élec
A. B
M.S.
M. G
B. Be
K. Z
MinistèThèm
ctroniqu
oufelfel
. Boumaz
GHERS
ennacer
Zanat
ère de l’enseM
Prés
Dépa Optme : Pro
ue des
SousProfes
a Profes
Profes
Profes
M.C.B
eignement sMémoir
senté à l’U
Faculté
artement de Filière tion : physi Par :opriétés
semi-co
s la directionsseur
U
sseur
U
sseur
U
sseur
U
B
U
supérieur etre de M
Université
é des MI
e : Sciences e : physique ique de la m : Diaf Chémagné
onducte
n de : Pr. M
JURY
Universit
Universit
Universit
Universit
Universit
t de la recheMagister
é de Guelm
I & SM
de la matiè e matière con rifétiques e
eurs ma
M.S. Bouma
té de Guel
té de Guel
té de Ann
té de Guel
té de Guel
erche scientima
ère ndenséeet struc
agnétiqu
azalma
P
lma
R
aba
E
lma
E
lma I
A ifiquecture
ues dilu
Président
Rapporteu
Examinate
Examinate
Invité
Année 2012ués
ur
eur
eur
2/2013Remerciements
Mes chaleureux remerciements à toutes les personnes que j’ai pu côtoyer au cours de ma thèse de magister et ceux qui ont permis son bon déroulement, mes premiers mots iront à mon encadreur, monsieur Le Professeur M.S. Boumaza, et monsieur le Docteur Zanat Kamel. Je tiens à leur exprimer toute ma
reconnaissance, tant pour leurs conseils, leurs intérêt et leurs vaste culture que pour la confiance qu’ils m’ont accordée pendant ce projet de recherche, merci de m’avoir ouvert les portes du monde passionnant de la recherche, et ce malgré tous ces aléas et difficultés.
La présente étude s’est déroulée au Laboratoire de physique de l’Université 08 Mai 1945 de Guelma. Je remercie le directeur, Monsieur le Professeur A.Boufelfel , de m’avoir accueilli au sein de son laboratoire d’avoir accepté de présider le jury de soutenance.
Mes remerciements vont particulièrement à Mme S.Djeroud président du comité scientifique du département . MEMBRES DE JURIE
Je remercie vivement, Mr. B.Bennacer, Professeur à l’université deGuelma et Mr.M.Ghers Professeur à l’université de Annaba d’avoir accepté de juger ce travail.
Je tiens à exprimer ma gratitude au responsable scientifique de cette post-graduation (membres du CPM) et au corps enseignant ayant assuré notre magister, à savoir : Pr N.Boukharrouba ,Mr Brakta, Mr Dr Kahlrass,.
Aussi , je tiens à exprimer toute ma gratitude à toute ma famille, plus particulièrement ma mère, ma femme mes enfants Heythem, Hala, Fouad et Chahine, mes frères, mes sœurs pour leur présence et leur affection permanentes, qui m’ont permis d’en arriver là.
Mes plus vifs remerciements à monsieur Boudjehem Hocine.
Je tiens à remercier pleinement mon ami monsieur Ferhani Kamel pour ses orientations et conseils. Mes chaleureux remerciements A mes collègues de promotion : R. Benchikh, M. Boualleg, W. Richi, F. Adjailia, M.Messiad, S. Fnides , et S.Karfaf à qui je souhaite finalisation de leur travaux de recherche. Merci enfin, à tous mes amis et plus particulièrement :
Dhafri Morad , Boulares samir, Bousaha Bouzid , Refes A/Rezzek, , Sedra Salah, amis sur qui je pourrai toujours compter.
ملاىىنا فبصولأ خُسُطبىغمنا صاىخنا و خُووشزكنلإا خُىجنا خساسد ىه ممعنا ازه هم فذهنا
خمعطمنا
بُسُطبىغم
. شصىعنا هم خَىئم خجسو ضَىعزث تكشمنا ازه ًهع بىهصحر ذمن In خطساىث شصىعنا هم داسر Mn . جمشجمنا بىهمعزسا خساسذنا يزه مجأ هم AKAI-KKR خمَشط و خفبثكهن لاوذنا خناد خَشظو ًهع ضكرشَ ٌزنا KKR خناذن .هَشغ مصلأا ملبىنا فصىن خُىجنا ذثاىث ًهع لىصحنا مر InSb خنلاذث خُهكنا خلبطهن يشغصنا خمُمنا هع ثحجنبث نصاىزنا ذىع .مجحنا كشر مك مجأ هم ضُ x ـن Mn تكشمهنو شصىع مكن خُهكنا خنبحنا خفبثك و خُنولأا خُهخنا ذثبث بىُع . خُزحر خمجط مك مجأ هم خنبحنا خفبثك دبُىحىم مسس كَشط هع خُووشزكنلإا صئبصخنا ذَذحر مر s ، p و d . ـن خُسُطبىغمنا صاىخنا ٌشُك حساشح خجسد و Tc ممحنا خَشظو لبمعزسبث بهذَذحر مر ضُكشر مك مجأ هم .طسىزمنا خ ُ ج َ ش م ز ن ا ق ش ط ن ا ب ى ه م ع ز س ا ، ة ب س ح ن ا ٍ ف L S D A ، L D A ، G G A و L M D . جئبزىنا خممحمنا .خَشظىنا جئبزىنا و خُجَشجزنا جئبزىنا عم قبفو ًهع ٍه . : ة د ا م ل ا خ ف ث ك م ن ا ح د ب م ن ا ةينبلا ةسارد وه لمعلا اذه نم فدهلا ملا صئاصخلا و ةينورتكللإا معطملا لقاونلا فاصنلأ ةيسيطانغ تاملك حيتافمتاملك
:حيتافم
باسح
ab initio – بُسُطبىغم معطم ملبو فصو – لاوذنا خناد خَشظو – خُووشزكنإ خُىث – ٌشُك حساشح خجسد curie ملا ةداـــ:لابقتسلإا ربخم
خعمبج ، ءبَضُفنا شجخم
80
ٌبم
54
خمنبل،
R E S U M E
Le but de ce travail est d’étudier la structure électronique et les propriétés magnétiques des semi-conducteurs magnétiques dilués .
On a obtenu ce composé en substituant certain pourcentage de l’élément In par des atomes de Mn.
Pour cette étude on a utilisé le code AKAI-KKR qui est basé sur la théorie de la fonctionnelle de la densité (DFT) et la méthode de KKR de la fonction de Green.
Les paramètres de structure du semiconducteur parent InSb sont obtenus à l’équilibre en minimisant l’énergie totale en fonction du volume.
Pour chaque concentration x du Mn on a déterminé le paramètre de maille et la densité d’état totale pour chaque élément et pour le composé .
Les propriétés électroniques sont déterminées par le traçage des courbes des densités d’état pour chaque sous couche s, p et d.
Les propriétés magnétiques de et la Tc pour chaque concentration (la variation de Tc en fonction de la concentration) sont déterminés en utilisant la théorie du champ moyen . Dans le calcul on a utilisé les approximations LSDA (approximation de la densité locale de spin), LDA (approximation de la densité locale) et la GGA (approximation du gradient généralisé).
Nous résultats sont en accord avec les données expérimentales et les calculs théoriques.
DISCIPLINE :
Matière condenséeMOTS-CLES :
Calcul ab initio, semi conducteur magnétique dilué, Théorie de la fonctionnelle de la densité, Structure électronique, Température de Curie.
LABORTOIRE D’ACCUEIL :
The intention of this work is to study the electronic structure and the magnetic properties of the diluted magnetic semiconductors .
We obtained this compound in substituent certain pourcentage of the element In by Mn atoms. For this study we used the code AKAI-KKR which is based on the theory of the functional of the density (DFT) and the method of KKR of the function of Green.
The structural parameters of the semiconductor InSb relative are obtained with balance by minimizing total energy according to volume.
For each concentration X of Mn We determined the cell parameter and the total density of state for each element and the compound .
The electronic properties are determined by the tracing of the curves of the densities of state for each under layer S, p and d.
The magnetic properties of and Tc for each concentration (Tc variation according to the concentration) are given by using the theory of the average field.
In calculation we used approximations LSDA (approximation of the local density of spin), LDA (approximation of the local density) and the GGA (approximation of the generalized gradient).
We results are in agreement with the experimental data and theoretical calculations.
DISCIPLINE :
Condensed matter
KEY WORDS :
Ab.initio calculation ; semi diluted magnetic conductor; density functional theory ; electronic structure; Curie Temperature
HOME LABORATORY:
Table des matières
Remerciments صــــــــخلـم Résumé Abstract
Liste des figures Liste des tableaux
Introduction générale……….
