatively low polarization at equilibrium compared to high-y
nuclei. Therefore, direct-polarization (DP) is relatively time-consuming to obtain spectra with
sufficient signal-to-noise for low-y nuclei. Instead of DP, the cross-polarization (CP) technique
can be used to reduce the time required to produce a spectrum for low-y nuclei with sufficient
signal-to-noise. Magnetization transfer from high-y ' H to nearby low-y nuclei, such as 1
3C and "N,
by rf irradiation is accomplished when the Hartmann-Hahn matching condition
(W13C - W1HOr)
is met. CP can not be used as a quantitative technique like DP can because the magnetization
transfer is different for every nucleus, based on the distance and relative motion between 'H and
low-y nuclei in molecules. CP preferentially detects rigid molecules, while DP detects all nuclei
uniformly. For this reason, CP is useful for identifying rigid portions of molecules.
Although 1D DP and CP allow us to examine overall conformation and dynamics of
membrane proteins of interest, the
13C natural abundance lipid signals overlap with
3C protein
signals, thus hampering unambiguous chemical shift assignments. In order to avoid the
3C signal
overlap of the proteins from natural abundance lipid 3C signals, a
13C- 3C double-quantum filter
(DQF) can be applied. The DQF suppresses signals from isolated spins, thus only leaving signals
from 3C nuclei with a neighboring 3C spin partner, which are not present in natural abundancelipids.
In chapter 4 and 5, this double-quantum technique is utilized in 2D J-1NADEQUATE
(8)
and double-quantum filtered (DQF) DIPSHIFT (9,
10)
experiments to obtain
13C-13C homonuclear
DQ coherences from directly bonded
3C spins and to selectively remove the natural abundance
lipid 13C resonances in the spectrum, respectively. For 13C-3C double-quantum recoupling, theSPC-5 recoupling sequence, which includes rotor-synchronized and spin-lock radiofrequency
irradiation, was employed. In general, C7 symmetry based DQ recoupling sequences require
13Crf field strengths to be matched to be sevenfold higher than the spinning frequency (7 x wr/21).
To avoid CP leakage, approximately 21 times the MAS frequency (3
x7
X Wr/2W)of 'H rf
decoupling strength is necessary to yield satisfactory decoupling. In contrast to C7 symmetry
recoupling sequences, SPC-5 reduced recoupling condition from seven- to fivefold
(11),
which
makes it possible to utilize the SPC-5 sequence at higher spinning frequencies without line
broadening and low sensitivity due to insufficient
1H decoupling strength.
(a) (b) (C)
n/2 n/2
SDD
H
LD
H
DD
7t/2 n/2 DQ filter
Figure 1.2 Pulse sequence schemes of ID NMR experiments. (a) Direct polarization (DP). (b) Cross polarization (CP). (c) Double-quantum filtered (DQF) CP.
1.6 Motional Amplitudes of Lipids and Membrane Proteins
Characterization of dynamic motions in biomolecules is essential to understanding their physical properties and mechanism for function. Here we introduce 2H quadrupolar coupling and 13C-1H dipolar coupling experiments to measure motional amplitudes of molecules in the fast motional regimes (r, < 1 s). As we recall the chemical shift frequency in the PAS, the chemical
shift frequency of a sample aligned parallel to BO can be expressed as (12):
S
a(6,
4);
6, 1)cs = Woo-aigned = S (3 COS2 0 - 1 - 17 sin2 6 cos 2q5) + wisoHowever, in the case of a mobile, unoriented proteoliposome sample, the motional axis is normally not parallel to B0 so, the resonance frequencies are entirely dependent on the motionally averaged anisotropy parameter, 9, which is identical to the chemical shift anisotropy frequency.
