Computational Analysis of Biochemical Networks For
Drug Target Identification and Therapeutic
Intervention Design
by
Nirmala Paudel
MEng, University of Oxford (2009)
MASSCHUSETTS INSW1 E
OF TECHNOLOGY
JUN 18 2014
LIBRARIES
Submitted to the Department of Biological Engineering
in partial fulfillment of the requirements for the degree of
Doctor of Philosophy
at the
MASSACHUSETTS INSTITUTE OF TECHNOLOGY
June 2014
©
Massachusetts Institute of Technology 2014. All rights reserved.
A uthor ...
Certified by ...
Signature redacted
Department of Biological Engineering
May 22, 2014
Signature redacted...
'I
Bruce Tidor
Professor of Biological Engineering and Computer Sciences
,rhesis Supervisor
Accepted by...Signature
redacted...
Forest M. White
Associate Professor of Biological Engineering
Chairman, Department Committee on Graduate Theses
Thesis Committee
Accepted
bySignature
redacted
K. Dane Wittrup
C.P. Dubbs Professor of Chemical and Biological Engineering
Chairman, Thesis Committee
Accepted by
....
.
...
Bruce Tidor
Professor of Biological Engineering and Computer Science
Signature redacted
Thesis Supervisor
A ccepted by ...
...
Domitilla Del Vecchio
W.M. Keck Career Development Professor in Biomedical Engineering
Computational Analysis of Biochemical Networks For Drug
Target Identification and Therapeutic Intervention Design
by
Nirmala Paudel
Submitted to the Department of Biological Engineering on May 22, 2014, in partial fulfillment of the
requirements for the degree of Doctor of Philosophy
Abstract
Identification of effective drug targets to intervene, either as single agent therapy or in com-bination, is a critical question in drug development. As complexity of disease like cancer is revealed, it has become clear that a holistic network approach is needed to identify drug targets that are specially positioned to provide desired leverage on disease phenotypes. In this thesis we develop a computational framework to exhaustively evaluate target behaviors in biochemical network, either as single agent or combination therapies. We present our single target therapy work as a problem of identifying good places to intervene in a network. We quantify a relationship between how interventions at different places in network affect an output of interest. We use this quantitative relationship between target inhibited and output of interest as a metric to compare targets. In network analyzed here, most targets show a sub-linear behavior where a large percentage of targeted molecule needs to be inhibited to see a small change on output. The other key observation is that targets at the top of the net-work exerted relatively small control compared to the targets at the bottom of the netnet-work. In the combination therapy work we study how combination of drug concentrations affect network output of interest compared to when one of the drugs was given alone at equivalent concentrations. By adapting the definitions of additive, synergistic, and antagonistic com-bination behaviors developed by Ting Chao-Chou (Chou TC, Talalay P (1984), Advances
in enzyme regulation 22: 27-55) for our system and systematically perturbing biochemical
pathway, we explore the range of combination behaviors for all plausible combination tar-gets. This holistic approach reveals that most target combinations show additive behaviors. Synergistic, and antagonistic behaviors are rare. Even when combinations are classified as synergistic or antagonistic, they show this behavior only in a small range of the inhibitor concentrations. This work is developed in a particular variant of the epidermal growth fac-tor (EGF) recepfac-tor pathway for which a detailed mathematical model was first proposed
by Schoeberl et al. Computational framework developed in this work is applicable to any
biochemical network.
Thesis Supervisor: Bruce Tidor
Acknowledgments
I would like to thank my advisor, Bruce Tidor, for all his support and guidance during
my time here at MIT. I particularly appreciate his role in helping me develop an insight into rigorous, hypothesis driven research with a strong emphasis on principled execution of scientific methods and effective communication of scientific findings.
I am equally grateful to all the members of the Tidor lab that I have had the privilege of
interacting with during my time here. A sincere thanks to David Hagen, Ishan Patel, Andrew Horning, David Flowers, Raja Srinivas, Brian Bonk, Kevin Shi, Nate Silver, Gil Kwak, Pradeep Ravindranath, Devanathan Raghunathan, Sudipta Samanta, and Sarah Guthrie for helpful scientific (and others) discussions and feedbacks. David Hagen deserves a special mention for maintaining the KorneckerBio toolbox and helping me understand mathematical formulations behind it in my early days in the lab. I would particularly like to thank Tina Toni, Yang Shen, Yuanyuan Cui, and Filipe Gracio for their friendship, encouragement, and support. I would also like to thank Nira Manokharan, Tidor lab administrator, for friendly chats, and stocked up stationery cupboard and tea counter.
I appreciate the contributions of my thesis committee members, Dane Wittrup and
Domi-tilla Del Vecchio, for their guidance and support. They have played an important role in encouraging me to ask important research questions and have provided helpful feedback
during committee meetings and beyond.
I would like to take this opportunity to specially mention two organizations, without
whose support my academic journey would not have come this far. PestalozziWorld and Pestalozzi International Village Trust changed the course of my life by financially supporting my education from primary school right up to undergraduate level. I will forever be indebted to them for this unparalleled opportunity.
