Sequence determinants in human polyadenylation site selection.
Texte intégral
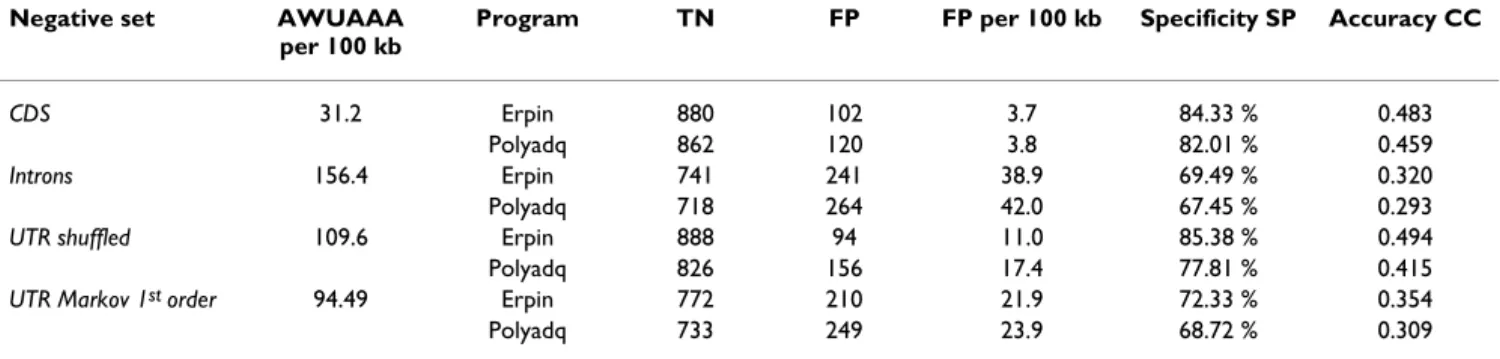
Figure


Documents relatifs
This article targets the accidental fault diagnosis problem for real-time embedded systems by proposing a modeling method based on hidden Markov models
Only UV possesses sufficient energy to break the primary bonds, and the only chemical effect of visible and infrared radiation is to speed up the rate of reactions that may
Contributions In this paper, we introduce PROS-N-KONS, a new OKL method that (1) achieves logarithmic regret for losses with directional curvature using second-order updates, and
ةﺮﻳﻮﺒﻟا ﺔﻳﻻو ﻲﻓ .(. ﺎﻬﺘﻟازإو ﺎﻬﻨﻣ ﺪﺤﻟا ﻢﻴﻠﻌﺘﻟا مﺎﻈﻧ ﻰﻠﻋ ﺐﺠﻴﻓ ﺔﻴﺒﻠﺴﻟا تﺎﻤﺴﻟا ﺎﻣأ ، ﺎﻬﺘﻴﻤﻨﺗو ﺔﻴﺑﺎﺠﻳﻻا تﺎﻤﺴﻟا.. 7 4 - ﺚﺤﺒﻟا ﺔﻴﻤهأ : تﺎﻤ ﺳ ﺪ ﻳﺪﺤﺘﻓ
A further differ- ence with respect to Experiment 1 is that in order to ensure that an eventual transposed- phoneme priming effect was not merely due to the fact that the
It is the first time, to our knowledge, that a decentralized SLAM using only vehicles equipped with a single camera and proprioceptive sensors is presented.. Data incest is
If the silicate mantles of Europa and Ganymede and the liquid sources of Titan and Enceladus are geologically active as on Earth, giving rise to the equivalent of
Après un deuxième bain tout habillé, cette fois dans l’eau de la rivière, nous avions profité d'un troisième pour nous remettre de nos émotions, mais celui-là, de soleil, de