1CHAPITRE I
La structure cristalline et energitique
I – La structure cristalline I.1. Introduction………. 3I.2.Réseaux cristallins……… ..3
I.3.Réseaux de Bravais ……… 4
I.4.Maille primitive et maille conventionnelle ……….. 6
I.5.Maille primitive de Wigner-Seitez ……… ………6
I.6. Le réseau réciproque ……… ………..…. 9
I.7. Zone de Brillouin ……….. 12
II-La structure énergitique II.1.Introduction ……….. 12
II.2.La théorie de bande ………. 14
II.3.Théorie de Bloch ……… 15
II.4.Equation de Schrodinger ………. 21
II.5.La densité d’état ………. 21
Références
CHAPITRE II
Les semi-conducteurs
I – Le semi-conducteur et les autres corps I.1.Le cristal intrinsèque ………. 22I.2. Les métaux ……….. 23
I.3. Les isolants ………. 24
I.6.2. le dopage de type N ………. 33
I.6.3.Le dopage de type P ………. 34
I.7.Les alliages……… ….35
II – Les semi-conducteurs magnétiques dilués ( DMS) II.1.Introduction ………. 37
II.2.Injection de porteurs polarisés en spin ……….. 38
II.3.Les densités d’états dans les ferromagnétiques ……… 40
II.4.La température de curie ……… 40
II.5 Les semi-conducteurs magnétiques dilués (DMS) ……… 41
II.6. L’alliage In1-x Mnx Sb ……… 42
II.6.1. L’indium In ………. 42
II.6.2. L’antimoine Sb ……….….. 42
II.6.3. Le Manganèse Mn ……… ………… 43
II.6.4.L’alliage Indium -Antimoine InSb ………. 45
II.6.5.L’injection de Mn dans l’InSb ………..…. 45
Référence
CHAPITRE III
Les méthodes de calculs
I. Théorie de la fonctionnelle de densité (DFT)……… ..…30I.1.Introduction ……… 30
I.2.La formulation de la DFT ……… ……….31
I.3.Théorème de Hohenberg-Kohn ………...33
I.4.Les équations de kohn-Sham ……….36
I.5.L’approximation de la densité locale (LDA) ………....37
I.6.L’approximation du gradient généralisé (GGA) ……….. 38
I.7. L’itération……… 39
II. La méthode de la fonction KKR-Green ……….. .39
II.1.La méthode KKR………. 39
II.2. Le problème de l’unique emplacement ………... 40
II.2.2.La forme asymptotique ……….41
II.2.3.Wronskian ………. 41
II.2.4.La fonction de Green dans l’espace libre ………...42
II.3.KKR traditionnel ………..43
II.3.1.Le potentiel Muffin-tin ………..43
II.3.2.La méthode linéaire des orbitales Muffin-tin (LMTO)………..44
II.3.3.L’expansion de la cellule centrée ………..45
II.3.4.La constante de la structure ……….. 45
II.3.5.La matrice de KKR ………..46
II.4.La fonction de Green en cristal ………...46
II.4.1.L’équation de l’intégrale ………. 46
II.4.2.Equation de Dyson ……….46
II.4.3. Etat de frontière périodique ……….…47
II.4.4.La densité des états ………. 47
II.4.5.Le contour d’intégration ( Dans le plan complexe )……… 49
II.5.L’approximation de potentiel cohérent (CPA)……… …49
II.6.Les subrotines………...51 Référence………53
CHAPITRE ІV
Résultats et discutions
Introduction……….
54 A)Première partie……….. 541-Manipulation du code AKAI-KKR……….. ..54
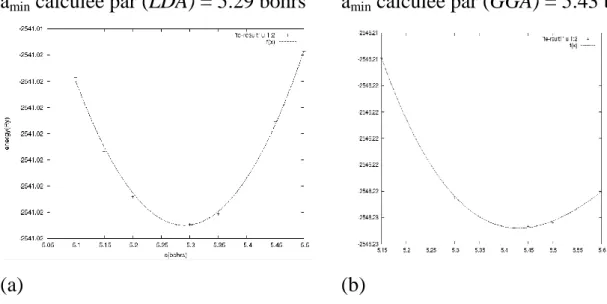
1-1.La configuration électronique et le paramètre cristallin de Fe (bcc)………… 54
1-2.Détermination du paramètre cristallin à l’ètat d’éqquilibre amin pour Fe (bcc) en utilisant la LDA et la GGA……….... 54
1-3. calcul de la densité d’état de Fe (bcc)……… .55
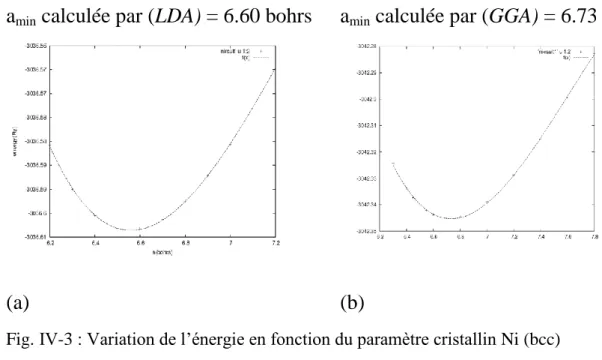
2-1. La configuration électronique et le paramètre cristallin de Ni(fcc)……… 56
2-2. Détermination du paramètre cristallin à l’ètat d’éqquilibre amin pour Ni (fcc) .56
2.3.Calcul de la densité d’état de Ni(fcc)……… …56
3.1. configuration électronique de Co(fcc)……… ..57
3.2. Détermination du paramètre cristallin à l’ètat d’éqquilibre amin pour Co (fcc)… .57
5.Calcul de la température de Curie de Fe,Ni et Co dans les états du désordre
des moments locaux (LMD)……… 64
B) Deuxième partie :Etude du semi-conducteur magnétique dilué le Mn/InSb……. 65
1.Etude du semi-conducteur InSb par le code AKAI-KKR……… 65
2.Le semi-conducteur magnétique dilué In1-xMnxSb ……… … 66
2.1. Le paramètre cristallin du In1-xMnxSb pour chaque concentration du Mn , en utilisant la LDA et la GGA……….... 66
2.2.La densité totale de In1-xMnxSb et la densité deMn(3d) pour chaque concentration……… .67
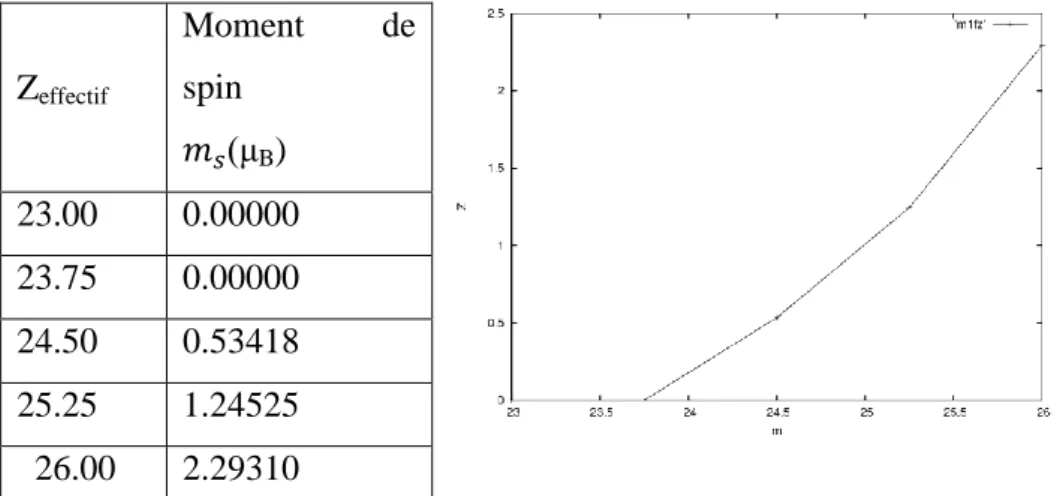
2.3.Détermination du moment magnétique de In1-xMnxSb pour chaque concentration De Mn%... .76
2.4.Détermination de la température de Curie de In1-xMnxSb pour chaque concentration de Mn en se basant sur la théorie du champ moyen(LDA)………... ..77
-Conclusion
……… 78-Perspectives
………. 80-Référence………..