Finally, I would like to thank my family for their love, support, and encouragement. The trust they have bestowed on me and the freedom they have provided me from an early age have been crucial in my exploration of opportunities across continents. I would also like to thank Suresh Sitaula for his support and encouragement.
Contents
1 Introduction 10
1.1 Background and Motivation . . . . 10
1.2 Biochemical Pathways . . . . 13
1.2.1 Epidermal Growth Factor (EGF) Receptor Pathway . . . . 14
1.3 Computational Modeling of Biochemical Pathways . . . . 15
1.3.1 Computational Models of EGFR Pathway . . . . 17
1.3.2 Variant Models of EGFR Pathway . . . . 21
1.4 Structure of This Thesis . . . . 22
2 A Framework for Evaluating Efficacies of Single Agent Therapy 24 2.1 Introduction . . . . 25
2.1.1 The Biochemical Model . . . . 26
2.1.2 M odel Variants . . . . 27
2.1.3 Format of Study . . . . 27
2.1.4 Summary of Findings . . . . 28
2.2 M ethods . . . . 28
2.2.1 The Normal Model . . . . 28
2.2.2 Cancer Variant Models . . . . 29
2.2.3 Drug Intervention Models . . . . 29
2.2.4 Target and Output Effect Metrics . . . . 32
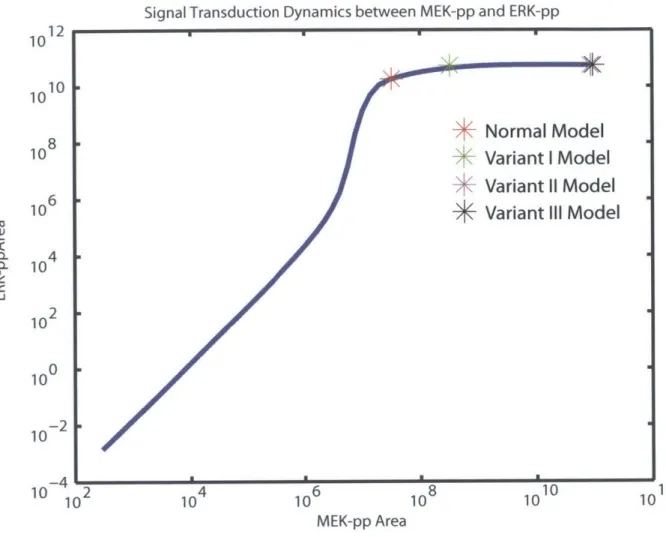
2.2.5 Signal Transduction between MEK and ERK . . . . 32
2.2.6 Parameter Variability Study . . . . 33
2.3.1 Intervention-free Models . . . . 34
2.3.2 Intervention Analysis . . . . 36
2.3.3 Normal Model - Target Comparisons . . . . 39
2.3.4 Cancer Variant Models . . . . 41
2.3.5 Signal Transduction Between MEK and ERK . . . . 42
2.3.6 Parameter Variability Analysis . . . . 44
2.4 Discussion . . . . 47
3 Computational Approach to Analyze Drug Combination for Synergy and Antagonism 51 3.1 Introduction . . . . 53 3.1.1 Biochemical Model . . . . 54 3.1.2 Format of Study . . . . 55 3.1.3 Summary of Results . . . . 56 3.2 M ethod . . . . 57
3.2.1 Combination Behavior Definitions . . . . 57
3.2.2 Drug Intervention Models . . . . 61
3.2.3 Target and Output Effect Metrics . . . . 63
3.2.4 Combination Summary Metric . . . . 64
3.2.5 Parameter Variability Analysis . . . . 65
3.3 R esults . . . . 66
3.3.1 General Trends . . . . 67
3.3.2 Additive Targets . . . . 68
3.3.3 Synergistic Targets . . . . 70
3.3.4 Antagonistic Targets . . . . 72
3.3.5 Parameter Variability Analysis . . . . 75
3.4 Discussion . . . . 76
4 Therapeutic Design Strategies for Safety and Efficacy 80 4.1 Introduction . . . . 81
4.2.1 Model Details and Setup . . . .
4.2.2 O bjectives . . . .
4.2.3 Design of Intervention Strategies and Optimization Framework . . . 4.2.4 Targets D esign . . . . 4.3 Analysis of the Optimized Designs . . . . 4.4 Sum m ary . . . .. . . . .. . 5 Summary and Future Directions
A Single Target Intervention
A.1 Target Effect Metric . . . . A.1.1 Single Substrate Enzymatic Reactions . . . . A.1.2 Two or More Substrate Enzymatic Reactions . . . .
A.2 Biochemical Network . . . . A.3 Molecular Identities of Targets Inhibited . . . .
B Combination Target Intervention
B.1 Molecular Targets Inhibited . . . .
C Effects Exerted by Interventions
C.1 Mathematical Basis . . . . C.1.1 Kinetically-Tuned Inhibitors . . . .
C.1.2 Feedback and Feed-Forward Loops . . . .