S
( = (o' -atigned - (Jso = 2 (3 cos2 6 - 1 - r7 sin2 0 cos 25)
In the presence of uniaxial rotation (ij = 0), the molecular orientation relative to the bilayer normal, expressed by 6, can be determined. The ratio of 9 to the rigid limit, 6, can be characterized with the order parameter, S, representing the motional amplitude of a bond of interest, in dipolar and quadrupolar interactions:
S (3 cos2 6 - 1)
S 2
2
H quadrupolar echo spectra
To determine the motional amplitudes of lipids, 2
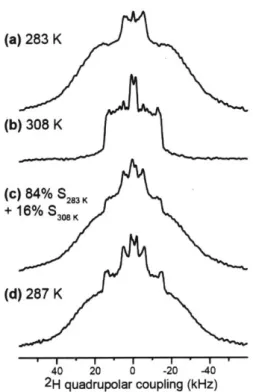
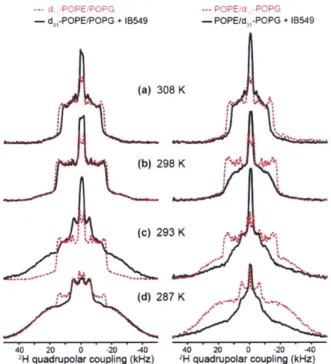
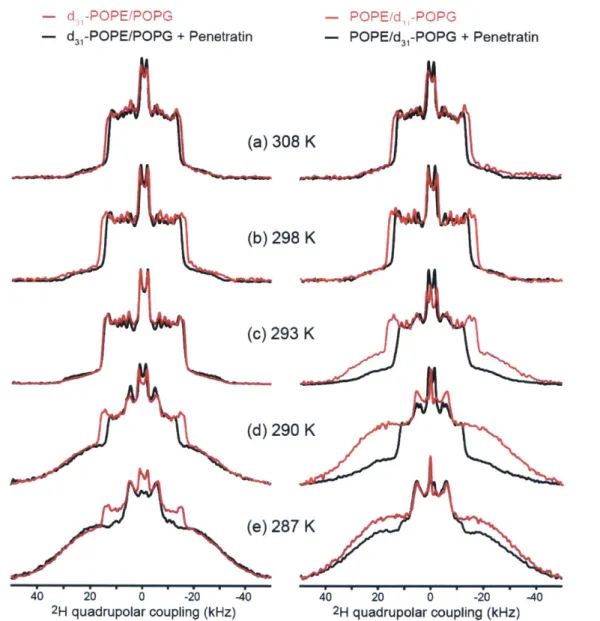
H quadrupolar couplings of deuterated lipid acyl chains can be measured (13, 14). 2H quadrupolar couplings reflect the time-averaged orientations of the C-H bonds in lipids with respect to the bilayer normal. Disordered lipids with large-amplitude motions possess small 2H quadrupolar couplings, indicating small C-H order parameters (S). The rigid limit 2H quadrupolar splitting of CD2 groups is about 127.5 kHz. This
quadrupolar splitting is much larger than 13C-1H dipolar rigid-limit coupling of 22.7 kHz. Figure 1.3a shows the 2H quadrupolar echo pulse sequence using echo delays of-40 ps (15).
90, 9O'
X 2
Figure 1.3 2H Qudrupolar echo pulse sequence.
Dipolar chemical shift correlation spectra
The measurement of heteronuclear dipolar couplings is a complementary approach used to probe motional amplitudes of membrane proteins with site-resolution; site-resolution is difficult to obtain for 2
H quadrupolar couplings. We have utilized dipolar chemical shift (DIPSHIFT) correlation experiments (16, 17) to detect dipolar couplings in the indirect dimension (W2) and chemical shifts in the direct dimension (wj). To measure the motionally averaged dipolar couplings (6D), proton magnetization is transferred to 13C through CP, and allowed to evolve under
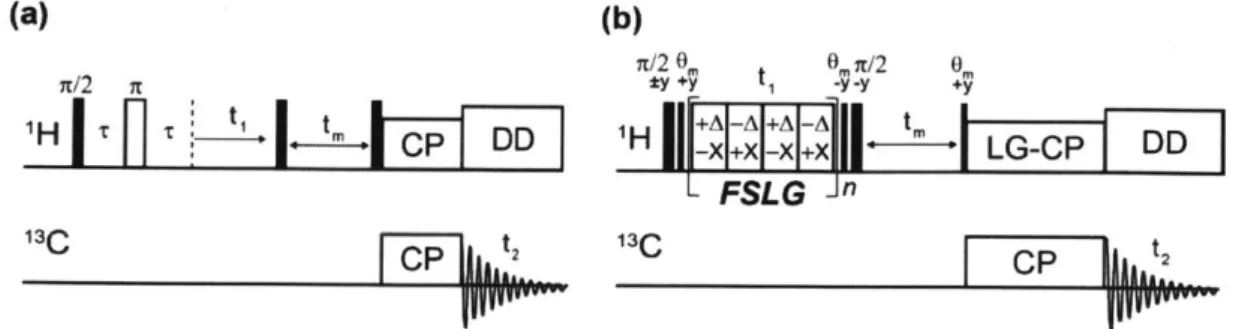
1H-1H homonuclear decoupling (MREV-8 (18) or FSLG (19)) to make certain that only 'H nuclei
directly bonded to 13C nuclei contribute to the measured dipolar couplings of 'H-13C. A '3C 1800
pulse is applied after a variable delay to refocus the dipolar interaction to a varying degree, giving rise to the 13C-'H dipolar dephasing curves and 13C isotropic chemical shifts in the indirect and
direct dimensions, respectively. To obtain the averaged dipolar coupling (6D), the apparent coupling extracted from the spectra needs to be divided by the scaling factor associated with the homonuclear decoupling sequence used. The scaling factors for MREV-8 and FSLG sequences are 0.47 and 0.577, respectively. The optimal homonuclear decoupling sequence depends on the spinning frequency used: MREV-8 or FSLG when o(r/2W < 7kHz or DUMBO when Or/21 > 7kHz. The dipolar order parameter (SCH) for a C-H bond is the ratio of the measured averaged dipolar coupling (6) to the rigid limt value (SD) and calculated by the following expression:
S1
SCH = - = -
(3
COS2 & -1)6D 2
Here 0 is the angle between the motional axis (bilayer normal) and C-H bond of interest. Spectral overlap of protein 13C resonances with natural abundance 13
C lipids signals makes site-resolved dipolar coupling measurements difficult or impossible for many nuclei. The DIPSHIFT experiment including Double-quantum filtered (DQF) is a unique and simple method to remove
the lipids signals by suppressing the single-quantum
13C coherences and only detecting
13C nuclei
with
3C-'
3C double quantum spin pairs.