81Liste des figures
Fig.I.1 vecteurs primitifs du réseau f.c.c. Le volume de la cellule primitive est au quart du volume
a3 de la cellule conventionnelle. Maille primitive (à l‘intérieure) et maille conventionnelle (à l’extérieur) du réseau cubique a faces centrées
5
Fig.I.2 Cellule de Wigner –Seitz du réseau de Bravais cubique face centrée (f.c.c. 5 Fig.I.3 La première zone de Brillouin de la structure zinc-blende 7 Fig.II.1 Branchement d’un cristal à une source continue 14 Fig.II.2 Influences extérieures sur la résistivité spécifique d’un métal 15 Fig.II.3 Influence de l’énergie extérieure sur la résistivité du semi-conducteur. 16 Fig.II.4 Schéma des bandes relatives aux conducteurs, semi-conducteurs et isolants. 17 Fig.II.5 Dopage d’un cristal semi-conducteur en conductibilité N 18 Fig.II.6 Dopage d’un cristal semi-conducteur en conductibilité P 19 Fig.II.7 Allure de la densité d’état dans un métal de transition ferromagnétique. Les niveaux
énergétiques EF Fe, EF Co et EF Ni indiquent la position relative du niveau de Fermi
respectivement dans le fer, le cobalt et le nickel par rapport aux différentes bandes présentes.
23
Fig.II.8 Densité d’état dans le fer. L’origine des énergies choisie correspond à la position du niveau de Fermi.
24 Fig.II.9 Densité d’état dans le cobalt. L’origine des énergies choisie correspond à la position du
niveau de Fermi.
25
Fig.II.10 Densité d’état dans le nickel. 25
Fig.II.11 Température de Curie calculée pour différents semi-conducteurs avec un dopage 5℅ de Mn et 3.5x1020 trous cm-3.
27 Fig.II.12 La maille conventionnelle de la structure zinc-blende d’InSb 30
Fig.III.1 .Densité d’état dans le cobalt. L’origine des énergies choisie correspond à la position du niveau de Fermi.
46 Fig.III.2 Intégration de contour de la fonction de Green 50
Fig.III.3 L’idée de CPA 52
Fig.III.4 L'organigramme du calcul de structure de bande 53 Fig.IV.1 Variation de l’énergie en fonction du paramètre cristallin pour Fe (bcc) 55 Fig.IV.2 Variation de la densité d’état avec les différents niveaux d’énergie Fe(bcc) 55 Fig.IV.3 Variation de l’énergie en fonction du paramètre cristallin Ni (bcc) 56 Fig.IV4 Variation de la densité d’état avec les différents niveaux d’énergie Ni (fcc) 57 Fig.IV.5 Variation de l’énergie en fonction du paramètre cristallin Ni (bcc) 58 Fig.IV.6 Variation de la densité d’état avec les différents niveaux d’énergie Co(bcc) 58 Fig.IV.7 Variation du moment de spin en fonction du nombre atomique FeNi (fcc) 59 Fig.IV.8 Variation du moment de spin en fonction du nombre atomique pour FeCo 60 Fig.IV.9 Variation du moment de spin en fonction du nombre atomique pour FeV (bcc) 60 Fig.IV.10 Variation du moment de spin en fonction du nombre atomique pour FeCr (bcc) 61 Fig.IV.11 Variation du moment de spin en fonction du nombre atomique pour FeMn (bcc) 61 Fig.IV.12 Variation du moment de spin en fonction du nombre atomique pour CoCr (hcp) 61 Fig.IV.13 Variation du moment de spin en fonction du nombre atomique pour CoMn 62 Fig.IV.15 Variation du moment de spin en fonction du nombre atomique pour CoNi 62 Fig.IV.16 Résultats de calcul de ms en fonction de Zeff (la courbe de Slater-Pauling) 62
Fig.IV.17 La courbe de moment de spin en fonction de Z 64
Fig.IV.18 Paramètre cristallin de InSb 65
Fig.IV.19 La densité totale de InSb 66
Fig.IV.20 Le paramètre cristallin en fonction de la concentration de Mn (LDA et GGA) 67 Fig.IV.21 La densité totale du Mn(3d) et la densité totale de In1-xMnxSb pour les concentrations 0%
à 50%
68
1-x x
et 6%
Fig.IV.25 La densité totale du Mn(3d) et la densité totale de In1-xMnxSb pour les concentrations 8%
et 10%
73
Fig.IV.26 La densité totale du Mn(3d) et la densité totale de In1-xMnxSb pour les concentrations 20%
et 30%
74
Fig.IV.27 La densité totale du Mn(3d) et la densité totale de In1-xMnxSb pour les concentrations 40%
et 50%
75
Fig.IV.28 Le moment magnétique de In1-xMnxSb en fonction des concentrations de Mn (0% à 50%) 76
Liste des tableaux
Tableau I.1 Certains points de haute symétrie 9
Tableau II.1: La température de Curie de quelques matériaux 26
Tableau II.2 Propriétés de l’In 28
Tableau II.3 Propriétés de Sb 28
Tableau II.4 Propriétés de Mn 29
Tableau III.1 les unités atomiques 38
Tableau III.2 Les subroutines dans le code KKR 54
Tableau IV.1 Calcul des moments magnétiques en fonction des paramètres cristallins des élément de transition (Fe,Ni,Co) calculés par la GGA et la LDA
59 Tableau IV.2 Calcul de la température de curie de Fe,Niet Co dans les états de désordre des
moments locaux(LMD)
64 Tableau IV.3 Calcul du moment magnétique de In1-xMnxSb pour différentes concentration de Mn 76
Tableau IV.4 Calcul de la température de Curie du In1-xMnxSb en fonction des concentratins de
Mn par la LSDA
Introduction générale
1
Introduction générale
La recherche dans le domaine des sciences des matériaux et en particulier des semi-conducteurs a pris une ampleur considérable grâce à l’enjeu économique du traitement de l’information. Un exemple éclatant est l’invention du transistor en 1947. Depuis les chercheurs n’ont cessé de miniaturiser les composants électroniques en donnant naissance à la microélectronique. Cette dernière repose toujours sur la manipulation des porteurs de charge électrique dans les semi-conducteurs. Dans ces applications, le spin porté par l’électron et son moment magnétique ne sont pas pris en compte. En revanche, les propriétés magnétiques sont exploitées dans des matériaux ferromagnétiques comme support pour le stockage de l’information (distiques magnétiques) et une « électronique de spin » se développe actuellement dans les assemblages de métaux magnétiques (têtes de lecture compactes et sensibles, mémoire vive d’ordinateur). Cette électronique de spin reste cependant difficile à intégrer dans l’électronique traditionnelle : pour cela , il faut fabriquer des structures associant métaux magnétiques et semi-conducteurs aussi proches que possible des semi-conducteurs usuels de la microélectronique, mais présentant en outre des propriétés magnétiques couplées aux propriétés électroniques, permettrait de conjuguer les avantages de l’électronique de spin et les possibilités de la microélectronique. C’est cette piste qu’ouvre le ferromagnétisme induit par les porteurs dans les semi-conducteurs magnétiques dilués (DMS) [1], [2], [3]. Cette possibilité intéresse des applications variées allant de l’électronique de spin (contrôle de spin ) jusqu'à l’ordinateur quantique (contrôle de la fonction d’onde ).
Le ferromagnétisme induit par les porteurs dans les semi-conducteurs magnétiques dilués résulte de l’introduction simultanée, dans un semi-conducteur standard, de spins localisés et de porteurs libres .L’interaction ferromagnétique entre spins est due aux porteurs libres ,et on peut donc utiliser les techniques de la micro –électroniques et de l’optoélectronique pour manipuler ces porteurs et ainsi contrôler les propriétés magnétiques (aimantation, anisotropie…)[4], [5]. De telles structures peuvent être obtenue en incorporant des éléments magnétiques (souvent le Mn) dans la matrice d’un semi-conducteur (Si,CdTe, GaAs, InAs…)[6], [7], [8] .
Pour le système Ga1-xMnxAs, des études expérimentales ont montré qu’il est ferromagnétique, sa température de Curie est relativement élevée (150°k) [9].
Pour In1-xMnxAs, il n’existe que des études théoriques [10] qui stipulent son caractère ferromagnétique.
Récemment, une étude a été effectuée, au laboratoire LPMS [11], sur la formation des DMS à base du manganèse : Mn/InAs(001).Les résultats obtenus dans cette étude ont montré que les propriétés physiques de ce système dépendent fortement du degré de dilution du Mn dans le semi-conducteur ainsi que les conditions de la préparation.
Notre objectif est d’éclaircir les différents phénomènes physiques induits lors du dopage de l’InSb par le Mn, cela pourrait permettre une compréhension du caractère ferromagnétique observé dans les DMS, un sujet qui n’est pas tout à fait compris à l’heure actuelle.