82 83 84 87 88 91 92 105 105 105 107 110 122 129 129 135 135 135 136
List of Figures
1-1 Schematic of epidermal growth factor receptor (EGFR) pathway . . . . 19
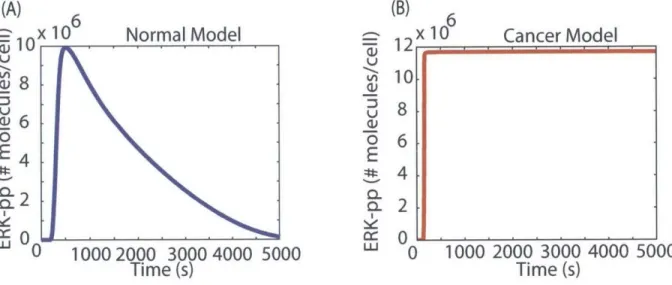
1-2 Definitions of normal and cancer phenotypes in terms of ERK-pp signal dy-n am ics . . . . 22
2-1 Overview of single target evaluation method . . . . 2-2 Schematic summary of three overstimulated (cancer) variants of EGFR pathway
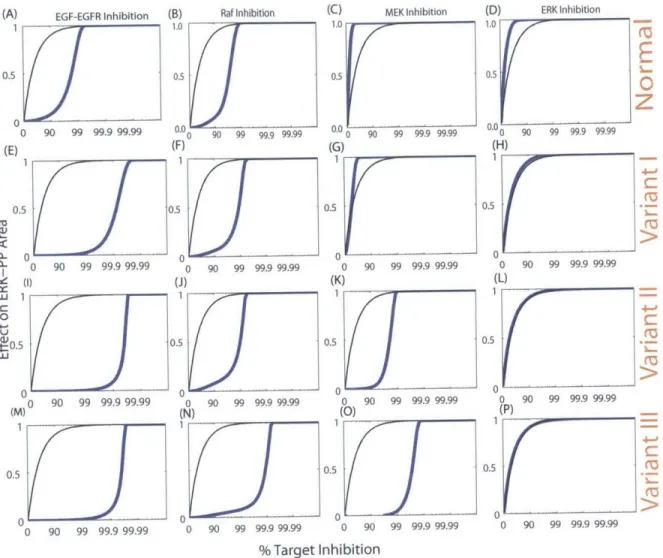
2-3 Overview of signal propagation dynamics in EGFR models . 2-4 Experimental comparison of target inhibition behavior . . . 2-5 Representative range of target behaviors in the normal and EGFR pathways . . . .
2-6 Signal transduction dynamics between MEK-pp and ERK-pp
2-7 Effects of parameter variability on target behaviors . . . . .
3-1 3-2 3-3
3-4
overstimulated
Overview of target combination evaluation method . . . . .
Definitions of combination target behaviors . . . . Schematic representation of combination metric . . . . Summary of all combination behaviors in EGFR pathway . .
3-5 Representative combinations showing additive and synergistic targets in EGFR
p athw ay . . . .
3-6 Representative combinations showing antagonistic behaviors in EGFR pathway
3-7 Model ensemble behaviors of combination targets . . . .
3-8 Combination behaviors in over-stimulated models . . . . 31 35 37 39 43 44 46 59 60 66 69 71 74 75 76
4-2 Schematics of therapeutic design strategies . . . . 85
4-3 Optimized designs for normal trajectory objective . . . . 89
Chapter 1
Introduction
1.1
Background and Motivation
Biology is increasingly turning into a quantitative science with advancements in experimental measurement techniques. The precision and the breadth with which cellular and molecular events in cells and tissues can be measured mean a detailed picture of the inner working principles of a cell, either in isolation or as a member of tissue, is emerging rapidly. Central to this endeavor is a quest to elucidate and understand the networks (be it at genetic, protein, or metabolic levels) that control cellular decisions and phenotypes both in normal physiological conditions and in pathology. In parallel, mathematical and computational methods are being developed to calibrate these networks based on the experimental measurements both at single-cell and population levels. One of the major goals of this endeavor is to use these quantitative models to make useful clinical predictions that can aid in drug design and discovery processes.
The goal of this thesis research is to develop methods to aid in drug design and discovery based on knowledge of biochemical networks as it becomes available. There are two questions that we address in this work. The first is "What makes a good target?" We develop and use holistic network-level strategies to find drug targets, either for single agent therapy or in combination, that are especially well positioned to exert suitably large effects on the very pathway the drug is trying to alter. This approach ties in with the popular idea of 'druggability of the target' that is well established in the drug design world and literature
[61]. The question of 'druggability of a target' has, so far, been looked at mainly from a molecular design perspective [22]. The question has been primarily interpreted as ease or difficulty with which a small molecule inhibitor can be designed to block a target of interest. Here, we take a step back and look at the problem from a network level, which adds an extra dimension to the problem and consequently its solution. The aim here is not to replace the idea of 'druggability' as it exists but to complement it with a holistic view of signal propagation in network of interest. The question we ask is, given the network of interactions required for a particular cellular phenotype, what are the most suitable targets in the pathway that are likely to produce desirable change at the output or output signature of interest. This framing differentiates the target at which drug acts from the output of interest.