(a) (b)
x/2 n/2
IH
I
p
iarDecuplig
1H
DDa
n n/2 DQ filter
CCP
3C
CP
exc
rec
I 2Figure 1.4
'3C-'H DIPSHIFT pulse sequences. (a) Single DIPSHIFT (b) Double-quantum filtered (DQF)
DIPSHIFT.
1.7 Conformation of Membrane-Bound Proteins
Homonuclear and heteronuclear correlation spectra
The starting point for determining membrane protein structures is a chemical shift analysis.
The chemical shifts of protein backbone nuclei such as N, Ca, C, and C' are sensitive to their
secondary structure, drug binding, and protein-ligand interaction. Therefore, assigning chemical
shifts is a crucial first step for structure analysis using NMR. Although 1 D NMR experiments are
helpful to delineate overall conformation and dynamics of membrane proteins, severe spectral
crowding of uniformly
13C, 1
5N-labeled membrane proteins with a molecular weight of 5
-10 kDa,
limits detailed residue-specific structure and dynamics information. Hence, multi-dimensional
NMR spectroscopy experiments are indispensable for characterizing even relatively small proteins.
"C-"C correlation spectra
Several 2D homonuclear 1
3C-
13C correlation techniques have been developed to obtain
well-resolved
13C resonances, providing secondary structure information on membrane proteins.
The homonuclear correlation experiments utilized in my research: proton driven spin diffusion
(PDSD), CP and DP versions of dipolar-assisted rotational resonance (DARR)
(20), water-edited
DARR
(21, 22),
and DP J-coupling-mediated incredible natural abundance double quantum
transfer exchange (J-INADEQUATE) will be briefly explained.
CP and DP
13C-
13C DARR experiments are modified versions of PDSD, where the
13C-
1H
dipolar coupling is recoupled by introducing continuous wave (cw) rf irradiation
(o,/27r)
on the
1
H
channel during the
13C-
13C mixing. The
13C magnetization evolves during tj, and is flipped
back to the z-direction by a 900 pulse for
13C-
13C spin diffusion under
13C-
1H dipolar recoupling
and then flipped back down to the transverse plane by a second 90' pulse for
13C detection. These
spin diffusion based '
3C-'
3C correlation techniques allow us to distinguish intra- and inter-residue
(or inter-molecular) correlations in the spectra depending on the length of the mixing time.
Relatively short mixing times
(-
50 ms) predominantly exhibit intra-residue cross peaks in the
spectrum, while longer mixing times of> 100 ms show inter-residue cross peaks as well.
(a) (b) 'H Dp D p 1H DDMlf Dipolar De ing 7/2 7/2 I1 13C 'T THT t2 1 CP (C) 1H Dipolar Decoupling n/2 n 13C
- n i.
t
(d) 7t/2 nIH
DD DD -7r/213C
r
ti t.2L
Figure 1.5 Pulse sequences used for '3C-'3
C correlation spectra. (a) DP DARR, (b) CP DARR, (c) DP J-INADEQUATE and (d) Water-edited DARR.
CP DARR preferentially detects rigid residues, while DP DARR detects all residues. Therefore, signals that are not present in CP DARR, but are present in DP DARR spectra must come from mobile residues. We use this technique to confirm the high mobility of the extra-cellular domain of M2 in chapter 4. In addition, the DP J-coupling-mediated INADEQUATE also preferentially observes the mobile species and gives rise to through-bond correlations. Compared with single-quantum "C-"C correlation spectra, double-quantum 13C-13
C spectra possess only signals from neighboring 13C-13
C spin pairs. In these asymmetric diagonal-free 2D double-quantum spectra, the sum of chemical shifts of coupled spins is in the indirect dimension (wj) and isotropic chemical shifts of single spins is in the direct dimension (W2). The pulse sequence for
coherence is excited by the fivefold symmetric SPC-5 recoupling sequence. The evolution of the
DQ coherence during t1 precedes the reconversion into the observable single-quantum coherence for detection.