Le travail de ce mémoire concerne l’étude des propriétés structurales, électroniques et magnétiques du système Mn/InSb. Le semi-conducteur InSb est intéressant tant sur le plan fondamental que technologique. Il est caractérisé par la plus faible bande interdite parmi tous les semi-conducteurs ІІІ/V (0.18 eV), une faible masse effective des électrons et par conséquent une forte mobilité (7x105cm2V-1s-1 à 77°k) [12], [13] . Il sert à de nombreuses
2
applications, on les trouve, par exemple, dans les capteurs infrarouges et les détecteurs à haute vitesse.
Dans la procédure de recherche de nouveaux matériaux, on peut soit suivre des protocoles expérimentaux, soit utiliser une étude théorique.
Cette dernière option constitue ce qui est appelé «la modélisation et la simulation des matériaux »
Les techniques de modélisation et de simulation peuvent être empiriques ou ab-initio (quantiques). Ces dernières sont connues pour êtres très précises et indépendantes des sources expérimentales, donc complètement prédictives.
Pour déterminer les propriétés magnétiques et structure électronique du système Mn/InSb, on a effectué une étude simulée en utilisant le code AKAI-KKR.
Ce mémoire comprend quatre chapitres avec une introduction et une conclusion générale. Le premier chapitre apporte des informations de base sur la structure cristalline et notions fondamentales sur la structure énergétique.
Le second chapitre est consacré aux semi-conducteurs et aux semi-conducteurs magnétiques dilués.
Les théories et les méthodes de calculs sur lesquelles est basé le code AKAI-KKR font l’objet du chapitre ІІІ.
Les résultats et discussions obtenus forment le chapitre ІV.
Introduction générale
3
Références
[1] H. Ohno, A. Shen, F. Matsukura, A. Endo, S. Katsumoto, Y. Lye,Appl. Phys. Lett. 69 (1996).
[2] H. Ohldag, V. Solinus, and F. U. Hillbrecht, J. B. Goadcoop, M. Finazzi, F. Matsukura, H. Ohno,Appl. Phys.Lett. 76,2928 (2000).
[3] A. Oiwa, S. Katsumoto, A. Endo, M. Hirasawa, Y. Iye, H. Ohno, F. Matsukura, A. Shen and Y. Sugawara, Solid State Commun. 103 (1997).
[4] G. Borghs, and J. De Boeck, Mater. Sci. Eng. B 84 (2001).
[5] H. J. Zhu, M. Ramsteiner, H. Kostial, M. Wassermeier, H. P. Schonherr, and K. H. Ploog, Phys. Rev. Lett. 87 (2001) .
[6] M. W. Ruckman, J. J. Joyce, and J. H. Weaver, Phys. Rev. B33 (1986).
[7] Y. B. Xu, E. T. M. Kernohan, D. J. Freeland, A. Ercole, M. Tselepi, and J.A. C. Bland, Phys.Rev. B58 (1998) .
[8] C. M. Teodorescu, F. Chevrier, R. Brochier, C. Richter, O. Heckmann, V. Ilakovac, P. De Padova and K. Hricovini. Surf. Sci. 482-485 (2001).
[9] K. C. Ku, S. J. Potashnik, R. F. Wang, S. H. Chun, P. Schiffer, N. Samarth, M. J. Seong, A. Mascarenhas, E. Johnston-Halperin, R. C. Myers, A. C. Gossard, and D. D. Awschalom, Appl. Phys. Lett. 82 (2003)2302.
[10] H. Akai, Phys. Rev. Lett. 81 (1998).
[11] Renaud Brochier, these de doctorat, Univ. Cergy-Pontoise (2002).
[12] D. M. Li. M. Atoji, M. Yamazaki, T Okamoto, T. Tambo, and C. Tatsuyama. Appl.
Surf. Sci. 130-132 (1998).
[13] K. Li, A. T. S. Wee, J. Lin, K. K. Lee, F. Watt, K. L. Tan, Z. C. Feng,and J.B.
4
La structure
cristalline et électronique
I.
LA STRUCTURE CRISTALLINE
I.1. INTRODUCTION
Ce premier chapitre apporte des informations de base sur la cristallographie qui doivent permettre de comprendre la structure et ses conséquences sur le comportement des électrons dans un cristal qui est un état ordonné de la matière. Autour de chaque atome, les voisins sont disposés suivant une configuration rigoureuse, constante dans tout le cristal .L’état
désordonné de la matière est représenté par les gaz [14]. Ce qui nous intéresse est le premier état qui est l’état cristallisé par le fait que les atomes sont arrangés aux nœuds d’un réseau périodique. Le résultat est un ensemble de noyaux et d’électrons liés entre eux par des forces essentiellement coulombiennes [15]. Il est utile d’examiner quelques propriétés géométriques les plus importantes des réseaux périodiques dans l’espace a trois dimensions.
I.2. RESEAUX CRISTALLINS
Un cristal est un arrangement périodique d’atomes répondant a un certain nombre de symétries : l’inversion, la rotation, la réflexion dans un parallélépipède.
On peut décrire la structure d’un solide parfait par un ensemble de points appelés « sites » ou « nœuds», dans l’espace de trois dimensions, le réseau est défini par trois vecteurs de
translation fondamentaux appelés vecteurs de base
( ⃗⃗⃗⃗ ⃗⃗⃗⃗ ⃗⃗⃗⃗ .
La position de chaque cellule de cristal est définie par un vecteur du réseau :
⃗ ⃗⃗⃗⃗ ⃗⃗⃗⃗ ⃗⃗⃗⃗ (І-1) Ou` u, v, w sont des nombres entiers.
I.3.RESEAUX DE BRAVAIS
On appelle réseau de Bravais un réseau dont tous les sites ont le même environnement [16]. Il spécifie l’ordre périodique dans lequel les unités élémentaire répétées du cristal sont disposées mais le réseau de Bravais représente seulement la géométrie de la structure périodique sans considérer la nature des unités (atomes uniques, des groupes d’atomes ou des molécules …) Le réseau de Bravais permet de distinguer les différents arrangements possibles des atomes pour former un cristal, pour cela on introduit la notion de la maille cristalline telle que selon les paramètres de cette maille on compte sept systèmes cristallins et quatorze types de réseaux de bravais [17].
CHAPITRE І Les structures cristalline et énergétique
5
I.4. MAILLE PRIMITIVE ET MAILLE CONVENTIONNELLE
Un volume de l’espace qui, translaté par tous les vecteurs d’un réseau de Bravais, remplit complètement l’espace sans se recouvrir lui-même ou laisser des vides est appelé une maille
primitive du réseau. On peut remplir cet espace avec des mailles non primitives ou mailles conventionnelles.
La maille conventionnelle est en général choisie de manière à être plus grande que la maille primitive.
La figure I.1 représente une maille primitive et une maille conventionnelle d’un réseau cubique à faces centrées.
Fig. I.1 : vecteurs primitifs du réseau f.c.c. Le volume de la cellule primitive est au quart du
volume a3 de la cellule conventionnelle. Maille primitive (à l‘intérieure) et maille conventionnelle (à l’extérieur) du réseau cubique a faces centrées [23].
6
la maille de Wigner – Seitz autour d’un point du réseau est la région de l’espace qui est plus proche de ce point que tout autre point du réseau .La maille de Wigner –Seitz est une maille primitive et elle est aussi symétrique que le réseau de Bravais .
La figure I.2 précise une maille de Wigner –Seitz du réseau de Bravais cubique à faces centrées.
I.6. LE RESEAU RECIPROQUE
D’après la définition du réseau de Bravais le réseau réciproque est lui-même un réseau de Bravais. Le réseau réciproque peut être engendré par les trois vecteurs primitifs :
⃗⃗⃗ ( ⃗⃗⃗⃗ ⃗⃗⃗⃗ ⃗⃗⃗⃗ ( ⃗⃗⃗⃗ ⃗⃗⃗⃗
⃗⃗⃗⃗ ( ⃗⃗⃗⃗ ⃗⃗⃗⃗ ⃗⃗⃗⃗ ( ⃗⃗⃗⃗ ⃗⃗⃗⃗ (І-2) ⃗⃗⃗⃗ ( ⃗⃗⃗⃗ ⃗⃗⃗⃗ ⃗⃗⃗⃗ ( ⃗⃗⃗⃗ ⃗⃗⃗⃗
Il est définit par rapport à un réseau de Bravais particulier. Pour un ensemble de vecteur ⃗ , un ensemble de ⃗ n’est appelé réseau réciproque que si l’ensemble des vecteurs ⃗ est un réseau de Bravais, tel que le vecteur position :
⃗ ⃗⃗⃗⃗ ⃗⃗⃗⃗ ⃗⃗⃗⃗ (І-3)
n1,n2,n3 : sont des entiers
⃗⃗⃗⃗ , ⃗⃗⃗⃗ , ⃗⃗⃗⃗ : sont trois vecteurs quelconques n’appartiennent pas à un même plan (vecteurs primitifs).