The motivation for this question comes from the fact that a 'druggable' target in the traditional sense has little meaning if intervening at this target does not affect the phenotype of interest. On the other hand, an intervention molecule that is difficult to design precisely at molecular level and hence only affects the target minimally, can still have a desired effect on the output of interest given the non-obvious way in which this effect propagates down the signaling cascade. This question would have been redundant if all biological networks had a single path for signal propagation with linear dynamics. In such a case, it would not really matter where one decides to intervene. A proportional effect would be observed at output. However, given the highly non-linear and branched nature of biological networks [44, 40, 102], we believe that this question can complement well the idea of 'druggability of the target' in designing suitable intervention molecules for suitable targets. It is important to point out that 'target identification' is a well established concept within drug development [19, 92]. But the question is framed differently. Most of the efforts so far has been to find mutated protein (resulting from mutated genes) [27, 80, 99, 29] that leads to the deregulation of the pathway activity and propose it as a drug target. Though this approach has been successful to an extent, it is our belief that a holistic network-based method, as we propose here, leads to an unbiased and potentially more fruitful approach to target selection. Mutated proteins of a particular pathway might not always be the suitable weak points that we might want to exploit in the design endeavor. However, if they truly are important nodes for drug
interventions, our analysis has a potential to identify those without any bias.
The second question that this thesis explores is intervention strategies that actively con-sider the multi-factorial, and often conflicting, objectives that drugs would ideally meet. The most common objective is selectivity, not just in terms of the molecules they target but also in terms of targeting only a fraction of the cells or tissues that are deregulated without affecting the ones in the vicinity that are functioning normally. An efficient way of doing this would be to target molecules specific to the pathology that are not present in normal cells. Despite immense effort in finding disease-specific molecules that can be targeted for interventions [21, 115], the picture that is emerging is that, in complex diseases like cancer, normal and diseased cells contain the same or very similar molecules, albeit in different quantities and functioning slightly differently [46, 106]. Hence, attempts to find specific disease markers for targeting have met with limited success. To complement this process, we take a systems approach and probe this question by considering classic, simple engineering design principles to explore the capabilities they provide in differentially regulat-ing the normal and disease phenotypes. The major motivation for this comes from the need to include adverse effects of drug as an active part of the design process. Drug side effects, such as those arising from chemotherapies in the case of cancer, not only limit the amount of drug that can be given to the patients but also affect greatly the quality of life of patients. An ability to address this aspect right at the design level has the potential to help identify intervention strategies with fewer side effects. While this kind of designs are not quite as common in drug development industry yet, emergence of field of synthetic biology with a goal of forward engineering biological components from known part is heading towards that directions. In this light, we computationally explore design principles that are little more complex than simple inhibitors that may also provide higher level of flexibility in terms of their design specifications.
In summary, we explore capabilities of three kinds of intervention strategies to evaluate their potentials and limitations in achieving two multi-factorial design objectives. The first objective is formulated to make both disease (cancer) and normal cells signal as normal cells. The second objective is formulated as a little more challenging problem of allowing normal cells to function as normal, but blocking signaling in disease (cancer) cells thus halting
their growth and proliferation. An important question that this part of thesis tries to raise is that, given the challenges posed by the design goals, can simple inhibitor molecules that regulate biology in almost boolean-logic like fashion (i.e., "on" and "off') meet multi-factorial objectives of minimizing the side effects and maximizing the efficacy of drugs, or are there inherent limitations in the designs and hence the need to think beyond inhibitors to more modular intervention strategies with advanced logical functionality?
Combined together, research work that is described in this thesis uses network level infor-mation of biochemical pathways to study where the better or worse drug targets are, where beneficial combination targets are, and what kind of design strategies need to be adopted and explored in order to design drugs that maximize the therapeutic window by actively maximizing for efficacy and minimizing for toxicity. Both computational and experimental approaches can be taken to evaluate these questions. In this thesis we use a computational framework. This allows us to look at the trends much more exhaustively and evaluate how the results depend on uncertainty about biochemical reactions in signaling pathways.
1.2
Biochemical Pathways
Cells, either in isolation or as a member of a tissue organization, can carry out large array of computations and functions. Most of these functions are achieved by interaction networks of proteins. These interaction networks can exist at genetic, proteomic, or metabolic levels. In genetic level networks, proteins or protein complexes are used to control when one or more genes are activated or inactivated. Most of the protein-protein interaction networks are used to transmit or integrate information from one part of the cell to another. At the metabolic level, proteins are used either to break down molecules from food, like sugar or fat, to energy units (adenosine triphosphates - ATPs) that cells can use or to utilize the resources available to synthesize macromolecules and other biomolecules that are needed for the cells to grow or divide properly. In this viewpoint almost all the cellular processes that range from sensing the environment, integrating large array of information, transmitting information from one part or compartment to another, making a cellular phenotypic decision and executing it is, dependent on the interaction networks that exist within cell.