Membrane proteins, such as influenza virus M2 and HIV- 1 gp4 1, contain distinct domains. The N-terminal ectodomain and C-terminal cytoplasmic tail domain of M2, and membrane-proximal external region (MPER) of gp41 are significant water-exposed domains that are anchored to their associated transmembrane domains (TM). The 2D 13C-13C DARR experiment is a useful
technique to obtain intra- and inter-residue correlations but it does not reveal any detailed water-protein contact information. Therefore, we introduced a 'H T2 filter step into the DARR pulse
sequence to preferentially select 'H magnetization of water, which has a relatively long T2
compared to that of proteins. Although lipids and water have very similar T2, transferring
magnetization from lipids to protein is very difficult, so it has negligible effects on water edited DARR spectra. The preferentially selected water magnetization then transfers to neighboring domains of membrane proteins via 'H-'H spin diffusion. Following 1H-1H spin diffusion, a
"C-3
C DARR mixing period is included before detection. Thus, in the resulting spectrum, only 13C signals of water-exposed residues of the proteins remain.
'sN-'
3C correlation spectra
Combining heteronuclear correlation with homonuclear correlation spectroscopy provides more detailed conformational and dynamic information about secondary structure and drug binding of membrane proteins than can be obtained through only using homonuclear correlation experiments. Although 15N nuclei have a low natural abundance and gyromagnetic ratio (y), the extreme
sensitivity of its chemical shifts to local conformation changes (e.g. drug binding) make spectra containing 5N valuable. Another hurdle to overcome for 15N spins is their narrow chemical shifts
range, 117 - 124 ppm in a-helical structure, except for glycine (-107 ppm). The heteronuclear
13C-'N correlation technique is essential for separating the "N chemical shifts into well resolved
peaks. In general, it is restricted to detect one or two bond correlation due to their weak 3C-'N
dipolar couplings but longer correlations using a 13C-13C mixing period after the "C-"N transfer
is also possible. After CP, the first series of a pulses generate anti-phase magnetization proportional to Iy,13CSz,15N frOm the initial in-phase coherence, Ix,13C
(23, 24).
A pair ofir/2
pulses on the '3C and 15coherence of "C to the anti-phase magnetization of "N (proportional
to IZ,13cSX,15N),which
evolves under the
15Nchemical shift. After t
1evolution, the second pair of 900 pulses converts the
anti-phase coherence of
5N to that of
"C (Iy,13cSz,15N) wherethe two
15Nand
13C 900pulses are
separated by -r to compensate for the rotor desynchronization resulted from the finite pulse
lengths, and then reconverts the coherence to observable in-phase Ix,13C magnetization by the
second r-pulse train before
13C detection during t
2.
1Hn/2
oP
Dipolar Decoupling
13
C
U
t
Ia
t2 15N
Rotor 0 1 2 3 4 5 6 7 8Period i Tmix - mix
Figure 1.6 2D "3C-"N HETCOR pulse sequence for a REDOR based 13
C-"N coherence transfer.
1.8 Depth of Insertion of Membrane-Bound Proteins in Lipid Bilayers
13C detected 1H spin diffusion
Determining the insertion depth of membrane proteins in the lipid bilayer is important for
understanding lipid-protein interactions. To do this, a variety of biophysical methods have been
utilized such as neutron diffraction
(25, 26),
fluorescence spectroscopy
(27, 28),
and electron
paramagnetic resonance (EPR) spectroscopy
(29).
NMR spectroscopy is a versatile tool to explore
the membrane immersion depth of proteins without perturbing the conformation of lipids and
membrane proteins. Solution and solid-state NMR spectroscopy using water-soluble or
lipid-soluble paramagnetic probes have been widely used to measure membrane immersion depth of
specific residues in membrane proteins. Membrane-embedded residues show reduced peak
intensities while increasing concentration of the lipid-soluble paramagnetic probes due to
paramagnetic relaxation enhancement (PRE) (30, 31). Another solid-sate NMR technique to
investigate the immersion depth of membrane proteins with semi-quantitative distance information
is a
13C-detected
1H spin diffusion experiment. In my thesis, two different types of the
13C-detected
'H spin diffusion experiment: liquid-crystalline (LC) phase (32-34) and gel-phase (35) 'H spin
diffusion heteronuclear correlation spectroscopy.
The first ssNMR approach is the LC-phase
13C detected 'H spin diffusion, which is shown
in Fig. 1.7a. The initial proton transverse magnetization is created by of r/2 pulse on
1H channel
and rigid 'H magnetization decays quickly during a 'H
T2filter, thus selecting for mobile
magnetization. In general, the magnetization from rigid proteins are removed in the
liquid-crystalline membrane system. The selected 'H chemical shifts are evolved during t
1and and
flipped to z-direction by
900
pulse for 'H-'H spin diffusion from magnetization source (e.g. 'H of
water and lipids) to magnetization sink (e.g. 'H of proteins) during mixing time (t). By the last
900
pulse, the 'H magnetization is flipped back to xy-plane and transferred by CP to
13C for
detection. The second approach is the gel-phase 'H spin diffusion technique at the temperature
below the phase transition temperature of lipids. If the membrane proteins experience
intermediate-time scale motions in the LC phase membrane around lipids phase transition
temperature, spectral line broadening and signal loss occur due to insufficient 'H decoupling and
polarization transfer. However, membrane proteins exhibit high
3C signals at low temperatures
below the phase transition temperature, thus the use of the gel-phase experiment becomes apparent.