Le réseau réciproque joue un rôle fondamental dans la plupart des études analytique
périodique. Soit un réseau de Bravais et une onde plane ⃗ pour un ⃗ quelconque ; une telle onde ne possède pas la périodicité du réseau de Bravais, mais, pour certain choix du vecteur d’onde, elle l’aura. L’ensemble de tous les vecteurs d’onde ⃗ donnent une onde plane de périodicité égale à celle d’un réseau de Bravais donné est appelé réseau de Bravais réciproque[18].
Si V est un volume d’une maille primitive dans le réseau direct, alors la maille primitive du réseau réciproque a un volume V*= 2π /V.
I.7.ZONE DE BRILLOUIN
La notion de Zone de Brillouin est nécessaire pour décrire les propriétés électriques d’un
cristal dans lequel la symétrie de translation joue un rôle essentiel [19]. Cette théorie permet de préciser la distribution de niveau d’énergie.
CHAPITRE І Les structures cristalline et énergétique
7
Dans l’espace réciproque, La première Zone de Brillouin est représenté avec la maille primitive de Wigner-Seitz (fig. I.2) .On définit des Zones de Brillouin d’ordre plus élevé qui apparaissent dans la théorie des bandes dans un potentiel périodique [20].
Sauf la première zone, chacune des autres zones comporte des morceaux dans les différentes directions. Toutes les zones ont le même volume tel que les états de la frontière de ces zones jouent un rôle important.
Les composées III-V ont une structure cristallographie de type zinc blende. Cette structure s’apparente à celle du diamant.
Le réseau réciproque du réseau de Bravais correspondant a la structure de zinc blende est un réseau cubique centré.
La première zone de Brillouin présente un centre de système a l’origine noté Γ et les axes de symétries sont Δ, Λ, et ∑. Les points de croisement de chacun de ces axes avec les frontières de la zone de Brillouin sont les points de haute symétrie et ils jouent un rôle primordial dans la structure de bande[21](fig.1.3).
Pour comprendre le fonctionnement et prévoir les performances des composants
électroniques, il est également nécessaire d’étudier la structure énergétique correspondante au milieu physique dans lequel se déplacent les porteurs de charge : c’est l’étude de la structure
électronique.
Fig. І.3:La première zone de Brillouin de la structure
8
spéciaux. Le tableau(I.1) qui suit présente quelques uns.
Tous ces points sont liés par des directions, elles-mêmes décrites par des symboles. Ces descriptions sont particulièrement utilisées lors de la caractérisation des propriétés électroniques d’un solide [22].
Symbole (k-point)
Description
k-vectors n1 n2 n3
Γ Centre de la zone de Brillouin 0 0 0
Cubique simple
M Milieu d'une arête ½ ½ 0
R Sommet ½ ½ ½
X Centre d'une face 0 0 1/2
cubique faces centrées
K Milieu d'une arête joignant deux
faces hexagonales ¾ 3/8 3/8
L Centre d'une face hexagonale ½ ½ 1/2
U Milieu d'une arête joignant une face
hexagonale et une face carrée (=K)
CHAPITRE І Les structures cristalline et énergétique
9
X Centre d'une face carrée ½ 0 1/2
Cubique centré
H Sommet joignant 4 arêtes ½ ½ -1/2
N Centre d'une face ½ 0 0
P Sommet joignant trois arêtes ½ ½ 1/4
II. LA STRUCTURE ELECTRONIQUE
II.1.INTRODUCTION
Dans un atome isolé, les niveaux d’énergie des électrons liés aux noyaux sont parfaitement localisés et régis par le principe de Pauli.
Dans les solides l’interpénétration de l’interaction des orbitales électroniques voisines entraine un élargissement des niveaux d’énergie, qui forme alors une bande d’énergie constituée de niveaux discrets.
L’origine physique de la différence entre un isolant, un conducteur et un semi-conducteur peut être donnée à l’aide de la théorie des bandes qui permet d’expliquer le comportement physique de certains matériaux.
II.2.LA THEORIE DE BANDES
Dans un solide, les atomes ne sont plus isolés mais liés d’une manière ou d’une autre (liaison covalente, ionique, métallique …) ; les orbitales s’hybrident souvent .On donne trois noms aux électrons selon leur « utilité » dans le matériau : les électrons de cœur, les
électrons de valence et les électrons de conduction.
Les niveaux d’énergie des électrons des couches de valence forment des bandes plus larges appelées bandes de valence, les bandes qui correspondent aux états excités des électrons des couches externes sont appelées bandes de conduction.
Ces bandes sont remplies par les électrons disponibles dans le matériau, en commençant par les plus basses énergies puis en complétant les bandes supérieures avec les électrons restants ;cela donne la configuration électronique de plus basse énergie , celle qu’a le matériau au zéro absolu (0 kelvin) : on parle d’état fondamentale .
A partir de cet état on peut définir le niveau de Fermi [25] qui est le dernier niveau le plus occupé.
10
II.3.THEOREME DE BLOCH
En 1928 [26], la théorie d’Arnold Sommerfeld représente l’état dynamique de l’électron par une fonction d’onde, l’électron ne peut pas être repéré par une trajectoire autour du noyau, mais par une probabilité de présence.
La même année, la théorie de Félix Bloch ; ou théorie des bandes a généralisé la précédente en s’appuyant sur le caractère périodique de la répartition des ions dans un métal.
Une conséquence de théorème de Bloch est l’apparition des bandes interdites par l’application d’un potentiel cristallin perturbatif périodique sur des électrons libres : c’est le modèle des
électrons presque libres.
pour décrire le mouvement des électrons qui sont supposé indépendants dans un potentiel périodique, on a besoin d’une fonction d’onde, la plus simple est celle de Bloch [27]. Les états propres solutions de l’équation de Schrödinger
* ( + ( ( (І-4) sont d’après Bloch de la forme suivante :
( ( ( ⃗ ) (І-5) Ou` ( est une fonction ayant la périodicité du réseau, c.à.d.
( ⃗ ) ( . (І-6) Pour tout ⃗ appartenant au réseau de Bravais.
Les électrons indépendants qui obéissent chacun à une équation de Schrödinger à un seul électron dans un potentiel périodique sont appelés électrons de Bloch (par opposition aux « électrons libres », qui sont les électrons dans un potentiel périodique partout nul). Soit l’hamiltonien :
( (І-7) Tel que ( est le potentiel périodique.
La fonction de Bloch est une fonction propre de H.
Physiquement, on peut dire que la fonction de Bloch (І-5) est la fonction d’onde d’un électron libre ⃗ modulée par le potentiel périodique du réseau d’ions à travers la fonction
( .
Le théorème de Bloch est parfois exprimé sous la forme alternative suivante :
Les états propres de H sont choisis de telle manière qu’à chaque Ψ soit associé un vecteur d’onde k tel que :
( ⃗ ) ⃗ ⃗ ( (І-8)
Pour tout ⃗ appartenant au réseau de Bravais.
Le théorème de Bloch montre que la fonction d’onde d’un électron est une onde plane
d’amplitude variable mais périodique ( . L’indice k indique que cette fonction dépend du vecteur d’onde k (la valeur propre de la périodicité du réseau). Le problème des bandes d’énergies est donc simplifié, il suffit de trouver les fonctions propres de l’électron dans la maille élémentaire ; ensuite utiliser le théorème de Bloch pour les déterminer dans les autre mailles du cristal.
CHAPITRE І Les structures cristalline et énergétique
11
II.4. EQUATION DE SCHRODINGER
Le problème théorique fondamental de la physique des solides est de comprendre
l’organisation intime des particules constituant un Cristal à l’origine de leurs propriétés. Donc il faut faire appel à la mécanique quantique puisque les notions de la mécanique classique sont insuffisantes.
La base de la mécanique quantique est la résolution de l’équation de Schrödinger
(І-9) Ou` H représente l’hamiltonien exacte de N corps.
: la fonction d’onde en fonction des cordonnées de tous les ions et les électrons. E : l’énergie du système
Les électrons et les ions sont les constituants des solides, donc l’Hamiltonien consiste de : L’énergie cinétique des électrons (Te) ; l’énergie cinétique des noyaux (Tn) ;
L’énergie de toutes les interactions entre ces particules : électron – électron, électron-noyau et noyau-noyau qui sont respectivement : Vee,Vne ,Vnn. Donc :
(I.10) L’équation fondamentale à résoudre pour décrire la structure électronique d’un système à plusieurs noyaux et électrons est l’équation établie par Schrödinger en 1925 [28] , et qui
s’écrit :
[ ∑ ∑ ∑ | ⃗ ⃗⃗ | ∑ | ⃗ ⃗ | ∑ | ⃗⃗ ⃗⃗ | ] (І-11) Les deux premier termes de l’Hamiltonien sont respectivement les opérateurs énergie
cinétique des N électron (indexés i) et des A noyaux atomiques (indexés I). Les trois autres termes représentent les différents potentiels d’interaction électron –noyau, électron –électron et noyau-noyau.