Understanding the network based interactions of proteins in normal cells should give a complete picture of how a cell functions. Study of these interactions in pathology should provide a guide to understand how these interactions are deregulated, paving ways to ra-tionally counteract these deregulations. However, given the complexity of studying all the interactions in a cell as whole, the task is made more tractable by studying interactions of small pathways present within larger networks in isolation. The hope is that detailed under-standing of the working principles of each pathway can be put together to understand the system as a whole. More importantly studying and understanding the currently tractable subsets of biochemical networks can give actionable insights that can be used to prolong life, or reduce the discomfort that result from disease process. For these actionable goals, we have to make decisions in the face of uncertainty. Understanding of small subset of interactions does reduce this uncertainty though cannot completely eliminate it.
1.2.1
Epidermal Growth Factor (EGF) Receptor Pathway
The epidermal growth factor (EGF) induced EGF receptor (EGFR) pathway is one such example of a cellular pathway that has been extensively studied to understand how cells sense the external environment and then transmit and integrate this information to make a phenotypic decisions [112, 83, 48, 26]. EGFR belongs to a family of receptors that have kinase activity leading to tyrosine phosphorylation at specific sites [59]. This receptor family is collectively called the receptor tyrosine kinase (RTK) family. EGFR is arguably the most well studied and understood system within the RTK family. This detailed understanding of EGFR receptor system has been crucial in elucidating functions, behaviors, and regulations of other members of the RTK family [112, 48]. This receptor system has been used in under-standing basic processes such as receptor-mediated endocytosis [96], oncogenesis [117, 52], mitogen-activated-protein-kinase (MAPK) signaling pathways, and receptor trans-activation
[18]. It is also the first system to emphasize the role of mechanistic mathematical modeling
to understand complex, integrated biological systems [65, 7]. Most of our current under-standing of receptor binding, internalization, and degradation is derived from quantitative models of these processes in EGFR system that were aided by related experimental measure-ments [37, 111]. Further, discoveries of mutations in some of the signaling proteins of this
cascade in a number of human epithelial cancers have provided us a basis for understanding processes like oncogenesis. Detailed understanding of the signal transduction in this path-way and deregulation of this signaling pathpath-way in various types of cancer has meant that proteins of this pathway have been successful candidates of some targeted cancer therapeutic strategies [91, 34].
In normal physiology, EGFR pathway is initiated by binding of EGF ligand to form EGF-EGFR monomer. The monomers come together to form dimers that are activated by auto-phosphorylation at number of tyrosine residues [75]. There are at least 20 auto-phosphorylation sites on the receptor [114], although exact detail of how many and what combinations of phosphorylation are needed for the activation or what the exact roles of these multiple phosphorylations sites are is unclear. The activated (phosphorylated) receptor dimer can signal and thus activate a number of downstream adaptor proteins that eventually lead to activation of important transcription factors that trans-locate into the nucleus to activate genes associated with growth, differentiation, or proliferation [83]. This pathway, which is normally associated with cellular phenotypes like cell growth and proliferation, [71, 66, 45, 93] has been found to be over-stimulated (signaling above the normal values) in large number of solid human cancers [116, 81]. A number of proteins that single in this pathway are mutated in cancers and are the sources of pathway over-stimulations [64, 105, 103]. Identification of this direct clinical relevance of this pathway has meant that it has been studied both in academic settings to discover the signaling principles and in pharmaceutical industries to understand the pathway such that suitable drugs can be designed to reverse the disease
(cancer) progression.
1.3
Computational Modeling of Biochemical Pathways
Towards the end of 20th century, molecular biology, which is concerned with study of net-work of interacting molecules that define cellular behaviors (and subsequently higher level interactions that define tissue, organ, and organism behaviors), started to see changes in the ways it was being studied and analyzed. The field slowly started moving away from reductionist way of studying molecular network one reaction at a time to a systems level
study which focuses on looking at molecular network as a whole by measuring and analyz-ing systemic level changes. This shift lead to an emergence of new field of biology called systems biology [54, 109, 110] that started to draw a lot parallels with studying and ana-lyzing man-made engineered systems. Unlike engineered systems, where there is a detailed knowledge of how parts are assembled together, components of these biological systems are largely unknown and have to be established through perturbation and inference [4, 47, 15]. This can be thought of as reverse engineering the cellular processes to understand how cells achieve their functions. In other words, it is framed as a problem of trying to understand a systems that was already 'engineered' (in this case evolved) but its blueprint was missing.
As the molecular networks started to be studied as a system rather than its component parts, the qualitative nature of analysis and interpretation of experimental data collected became cumbersome and non-intuitive paving a way for quantitative modeling, analysis, interpretation of the results in process giving rise to a sub -field of computational systems biology [63, 104, 54, 112] that emphasized on mathematical modeling of biochemical inter-actions. Mathematical models had been used to understand, interpret, and model biological processes much earlier [67, 42, 72]. However, their use in study of molecular network biol-ogy really took off towards the beginning of the 21st century. A crucial contribution in the emergence of this field comes from technological advances that lead to high-throughput data collection possible.