The fact that uniaxially-rotating mobile lipids and proteins undergoing rotational diffusion under
moderately fast spinning frequencies ensures high-resolution 'H spectra, but
1H-'H homonuclear
dipolar decoupling is required to obtain high-resolution 'H spectra in the gel-phase experiment due
to the immobile proteins and lipids at the low temperature. Figure 1.7b represents gel-phase 'H
spin diffusion pulse sequence with FSLG 'H homonuclear decoupling sequence during the t
1evolution period.
(a)
(b)
n/2 O 0mn/2 0 n/2 n *y +'y -y -y +'y T -ryty1H
t1 ,
IDD
H
+
I
"
[D
FSLG n
13C 2 1C 2Figure 1.7 2D 11C detected 'H spin diffusion experiment pulse sequences. (a) Liquid-crystalline phase and
1.9 Heteronuclear and Homonuclear Distance Measurements for Membrane Proteins The ssNMR techniques described in the previous section provide the secondary structure and dynamics of membrane proteins but the lack of quantitatively measured intra- and intermolecular distances and oligomeric assembly prevent elucidation of tertiary structure. To make up for this issue, Rotational-Echo Double Resonance (REDOR) (36-38) for long-range heteronuclear distance measurements and Centerband-Only Detection of Exchange (CODEX) (39, 40) experiment for homonuclear intermolecular distance and oligomerization measurements were utilized in my thesis work.
13C-1 9F REDOR
Heteronuclear dipolar recoupling under MAS by a series of TT pulses was introduced to
measure dipolar coupling for obtaining internuclear distance information (Wd/ 2 r oc 1/r3). The truncated heteronuclear dipolar coupling Hamiltonian (5) is:
_(O)
110
1
1
CS
Hif =s 1s(t)- 21zz = -h y1ysT-(3 s 1) - 21zz
where, wis is a spatial part of the Hamiltonian and affected by MAS and sample orientation in molecules and 2192 is a spin part influenced by rf pulses. This is rewritten by Average
Hamiltonian Theory (AHT) analysis:
H' s(t) = &)ns(t) -2IzS
4-hw -It [sin2 f cos 2(y + cort) - N5 sin 2/? cos(y + wrt)] - 21z 4r ris3 2
The y and a are defined as azimuthal and polar angles, respectively, and Or is a MAS frequency. The fact that MAS averages the dipolar coupling out to zero over one rotor period, ftr jis(t)dt = 0. So, the following equation is accomplished:
tr/2s(dt + iRs(t)dt = t Ris(t)dt
= ws(t) - 2!zgzdt = Ot, -21z z
fo ft /2 fo f
where, #tr =
fw
ws(t)dt, and the density operator at the end of one rotor period with a X pulse in the middle of the rotor period such as, tr/2 ~ 7x - tr/2 is:p(tr) = Ix cos 24 + 2IyS, sin 245
and repeat the same sequence with alternate x and y pulses, tr/2 - 'Tx - tr/2 - tr/2 Y - tr/2
is:
p(2tr) = -I, cos2 24) -2IySz cos 24) sin2
4)
+ 2IySz sin2q5
cos 24) - I, sin2 24= -I; =
-p(O)
indicating that the density operator returns to the initial state. Thus, there is no net dipolar phase accumulation, no recoupling at the end of two rotor periods. In order to accumulate the dipolar phase, an application of i-pulses at every half-rotor period is necessary on the dephased-spin channel. However, dipolar recoupling of two spins is accompanied by recoupling of chemical shift anisotropy (CSA) of dephased-spins; a single i pulse in the middle of the REDOR n pulse train is thus used to refocus chemical shift evolution on the detection channel. As long as there are two i pulses per rotor period on the two channels, it is possible to recouple the heteronuclear dipolar coupling, remove dephased-spin CSA recoupling, and refocus detected-spin isotropic chemical shifts. Figure 1.8a shows frequency selective 13C-detected 19F-dephased REDOR (sel-REDOR)