12
Référence :
[ 1 ] M.Brousseau, physique du solide : propriétés électroniques, Masson, Paris ( 1992).
[ 2 ] Hanry Mathien. Physique des semi-conducteurs et des composants
électroniques. Dunod, Paris (2004).
[ 3 ] Hung T.Diep, physique de la matière condensée,Dunod, Paris (2003). [ 4 ] Charle Kittel, physique de l’état solide, Dunod, Paris (2007).
[ 5 ] I. Lakhtine, Métallographie et traitement thermiques des métaux, Edition MIR.Mouscou (1986).
[ 6 ] M.Brousseau, Physique du solide, Masson,Paris (1992).
[ 7 ] Neil W.Ashcroft et N.David Mermin (traduction par Frank Biet et Hamid Kachkachi ), physique des solides,EDP sciences (2002).
[ 8 ] Tuong Nguen Quang, effet de spin dans les nanostructures semi-conductrices :
modélisation et expériences de magnéto-transport (2006).
[9 ] http://fr.wikipedia.org/wiki/Zone_de_Brillouin [10] Un article de Wikipédia, l'encyclopédie libre. [11] www.unine.ch/phys/...2004/.../node4.html [12] E. Fermi, Zeits. F.Physic 48,73(1928).
13] Neil W.Ashcroft et N.david Mermin (traduction par Franck Biet et Hamid Kachkachi ),physique des solides
[14] Rapport CEA –R-Nicolas Richard. Artinies et terres rares sous pression : Approche Pseudopotentiel.
[15] E. Schrodinger, “ An Undulatory theory of mechanics of atoms and
molecule”.Phys.Rev.vol.28 , 1926,p.1049-1070.
[16] G.C. Flectcher , the electron theory of solids, Manash university Clayton ,Australia(1997).
CHAPITRE І Les structures cristalline et énergétique
14
CHAPITRE II
Les semi-conducteurs
I. CONDUCTION ELECTRIQUE
Dans le modèle classique, un corps est isolant s’il ne contient pas d’électrons mobiles. Dans un conducteur, des électrons sont peu liés aux noyaux et peuvent se déplacer dans le réseau cristallin.
Si n est la densité des électrons libres, v leur vitesse moyenne, dans une barre de
longueur L, de section S avec une tension V entre les extrémités, la densité de courant J = I/S est égale à J = n.e.v.
La vitesse des électrons est proportionnelle à la force à laquelle ils sont soumis donc au champ électrique E = V/L. Si μ désigne la mobilité, on a : v = μ.E
(……..) Le modèle classique a été remplacé par le modèle quantique des bandes d’énergie. Dans l’atome isolé les électrons occupent des niveaux d’énergie discrets. Dans un cristal, par suite des interactions entre les atomes, ces niveaux discrets s’élargissent et les électrons occupent des bandes d’énergie permises séparées par des bandes interdites.
La répartition des électrons dans les niveaux obéit aux lois de la thermodynamique statistique. Au zéro absolu, seuls sont peuplés les niveaux de plus basse énergie. Dans les isolants ( Fig.ІІ-1), les bandes d’énergie les plus faibles sont entièrement pleines. La hauteur de la bande interdite est grande (≈5 eV). Il n’y a pas de niveaux d’énergie accessibles et pas de conduction. Par exemple, la résistivité du diamant est et celle du mica varie entre 1010 et 1015 .
Dans les conducteurs, la dernière bande occupée est partiellement remplie : il existe beaucoup de niveaux disponibles et la conduction est grande.
Pour des métaux bons conducteurs, on obtient : ;
;
Pour les semi-conducteurs, le taux de remplissage de la dernière bande occupée est soit très faible soit très important. La hauteur de la bande interdite est faible (≈1 eV).
La conduction est faible et varie beaucoup avec la température. Pour le silicium et le germanium, on mesure à 300 K :
;
CHAPITRE II : Les semi-conducteurs
15
IІ - SEMI - CONDUCTEURS
IІ -1-
Structure des semi-conducteurs
La structure du silicium et du germanium est la même que celle du diamant (Fig.ІІ-2). Chaque atome est lié à 4 voisins placés aux sommets d’un tétraèdre par une liaison covalente : Ces éléments sont « tétravalent ».
Fig.ІІ-2-Diamant
La Fig.ІІ-3 correspond à une représentation sur un plan de la structure. Les traits figurent les électrons de valence.
La théorie des bandes appliquée aux semi-conducteurs amène à considérer une bande de
valence entièrement pleine qui es t séparée d’une bande de conduction par une bande interdite distante de l’énergie ΔE.
Fig.ІІ-3
Si on apporte une énergie thermique ou lumineuse suffisante à un électron, il peut passer de la bande de valence à la bande de conduction avec une probabilité P proportionnelle à : P α exp (–ΔE / kT) ΔE est l’écart en énergie séparant les deux bandes.
T la température absolue. est la constante de Boltzmann. Pour T = 300 K , kT = 0,0025 eV
Diamant ΔE = 7 eV ; Silicium ΔE = 1,12 eV ; Germanium ΔE = 0,7 eV.
Dans un semi-conducteur, ΔE est assez faible pour autoriser, à température ambiante, le passage d’un petit nombre d’électrons de la bande de valence vers la bande de conduction.
14
CHAPITRE II
Les semi-conducteurs
I. CONDUCTION ELECTRIQUE
Dans le modèle classique, un corps est isolant s’il ne contient pas d’électrons mobiles. Dans un conducteur, des électrons sont peu liés aux noyaux et peuvent se déplacer dans le réseau cristallin.
Si n est la densité des électrons libres, v leur vitesse moyenne, dans une barre de
longueur L, de section S avec une tension V entre les extrémités, la densité de courant J = I/S est égale à J = n.e.v.
La vitesse des électrons est proportionnelle à la force à laquelle ils sont soumis donc au champ électrique E = V/L. Si μ désigne la mobilité, on a : v = μ.E
(……..) Le modèle classique a été remplacé par le modèle quantique des bandes d’énergie. Dans l’atome isolé les électrons occupent des niveaux d’énergie discrets. Dans un cristal, par suite des interactions entre les atomes, ces niveaux discrets s’élargissent et les électrons occupent des bandes d’énergie permises séparées par des bandes interdites.
La répartition des électrons dans les niveaux obéit aux lois de la thermodynamique statistique. Au zéro absolu, seuls sont peuplés les niveaux de plus basse énergie. Dans les isolants ( Fig.ІІ-1), les bandes d’énergie les plus faibles sont entièrement pleines. La hauteur de la bande interdite est grande (≈5 eV). Il n’y a pas de niveaux d’énergie accessibles et pas de conduction. Par exemple, la résistivité du diamant est et celle du mica varie entre 1010 et 1015 .
Dans les conducteurs, la dernière bande occupée est partiellement remplie : il existe beaucoup de niveaux disponibles et la conduction est grande.
Pour des métaux bons conducteurs, on obtient : ;
;
Pour les semi-conducteurs, le taux de remplissage de la dernière bande occupée est soit très faible soit très important. La hauteur de la bande interdite est faible (≈1 eV).
La conduction est faible et varie beaucoup avec la température. Pour le silicium et le germanium, on mesure à 300 K :
;
CHAPITRE II : Les semi-conducteurs
15
IІ - SEMI - CONDUCTEURS
IІ -1-
Structure des semi-conducteurs
La structure du silicium et du germanium est la même que celle du diamant (Fig.ІІ-2). Chaque atome est lié à 4 voisins placés aux sommets d’un tétraèdre par une liaison covalente : Ces éléments sont « tétravalent ».
Fig.ІІ-2-Diamant
La Fig.ІІ-3 correspond à une représentation sur un plan de la structure. Les traits figurent les électrons de valence.
La théorie des bandes appliquée aux semi-conducteurs amène à considérer une bande de
valence entièrement pleine qui es t séparée d’une bande de conduction par une bande interdite distante de l’énergie ΔE.
Fig.ІІ-3
Si on apporte une énergie thermique ou lumineuse suffisante à un électron, il peut passer de la bande de valence à la bande de conduction avec une probabilité P proportionnelle à : P α exp (–ΔE / kT) ΔE est l’écart en énergie séparant les deux bandes.
T la température absolue. est la constante de Boltzmann. Pour T = 300 K , kT = 0,0025 eV
Diamant ΔE = 7 eV ; Silicium ΔE = 1,12 eV ; Germanium ΔE = 0,7 eV.