Despite the role of computations and bioinformatics in completing the human genome project, computational biology is still a young field. The most popularly known form of computational biology refers to a subfield of bioinformatics. This mostly comprises of using computer science algorithms to shift through, align, and stitch together vast amount of genomics data. There is a second field of computational biology, sometimes referred to as computational systems biology, that is involved with modeling and understanding molecular interaction networks (be it at genetic, proteomic, or metabolic levels) as dynamic systems. The structure of these models are derived from the knowledge of the underlying biology which they aim to quantify. Broadly speaking, one can think of bioinformatics as a process of assembling a static picture of the cellular make up, and dynamic modeling as way of understanding how these systems respond to changes or perturbations in their environments.
The second class of computational biology that aims to build dynamic molecular networks of biological systems (sometimes called computational systems biology) forms a basis for this thesis.
The work described here builds up on the mathematical dynamic models of molecular networks to get insights on how they behave in response to stimuli. How this dynamic network level understanding of biology can be exploited to find suitable drug targets for single and combination therapies is one of the goals of the work presented here. We also exploit how these network biology models can be utilized in designing suitable interventions strategies to maximize efficacy and minimize toxicity. There are number of mathematical techniques that are used to model molecular network dynamics. Some of the more prevalent techniques are deterministic kinetic models described by systems of ordinary differential equations [5, 86,
102], stochastic kinetic models [100, 101], fuzzy logics [3, 77], and agent-based models [85,
1071. These modeling techniques have played a crucial role in understanding the underlying
mechanism of biological and cellular functions.
1.3.1
Computational Models of EGFR Pathway
For the purpose of this thesis project that is concerned with development of system level methods for target identification and therapeutic intervention design, EGFR (section 1.2) pathway provides a natural place to start. Given a detailed biological understanding of the system, well developed mathematical models, and its deregulation in number of human epithelial cancers, it lays the right background for us to ask questions that go beyond model calibration. Further, key topological features like non-linearity arising from the bimolecular nature of interactions, the branched nature of signal transduction, and the signaling events that expand across multiple compartments encompass the key aspects of signaling pathways that we want to train my methods on.
Among various available mathematical models of this pathway [50, 62, 20, 90] we choose to start with the model proposed by Schoeberl et al. [94] which incorporates signaling events associated with this pathway from binding of EGF ligand to EGFR eventually leading to the activation mitogen activated protein kinase (MAPK) called extracellular-regulated kinase (ERK). The choice is mainly influenced by the level of details incorporated in the model,
actual size of the model, and key topological features like the branched nature of the signal
transduction and signaling events that expands across multiple compartments, namely
-extracellular matrix, cytosol, and endosome. Easy access to the model in our group and familiarity with it in the context of other related projects were also factors that contributed to this model choice. It is important to point out that while this model will be used as basis for developing our methods, the methods themselves are generalizable to other variants of the pathway or models of other biochemical processes.
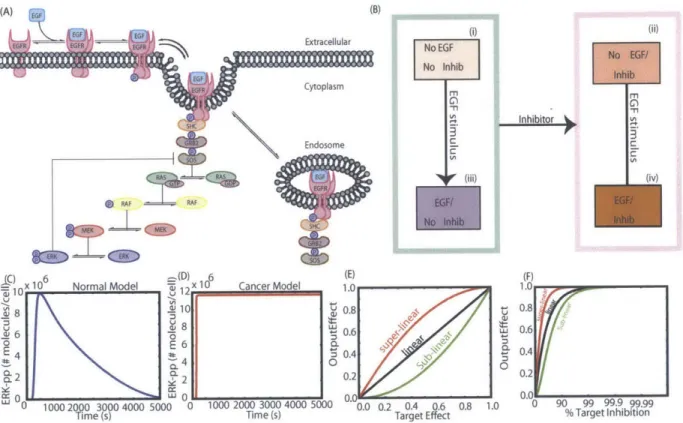
Biochemical model of EGFR pathway proposed by Schoeberl et al. [94] is a deterministic mass-action model represented by a system of ordinary differential equations (ODEs). The exact variant of the model that we use is the one that was modified by Apgar et al. [6]. The modification accounts for the synthesis of the adaptor proteins that are degraded along with the receptor complexes. In the original model, when the receptor complexes are degraded, the receptors are synthesized to return the cell to the pre-stimulus state so that it can respond to future stimuli, but the adaptor proteins that were degraded along with receptors were not synthesized. So, although an attempt was made to return the cells to pre-stimulus state, this was not achieved as some of the adaptor proteins that were degraded were not synthesized. Apgar et al. [6] updated this aspect of the model by including synthesis and degradation for adaptor proteins such that the model does return to the pre-stimulus state ready to respond to the next set of input signals. A schematic of this biochemical pathway is shown in Figure
1-1. Only key reaction stages are shown in the schematic for clarity. The detailed model
contains 101 species (protein or protein complexes), that interact in 148 different chemical reactions and are characterized by 107 zeroth, first, or second order rate constants.