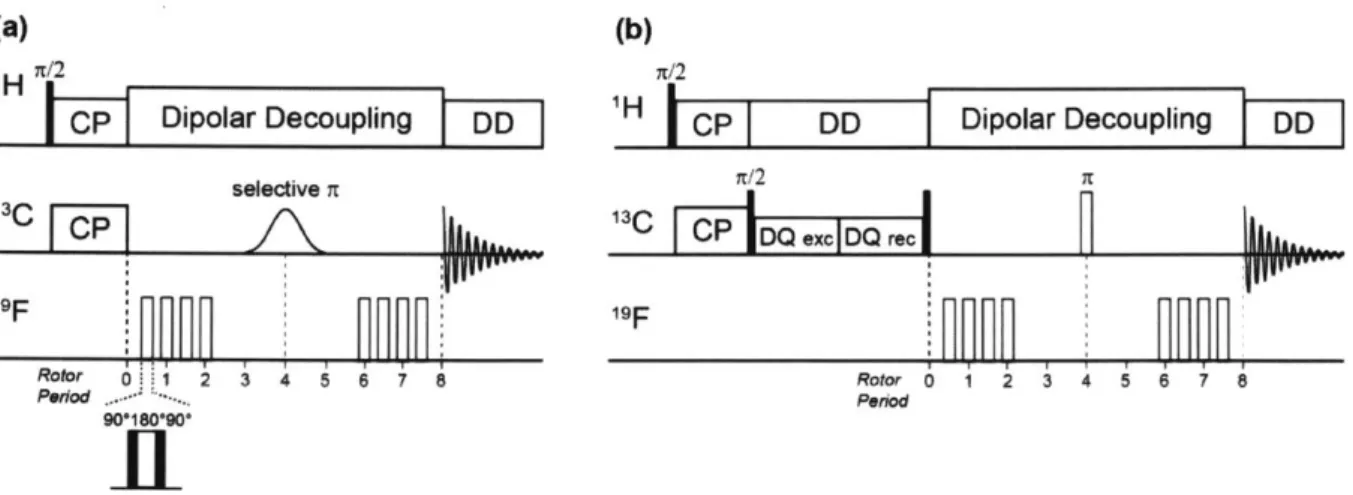
(41) and double-quantum filtered (DQF) REDOR pulse sequences.
(a) (b)
n/2 n/2
H
DipoIar ecoupIi n g
6
H CP
DD
Dipolar Decoupling
DD
selective 7T n/2 X 19F 19F Rotor --1 2 3 4 i 6 7 8 o 0 1 2 3 4 5 6 7 8 Period.Peio 90018090, Figure 1.7 13
C- '9F REDOR pulse sequences. (a) J-decoupled frequency-selective REDOR with soft Gaussian selective t pulse. The n-pulse trains in the '9F-dephased channel to recouple the 13C-1 9F dipolar interaction in the S experiment. Composite n pulses are used on the '9F channel to compensate pulse imperfections. (b) Double-quantum filtered (DQF) REDOR to remove "C resonances from lipids.
The rotor-synchronized soft Gaussian 71 pulse was applied at the center of the I3C channel to remove 13C-13C J-couplings between "C spins that are directly bonded each other, which can interfere with weak 3C-19F dipolar couplings in the range of 30 ~ 60 Hz. The 71 pulses on the 19F
channel can be substituted with composite 9001800900 pulses to reduce the pulse imperfections and enhance the distance accuracy (42). One distinctive feature of REDOR experiment is that this experiment requires a pair of experiments, a control experiment (SO) without dephasing pulses on
19F channel and a dephased experiment (S) with dephasing pulses on the 19F channel to compensate
for dephasing arising from T2 relaxation during the measurement. The normalized dephasing S/So intensities as a function of tm give the 1 3C-19F dipolar couplings where the relevant distance information can be extracted by fitting the experimentally determined S/So intensities to the numerical REDOR simulation curve using SIMPSON program (43).
1 9F-19F CODEX
In conjunction with REDOR experiments, 1H-driven X-spin anisotropic spin diffusion technique, CODEX is essential to give insight into determining atomic-resolution quaternary structure of an oligomeric assembly and intermolecular distances of membrane proteins. CODEX was originally developed for characterizing slow segmental reorientations in amorphous polymers, but we adopted the technique to detect the oligomeric assembly of membrane proteins exclusively arising from the spin diffusion mechanism by decreasing the temperature to ~ 235 K to remove molecular motions.
In the same way as REDOR, CODEX also requires a pair experiments, S and So. For the control experiment (So) the spin diffusion mixing time (tsD) is short while the z-filter delay (tz) is long. For the dephased experiment (S) the tSD and t, are switched each other. The total delayed
time ( ttotai = tSD + tz) need to be constant to compensate for Ti relaxation during the
measurements.
After CP step, a series of rotor-synchronized 7r-pulses spaced every half-rotor period on the X-channel, which is similar to REDOR r-pulse train, was applied to recouple the X-spin chemical shift anisotropy (CSA). Then, the magnetization is returned to the z-direction by 90' pulse for spin diffusion during tSD. If no magnetization exchange (spin diffusion) occurred between chemically equivalent and orientationly inequivalent spins during tSD, there is no CSA frequency perturbation, resuting in the complete refocusing of the chemical shift evolution after a read-out 90' pulse and
second a-pulse train. However, in the presence of magnetization exchange between the two spins
during the mixing time, the orientation-dependent CSA frequency changes and the CSA cannot be
completely refocused, resulting in loss of signal intensity.