Dans un semi-conducteur, ΔE est assez faible pour autoriser, à température ambiante, le passage d’un petit nombre d’électrons de la bande de valence vers la bande de conduction.
16
IІ -2-
Conduction par électron et par trou
Si une liaison de valence est brisée (agitation thermique, photon ...) l’électron devient mobile : il laisse un excès de charge positive le « trou » (symbolisé par un + dans un carré)
(Fig.ІІ-4).
Cette lacune va être comblée par un électron voisin libéré par agitation thermique et qui va à son tour laisser un trou : ceux-ci semblent se déplacer dans le réseau. Aux électrons (masse positive, charge négative) correspondent des trous (masse négative, charge positive). Le déplacement des trous étant un processus à deux étapes, leur mobilité dans le réseau est plus faible que celle des électrons.
Trous et électrons constituent les porteurs libres intrinsèques dont le nombre est fonction de la température. La neutralité électrique du matériau impose que les trous et les électrons soient en nombres identiques (ni et pi).
Pour le silicium pur à 300 K, on mesure : ni = pi = 1,5.1010.cm-3 .Ce nombre est très faible si on le compare au nombre des atomes.
Toujours pour le silicium pur à 300 K, les mobilités sont :
et
La conductivité intrinsèque du matériau est très faible.
Fig.ІІ-4
IІ -3-
Semi-conducteurs dopés ou extrinsèques
Dans un matériau pur, on introduit des impuretés par dopage. Pour que celui-ci soit
contrôlable, il faut que le degré de pureté initial global soit supérieur au taux du dopage. Les taux de dopage utilisés sont de l’ordre de 10-8 à 10-11. Une mole de silicium (28 g)
correspond à 6,023.1023 atomes et la densité du silicium est voisine de 7 : 1 cm3 de silicium contient donc environ 1,5.1023 atomes. Avec un taux de dopage de l’ordre 10-10, il y a environ 1,5.1013 atomes d’impureté par cm3.
* – Dopage de type N (Fig.ІІ-5)
CHAPITRE II : Les semi-conducteurs
17
On introduit dans la matrice de silicium des atomes d’impuretés pentavalents tels que le
phosphore P, l’arsenic As et l’antimoine Sb.
Chaque atome d’impureté amène un électron de valence supplémentaire. Cet électron est peu lié au noyau (E ≈ 0,01 eV) et passe aisément dans la bande de conduction.
La conductivité du matériau (conductivité extrinsèque) devient à cause du taux de dopage, très supérieure à celle du matériau pur. Les atomes pentavalents ou donneurs deviennent des ions positifs après le passage des électrons excédentaires dans la bande de conduction.
Les données numériques précédentes montrent que le nombre des électrons dans le matériau, fonction du taux de dopage, est supérieur au nombre des trous, fonction de la température, d’un facteur supérieur à 103
. La conduction dite de type N (négative) est assurée par des électrons. Les électrons sont les porteurs majoritaires.
*– Dopage de Type P
On introduit dans le réseau une impureté trivalente : bore B, aluminium Al, gallium Ga, indium In. Il manque à l’impureté un électron de valence pour assurer les 4 liaisons avec les atomes de silicium voisins. Un faible apport d’énergie (≈ 0,05 eV) suffit pour qu’un électron d’un silicium voisin soit capté par l’impureté : il y a formation d’un
trou peu lié et donc mobile. Les atomes trivalents (accepteurs) deviennent des ions
négatifs par capture d’un électron. Compte tenu des taux de dopage, ces trous sont beaucoup plus nombreux que les porteurs intrinsèques du cristal pur. La conduction de
type P (positive) est assurée par des trous. Les trous sont les porteurs majoritaires.
IІ -4-
Bandes d’énergie des semi-conducteurs dopés Type N Type P
Les atomes de pentavalents (donneurs) introduisent des charges positives dans le réseau, charges qui attirent les électrons en créant ainsi de nouveaux niveaux dont l’énergie est légèrement inférieure à ceux de la bande de conduction du matériau pur. Si on élève la température, ces électrons peuvent passer dans la bande de conduction.
Les atomes de trivalents (accepteurs), introduisent des trous dans la bande de valence. Si on élève la température, ces trous se comportent comme des charges positives libres.
18
IІ -4-
Les gaps d’énergie des semi-conducteurs
[2].Matériau gap d’energie (eV)
0K 300K Si 1.17 1.11 Ge 0.74 0.66 InSb 0.23 0.17 InAs 0.43 0.36 InP 1.42 1.27 GaP 2.32 2.25 GaAs 1.52 1.43 GaSb 0.81 0.68 CdSe 1.84 1.74 CdTe 1.61 1.44 ZnO 3.44 3.2 ZnS 3.91 3.6
Tab. ІІ-1 :Gap d’énergie pour quelques SC
IІІ- LES ALLIAGES
La construction d’alliage à partir des 70 (ou à peu près) métaux primitifs est ensoi un immense sujet. Bien qu’il n’y ait aucune garantie que deux métaux puisse se dissoudre l’un dans l’autre (l’indium, par exemple, ne se dissout pas dans le gallium), la plupart des pairs forment ce que l’on appelle des alliages binaires sur de larges domaines de
concentrations. Des alliages ternaires (à trois composants), quaternaires ( à quatre composants), et ainsi de suite sont aussi réalisés et étudiés.
Les alliages se regroupent commodément en deux grandes classes : ordonnés et désordonnés. Les alliages ordonnés, parfois aussi appelés stœchiométrique, possèdent la symétrie de translation d’un réseau de Bravais. Leur structure est obtenu en plaçant sur chaque site du réseau de Bravais un motif polyatomique.
L’étude théorique des alliages désordonnés est un sujet beaucoup plus difficile. En raison de l’assignation aléatoire des atomes aux sites, il n’existe pas de théorème de Bloch, et sans le nombre quantique k, on ne peut décrire aucune propriété électronique.
CHAPITRE II : Les semi-conducteurs
19
Dans un alliage désordonné, la périodicité est détruite, et l’analyse semi-classique qui conduit à des courants non dégradés en l’absence de mécanisme de diffusion, n’est plus valable [3].
IV. LES SEMI-CONDUCTEURS MAGNETIQUES DILUES (DMS)
IV.1.INTRODUCTION
Depuis l’invention du transistor au milieu du xxe
siècle, l’électronique repose sur la manipulation de porteurs de charge électrique dans des assemblages de matériaux semi-conducteurs sur lesquels on applique des champs électriques. L’électron porte non seulement une charge mais aussi un spin et un moment magnétique associé :ce moment magnétique de chaque électron est sensible à l’application d’un champ magnétique, mais les énergies mises en jeu sont faibles devant les fluctuations thermiques à température ambiante. Ce spin n’intervient donc pas en électronique.
Dans un matériau ferromagnétique, l’interaction entre les spins individuels permet de les associer dans des domaines magnétiques stables à haute température .Les propriétés magnétiques de tels matériaux sont utilisées depuis longtemps comme support pour le stockage de l’information (disques magnétiques par exemple ).Plus récemment, la découverte d’assemblage artificiels de matériaux présentant des propriétés de
magnétorésistance géante (forte variation de la résistance électrique sous application d’un champ magnétique ) a suscité le développement d’une électronique de spin à base de métaux magnétiques. Ainsi, des têtes de lecture compactes et sensibles sont déjà commercialisées. Cette électronique de spin reste cependant difficile à intégrer dans l’électronique traditionnelle : pour cela, il faut fabriquer des structures et des composants hybrides, associant métaux magnétiques et semi-conducteurs.
Elaborer des semi-conducteurs aussi proches que possible des semi-conducteurs usuels de la microélectronique, mais présentant en outre des propriétés magnétiques très couplées aux propriétés électroniques, permettrait de conjuguer les avantages de
l’électronique de spin et les possibilités de la microélectronique. C’est cette piste qu’ouvre le ferromagnétisme induit par les porteurs dans les semi-conducteurs magnétiques dilués
20
IV.2.INJECTION DE PORTEURS POLARISES EN SPIN
Dans les semi-conducteurs ІІ-ІV, on peut remplacer une partie des atomes métalliques par des atomes de transition : par exemple, les atomes Zn de ZnTe par des atomes Mn ( de configuration 3d54s2).
L’alliage obtenu, Zn1-xMnxTe, est un semi-conducteur magnétique dilué.