For both the drug target identification and intervention strategy design parts of the project, the model system that we are using is Epidermal Growth Factor (EGF) induced
EGF Receptor (EGFR) pathway proposed by Schoeberl [94], Hornberg [50] and modified by
Apgar et al [6]. This pathway is activated in the presence of extracellular ligand EGF which can bind to the EGF Receptor (EGFR) on the cell membrane resulting in its dimerization and cross phosphorylation. The ligand bound phosphorylated receptor dimer recruits and activates a number of Adaptor Proteins in the cell eventually leading to the activation of Mitogen Activated Protein Kinase (MAPK) called Extracellular Regulated Kinase (ERK)
EGV
gExtracellular
Cytoplasm
Endosorm
Figure 1-1: Schematic biochemical pathway of EGF induced EGFR system [6].
by phosphorylations at two different residues. This pathway captures most of the essential
features of a biological signal transduction pathway. The pathway is highly non-linear with bimolecular nature of interactions. The signal can flow in a number of parallel branches down the cascade and the signaling can take place both in the cytosol and in endosome before the endocytosed molecules are either degraded or recycled back to the membrane. This is an important pathway that has been well studied experimentally and has been extensively calibrated making it a suitable model choice for us to ask some of the questions that go beyond model calibration and are the focus of this thesis project. Further, besides having the suitable topological features to develop our methods, the pathway is of huge interest in the academic and pharmaceutical community as it is deregulated in a large fraction of human cancers of epithelial origin [82, 97]. The common mutations that are widely observed in cancers that are associated with deregulations of this pathway are: (1) EGFR mutation or over-expression [57, 87, 1] (2) mutation of Ras protein [12, 13] and (3) mutation of the Raf protein [13, 30, 33, 74]. EGF is usually taken as the input to this pathway and activated (doubly phosphorylated) ERK is taken as its output. We adopt the same convention in this project. The schematic representation of the pathway is shown in figure 1-1. Only a part of the biochemical interactions are shown in the figure for clarity reasons. The detailed model contains 148 chemical reactions between 101 reacting species and are modeled by 107 kinetic rate constants which are either zeroth, first or second order.
This biochemical network is modeled using systems of Ordinary Differential Equations (ODEs). A toy model is shown here as an example of setting up the ODE model from biochemical network of reactions. In this model, species A and B react with second order (bimolecular) rate constant k1 to produce C, C can dissociate back into A and B with first order (unimolecular) rate constant k2 or can go on to form species D with a first order rate (unimolecular) constant k3. Equation (1.1) here represents the biochemical process and equations (1.2),(1.3),(1.4) and (1.5) show how the species A,B, C, and D in the system evolve over time. k1 k3 A+B C- >D (1.1) k2
d[ A]
dt -kl [ A] [B] + k2[C]
(1.2)
dt
d[B]
d
-kl[ A][B] + k2[C]
(1.3)
dt
d[]
- k2[C] (1.4)dt
d[D]
=t k3[C] (1.5)A generic rate law for all the species in a system can be represented as shown below in
equation (1.6):
S= Aix +
A2x Ox +Blu+
B2U o x+
B3U& u + k
(1.6)dt
Here, x is the vector of all the species in the biochemical system, u represents the input vectors and 9 represents the Kronecker product. Kronecker product between vectors a and
b is a vector with all the possible product combinations of elements in a and elements in b.
A
1,
A2,
B1, B2,
B3 contain the suitable coefficients for the reactions which are a combination of stoichiometric matrix, unimolecular, and bimolecular rate constants. k is a vector with constants which is used to capture the fixed production rate of one or more of the species inx.
(ODEs) like the well mixed compartment (mainly resulting from large number of protein involved) is taken to be valid for the system within a given reaction compartment. There are at least 10,000 copies (~ 20nM) of each protein present in this pathway in a cell. The model includes three compartments: (1) the extracellular environment where the ligand is present (2) cytosol where the majority of the signaling events take place (3) endosome where some signaling can still take place before the proteins/molecules are either degraded or recycled back to the plasma membrane. The relative differences in the volumes in these compartments is introduced implicitly through the rate constants of the reactions and hence no explicit correction volume ratio is present in the reaction equations. The ODEs are integrated using odel5s function in matlab 2009a (Mathworks) to evaluate the temporal dynamics of all the proteins and the complexes in the system. The stiff integration provided by the ode15s is necessary for the simulation of the model because the rate constants associated with this model vary across several orders of magnitude. Design, simulation, and optimization of the model parameters as appropriate is core to both the questions that we are trying to explore in this project and these aspects will be discussed in greater details in the sections to follow.