(a)
//21F
p
InnS
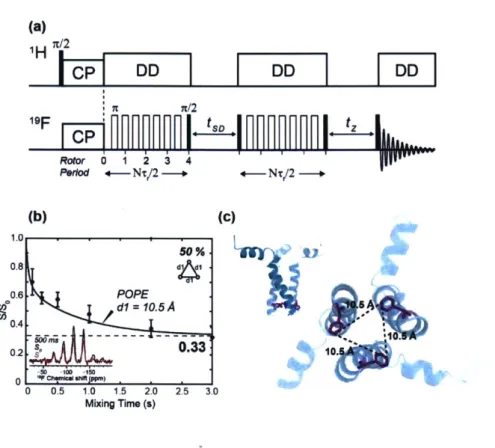
Rotor 0 1 2 3 4 Peod 4-Nt/2 -- +-NT/2 --(b) (c) 1.0 50 %1 0.8 0.6. POPE *dl = 10.5 A 50 0.4 0.2 s.3 10. 0 0.5 1.0 1.5 2.0 2.5 3.0 Mixing Time (s)Figure 1.8
19F-
19F CODEX distance measurement of 4-
19F-F699 labeled gp4l MPER-TMD in
POPE. (a) CODEX pulse sequence. (b) Time-dependent S/So dephasing value of 4-
19F-F699
equilibrated to 0.33 at a mixing time of 3 s, indicating the trimerized transmembrane domain of
the gp4l (inset: Representative S and So spectra at a mixing time of 500 ms). (c) Bottom view of
the intermolecular
19F-
19F distances of 10.5 A between the F699 (magenta) in the POPE membrane.
(Inset: Side view of the trimer).
The experimentally measured S/So intensities are plotted as a function of the spin diffusion
mixing time
(tsD)and fitted to the exponential decay (single or double) curve generated from
MATLAB program using the exchange matrix, which describes the spin system. The 3 x 3
exchange matrix contains the rate constant (kij) that are proportional to an overlap integral, Fi
1(0)
and the square of homonuclear dipolar couplings (owij). Based on 5-19F-Trp results, the overall integral values Fij(0) are 37 us and 41 us for 8 kHz and 10 kHz MAS, respectively.
kii = 0.5ir>?; Fjj(0)
.y
2 1 (3 cos
2 6 _ 1)
47r ri, 2
Where the Oij indicates the angle between the internuclear vector and BO. Since the initial magnetization is equally transferred and equilibrated among M spins, the equilibrium dephasing value approaches 1/M at long exchange mixing times. For example, when the equilibrium dephasing value approaches 1/3 at long exchange mixing time, the oligomeric number is 3,
1.10 References
1. X. C. Bai et al., An atomic structure of human gamma-secretase. Nature 525, 212 (Sep 10,
2015).
2. B. Y. Liang, L. K. Tamm, NMR as a tool to investigate the structure, dynamics and function of membrane proteins. Nat Struct Mol Biol 23, 468 (Jun, 2016).
3. E. Gabellieri, G. B. Strambini, Perturbation of protein tertiary structure in frozen solutions revealed by I -anilino-8-naphthalene sulfonate fluorescence. Biophys J 85, 3214 (Nov 1, 2003).
4. W. D. Van Horn, A. K. Simorellis, P. F. Flynn, Low-temperature studies of encapsulated proteins. JAm Chem Soc 127, 13553 (Oct 5, 2005).
5. K. Schmidt-Rohr, H. W. Spiess, Multidimensional Solid-State NMR and Polymers. (Elsevier Science, Burlington, 2012).
6. M. H. Levitt, Spin dynamics : basics of nuclear magnetic resonance. (Wiley, Chichester, 2015).
7. I. J. Lowe, Free Induction Decays of Rotating Solids. Physical review letters 2, 285 (1959).
8. M. Hong, Solid-state dipolar INADEQUATE NMR spectroscopy with a large double-quantum spectral width. JMagn Reson 136, 86 (Jan, 1999).
9. D. Huster, L. S. Xiao, M. Hong, Solid-state NMR investigation of the dynamics of the soluble and membrane-bound colicin Ia channel-forming domain. Biochemistry-Us 40, 7662 (Jun 26, 2001).
10. M. Tang, A. J. Waring, M. Hong, Arginine dynamics in a membrane-bound cationic beta-hairpin peptide from solid-state NMR. Chembiochem 9, 1487 (Jun 16, 2008).
11. M. Hohwy, C. M. Rienstra, C. P. Jaroniec, R. G. Griffin, Fivefold symmetric homonuclear dipolar recoupling in rotating solids: Application to double quantum spectroscopy. J Chem
Phys 110, 7983 (Apr 22, 1999).