Les deux électrons s du manganèse peuplent la bande de valence du semi-conducteur comme l’auraient fait ceux du zinc, si bien que l’impureté ne dope pas le matériau. Les électrons d restent localisés sur l’impureté. Cette couche d étant incomplète, l’ion de transition est porteur d’un spin (5/2 dans le cas Mn) et donc avec un moment magnétique. On s’attend alors à mesurer une aimantation MMn de type paramagnétique, caractéristique d’un ensemble de spins indépendants. A faible champ, l’aimantation d’un tel système est donné par ou` la susceptibilité est proportionnelle à la densité de spins et
inversement proportionnelle à la température :
(Loi de Curie) (II.1) En résumé, l’incorporation d’impuretés magnétiques comme le manganèse permet d’introduire des spins localisés. Ceux-ci apportent une aimantation qui ne s’établit qu’à basse température et est réduite par les interactions antiferromagnétiques à courte distance, mais peut être grandes (jusqu’à 1021ions Mn non appariés par cm3 et un spin 5/2 par ion). Surtout, les spins localisés sont en forte interaction avec les électrons des bandes du semi-conducteur [5]
IV.3. DENSITE D’ETAT DANS LES FERROMAGNETIQUES
Afin d’expliquer de manière phénoménologique l’apparition d’un moment magnétique spontané dans les métaux ferromagnétiques, Weiss a supposé au début du siècle l’existante dans ces matériaux d’un champ magnétique interne, dit champ moléculaire ou d’échange, qui a tendance à orienter parallèlement les spins électroniques
[5] .
Ce champ est une représentation approximative de l’interaction d’échange intra-atomique en mécanique quantique. Une interaction Uéch d’origine électrostatique lie en
effet deux électrons de spin Si et Sj présents sur des orbitales atomiques d’un même atome : (II.1) Ou` Jéch est l’intégrale d’échange, dont la valeur est liée au recouvrement des
CHAPITRE II : Les semi-conducteurs
21
Cette interaction trouve son origine dans la répulsion coulombienne existant entre électrons et dans le principe d’exclusion de Pauli. Ainsi, deux électrons de même spin étant forcément sur deux orbitales différentes se repoussent moins que deux électrons de spins opposés placés sur la même orbitale. Pour minimiser l’énergie de répulsion colombienne, il est plus favorable d’avoir le plus grand nombre de spins parallèles, ce qui correspond à la règle de Hund.
Si l’on considère maintenant le cas d’un atome de spin Si parmi N autres, on peut modéliser le couplage entre cet état et N autres par l’interaction d’échange suivante [6]: (II.2)
Où γ est le rapport gyromagnétique liant le spin de l’électron au moment magnétique qu’il lui est associé, et est le champ moléculaire.
∑ é (II.3) Si Jéch est strictement positif, l’interaction est maximale pour les orientations de spin parallèles. L’interaction d’échange favorise donc la mise en parallèle des orientations de spin et on a un matériau ferromagnétique (Fe, Co, Ni). Au contraire, si Jéch est strictement négatif, le matériau est antiferromagnétique car le cas le plus favorable est alors celui d’orientations de spin opposées. Le chrome et le manganèse sont des exemples de tels matériaux.
Enfin, dans le cas d’une description type de bande électronique, l’utilisation du concept de champ moléculaire prévoit un décalage des densités d’états des électrons d suivant leur orientation de spin (phénomène de spin splitting). Ce spin splitting (ΔE de 0,5 à 1eV) est en effet favorable à la minimisation de l’énergie d’un cristal ferromagnétique. Dans le cas du fer, du cobalt et du nickel, la forme globale de la densité d’états peut se résumer à l’allure schématisée sur la (figure ІІ.6.) Sur cette figure deux types de bande d’énergie apparaissent : celui correspondant à des électrons de type s, qui est indépendant à l’orientation de spin, et celui correspondant à des électrons de type d, qui est caractérisé par un phénomène de spin splitting. Le fer, le cobalt et le nickel se distinguent par la position relative du niveau de Fermi (indiquée respectivement par EF Fer, EF Co, EF Ni,
(Fig. ІІ.6) par rapport à ces bandes.
Dans le cas du Ni et du Co, la bande des électrons de type d à spin vers le haut (Fig.1, à droite) est entièrement sous le niveau de Fermi. Elle est donc complètement remplie et les électrons qu’ils occupent sont dits majoritaires. Le niveau de Fermi traverse
22
en revanche la bande des électrons à spin orienté vers le bas (Fig. ІІ.1, à gauche).Cette bande n’est donc pas complètement remplie et les électrons qui l’occupent sont dits minoritaires.
Ainsi, la bande d du Ni et du Co apparait fortement polarisée en spin au niveau de Fermi : le nombre d’états à spin down y est nettement supérieur au nombre d’électrons à spin up.
Dans le cas du Fe, la position du niveau de Fermi par rapport à la bande d est assez Différente : EF Fe ne se situe pas au dessus des états d à spin vers le haut mais traverse cette bande. De ce fait, la polarisation du spin à ce niveau énergétique est plus faible en valeur absolue que dans le cobalt et le nickel.
Sur les figures 2, 3 et 4, nous avons reproduit les densités d’états dans le fer, dans le cobalt et enfin dans le nickel. La polarisation de spin au niveau de fermi est bien fortement négative dans le nickel et le cobalt, elle est faiblement positive dans le fer.
Les bandes d, correspondant aux différents extrema apparaissant sur les figures 2, 3 et 4, sont assez étroites en énergie et représentent une densité d’états très grande par rapport à la bande des électrons s. La bande s s’étend sur environ 18 eV alors que les bandes d ne s’étendent que sur environ 6 eV chacune. La densité d’états dans les bandes d dépasse celle dans les bandes s d’environ un ordre de grandeur. En fait les électrons d sont considérés comme des électrons localisés que l’on modélise plutôt par une approche du type liaison forte, alors que les électrons s sont généralement considérés comme des électrons quasi libres. Par exemple dans le cas du cobalt la masse effective des électrons s
Fig. ІІ.6. Allure de la densité d’état dans un métal de transition ferromagnétique.
Les niveaux énergétiques EF Fe, EF Co et EF Ni indiquent la position relative du niveau
de Fermi respectivement dans le fer, le cobalt et le nickel par rapport aux différentes bandes présentes.
CHAPITRE II : Les semi-conducteurs
23
est à peu près égale à celle des électrons dans le vide (m0) alors que la masse effective des électrons d est de l’ordre de 5m0.
Fig. ІІ.7. Densité d’état dans le fer. L’origine des énergies choisie correspond à la
position du niveau de Fermi.
Fig. ІІ.8.Densité d’état dans le cobalt. L’origine des énergies choisie correspond à la position
du niveau de Fermi.
24
IV.4. LA TEMPERATURE DE CURIE
Tout matériau magnétique qui est aimanté est caractérisé par un certain ordre des petits
aimants atomiques. Chauffer un matériau est une façon de perturber cet ordre magnétique et de provoquer le désordre. Ainsi, pour chaque matériau magnétique, il existe une température de Curie (de Pierre Curie prix Nobel de physique). Au delà de cette température les matériaux sont dans un état désordonné dit paramagnétique [7].
Les températures de Curie peuvent être variées selon les matériaux :
Plus de 1100°c pour le cobalt qui peut rester aimanté jusqu’à haute température, 770°c pour le fer mais aussi moins 270°c pour d’autres matériaux qui n’ont alors que peut d’applications. Matériau Tc (K) Co 1 388 Fe 1 043 Fe2B 1 015 FeOFe2O3 858 NiOFe2O3 858 CuOFe2O3 728 MgOFe2O3 713 MnBi 630 Cu2MnAl 630 Ni 627 MnSb 587 MnB 578 Matériau Tc (K) MnOFe2O3 573 Y3Fe5O12 560 Cu2MnIn 500 CrO2 386 MnAs 318 Gd 292 Au2MnAl 200 Dy 88 EuO 69 CrBr3 37 EuS 16,5 GdCl3 2,2
![Fig. I.2 : Cellule de Wigner –Seitz du réseau de Bravais cubique face centrée (f.c.c.) [23]](https://thumb-eu.123doks.com/thumbv2/123doknet/14935404.665726/16.892.154.621.791.1091/fig-cellule-wigner-seitz-réseau-bravais-cubique-centrée.webp)
![Fig. І.3:La première zone de Brillouin de la structure zinc- zinc-blende [24 ]](https://thumb-eu.123doks.com/thumbv2/123doknet/14935404.665726/18.892.241.698.446.822/fig-première-zone-brillouin-structure-zinc-zinc-blende.webp)

![Fig. ІІ-10. Température de Curie calculée pour différents semi- semi-conducteurs avec un dopage 5℅ de Mn et 3.5x10 20 trous cm -3 [9]](https://thumb-eu.123doks.com/thumbv2/123doknet/14935404.665726/38.892.222.692.707.982/fig-іі-température-curie-calculée-conducteurs-dopage-trous.webp)