12. M. Hong, T. Doherty, Orientation determination of membrane-disruptive proteins using powder samples and rotational diffusion: A simple solid-state NMR approach. Chem Phys
Lett 432, 296 (Dec 4, 2006).
13. J. H. Davis, K. R. Jeffrey, M. Bloom, M. I. Valic, T. P. Higgs, Quadrupolar Echo Deuteron Magnetic-Resonance Spectroscopy in Ordered Hydrocarbon Chains. Chem Phys Lett 42, 390 (1976).
14. B. Kwon, A. J. Waring, M. Hong, A 2H solid-state NMR study of lipid clustering by cationic antimicrobial and cell-penetrating peptides in model bacterial membranes.
Biophys J105, 2333 (Nov 19, 2013).
15. R. Hentschel, H. W. Spiess, Deuterium Fourier-Transform Nmr in Solids and Solid Polymers. JMagn Reson 35, 157 (1979).
16. M. G. Munowitz, R. G. Griffin, G. Bodenhausen, T. H. Huang, Two-Dimensional Rotational Spin-Echo Nuclear Magnetic-Resonance in Solids - Correlation of
Chemical-Shift and Dipolar Interactions. JAm Chem Soc 103, 2529 (1981).
17. M. Hong et al., Coupling amplification in 2D MAS NMR and its application to torsion angle determination in peptides. JMagn Reson 129, 85 (Nov, 1997).
18.
P. Mansfield, M. J. Orchard, D. C. Stalker, K. H. Richards, Symmetrized Multipulse
Nuclear-Magnetic-Resonance Experiments in Solids
-
Measurement of Chemical-Shift
Shielding Tensor in Some Compounds. Phys Rev B 7, 90 (1973).
19.
J. W. Bielecki, J. Dlugosz, E. Pawlicka, A. Gabryelewicz, The Effect of Pancreatitis
Associated Ascitic Fluid on Some Functions of Rat-Liver Mitochondria
-
a Possible
Mechanism of the Damage to the Liver in Acute-Pancreatitis. Int JPancreatol
5, 145 (Sep,
1989).
20.
K. Takegoshi, S. Nakamura, T. Terao,
13C-H dipolar-assisted rotational resonance in
magic-angle spinning NMR. Chem. Phys. Lett. 344, 631 (Aug 31, 2001).
21.
0. C. Andronesi et al., Determination of membrane protein structure and dynamics by
magic-angle-spinning solid-state NMR spectroscopy. JAm Chem Soc 127, 12965 (Sep 21,
2005).
22.
S. Y. Liao, K. J. Fritzsching, M. Hong, Conformational analysis of the full-length M2
protein of the influenza A virus using solid-state NMR. Protein Sci 22, 1623 (Nov, 2013).
23.
C. A. Michal, L. W. Jelinski, REDOR 3D: Heteronuclear distance measurements in
uniformly labeled and natural abundance solids. JAm Chem Soc 119, 9059 (Sep 24, 1997).
24.
M. Hong, R. G. Griffin, Resonance assignments for solid peptides by dipolar-mediated
C-13/N-15 correlation solid-state NMR. JAm Chem Soc 120, 7113 (Jul 22, 1998).
25.
A. Chenal et al., Deciphering Membrane Insertion of the Diphtheria Toxin T Domain by
Specular Neutron Reflectometry and Solid-State NMR Spectroscopy (vol 391, pg 872,
2009). JMol Biol 394, 587 (Dec 4, 2009).
26.
J. P. Bradshaw, S. M. A. Davies, T. Hauss, Interaction of substance P with phospholipid
bilayers: A neutron diffraction study. Biophys J75, 889 (Aug, 1998).
27.
J. H. Kleinschmidt, L. K. Tamm, Folding intermediates of a beta-barrel membrane protein.
Kinetic evidence for a multi-step membrane insertion mechanism. Biochemistry-Us 35,
12993 (Oct 8, 1996).
28.
M. Zoonens, Y. K. Reshetnyak, D. M. Engelman, Bilayer interactions of pHLIP, a peptide
that can deliver drugs and target tumors. Biophys J95, 225 (Jul 1, 2008).
29.
Z. Y. Sun et al., HIV- 1 broadly neutralizing antibody extracts its epitope from a kinked
gp41 ectodomain region on the viral membrane. Immunity 28, 52 (Jan, 2008).
30.
I. Solomon, Relaxation Processes in a System of 2 Spins. Phys Rev 99, 559 (1955).
31.
J. Dev et al., Structural basis for membrane anchoring of HIV-1 envelope spike. Science
353, 172 (Jul 8, 2016).
32.
K. K. Kumashiro et al., A novel tool for probing membrane protein structure: Solid-state
NMR with proton spin diffusion and X-nucleus detection. JAm Chem Soc 120, 5043 (May
27, 1998).