1.3.2
Variant Models of EGFR Pathway
Three variants of the model, besides the 'Original' model (sometimes also referred to as 'normal') proposed by schoeberl [94], Hornberg [50] and modified by Apgar et al [6], are to be used in both parts of the project. These variants are modeled by introducing one of the three common mutations associated with this pathway to obtain an aberrant signaling of the output protein ERKpp. This aberrant ERKpp dynamics will be considered to be the 'cancerous' phenotype of the model. The three mutations considered are: (1) EGFR overexpression and Endocytosis defect, sometimes referred to as 'Cancer 1' (2) Defective form of Ras GTP that has impaired GTPase activity and stabilizes in the GTP bound state, sometimes referred to as 'Cancer 2' and (3) Defective form of Raf protein that cannot be deactivated (dephosphorylated) once it has been activated (through phosphorylation) by up stream GTP, sometimes referred to as 'Cancer 3'. It is important to note here that Ras-GTP is not a kinase as such, but most of the modeling work of this network is implemented as if this were the case because the direct kinase that phosphorylates Raf protein is not known
yet. To the best of our knowledge, we are not aware of experimental data that supports whether each one of these mutations alone is able to transform a normal cell to a cancer cell. Further, we do not know what temporal dynamics of the ERKpp signal can be classified as a cancer phenotype. So, for the purpose of modeling these mutations here, we will consider the transient response (Figure 1-2) as a normal phenotype and sustained high signal as a
cancer phenotype.
(A)
6
(B)
6
X
106
Normal Model
X
106
Cancer Model
U U
38
0
E 4
E
2
0-cL-
1-10
8
6
4
2
"'0
1000 2000 3000 4000 5000
W0
1000 2000 3000 4000 5000
Time (s)
Time (s)
Figure 1-2: Definitions of normal and cancer phenotypes in terms of ERK-pp signaling dynamics
1.4
Structure of This Thesis
This thesis, over a span of next three chapters (Chapter 2, Chapter 3, Chapter 4), explores two key questions of early stage of drug discovery. The first question is concerned with identifying the best places for intervention, either as single agent therapies or combination therapies. Chapter 2 explores the effectiveness of single agent therapies at different places in the network to evaluate where the best places for intervention are. Chapter 3 evaluates the
same question for combination therapies. Specifically, all plausible target combinations are evaluated to quantify an effect they exert on an output of interest in combination compared to when one of the drug was given alone at equivalent concentrations. Chapter 4 explores the capabilities and limitations of inhibitor therapies that mostly work as "on" or "off switches" for safety and efficacy when they affect both normal and deregulated signaling networks
as is commonly the case in disease site like cancer. We then explore some protein based intervention strategies that may be better suited for multi-factorial objectives that drugs should ideally meet.
Chapter 2
A Framework for Evaluating Efficacies
of Single Agent Therapy
Abstract
Mechanistic systems biology models describe normal and diseased processes of cellular events and serve to represent our current state of knowledge of the relationship between biology and disease. A key goal of this endeavor is to inform clinical decision-making and drug discovery to improve therapeutic approaches using a systems-level view. In this work we focus on the important challenge of selecting effective drug targets. We develop a computational approach that uses network-level information and simulation methodology to probe for the optimal places for intervention. Our method evaluates the amount of control provided by each potential target over network output, thus identifying proteins best poised for intervention. We apply the method to signal transduction in the epidermal growth factor receptor pathway, in which aberrant behavior has been linked to many cancer processes. The results exhibit a wide range in the level of control exerted by different potential targets. Targets near the top of the pathway exert relatively weak control, consistent with known experimental results; some targets near the bottom of the pathway exert much stronger control due to network properties that are analyzed. These behaviors observed are robust to details of the parameterization of the model, suggesting that the specific results obtained here will not be strongly affected by model uncertainty. Taken together, the results of this study provide strong evidence that effects of network structure and dynamics can have a strong influence on drug target effectiveness.
2.1
Introduction
Significant work in cell biology is focused on elucidating the networks of protein interactions
responsible for key cellular processes and that lead to individual phenotypes [53, 10, 8]. An
emerging picture is that these interactions are deregulated to some extent in certain diseases
such as cancer [46, 55], leading to studies undertaken to understand networks in the contexts
of both normal and abnormal physiology [41, 56, 51, 68]. Furthermore, computational and
mathematical approaches are being applied to quantitatively represent these biochemical
networks with different levels of mathematical abstraction [83, 63, 39, 53, 77]. In order to
understand the mechanistic details of signal transduction in these pathways, a class of models
represented by systems of ordinary differential equations (ODEs) is being widely developed
and calibrated using experimental data [94, 14, 2, 20]. An additional benefit from this type
of endeavor could be to develop therapeutic intervention strategies able to address network
deregulation problems from a holistic viewpoint.
A key challenge within this framework is to find nodes (protein or protein complexes) in
a network that are most suited to alter the deregulated network behavior in a desired way
[38, 49, 84, 19]. This challenge, popularly referred to as the 'Target Identification' problem,
has been dominated by molecular-level perspectives. The question of what makes a good drug
target has typically been addressed by identifying proteins whose active sites are especially
amenable to tight-binding by molecules with the size, shape, relative hydrophilicity, and other
properties matching those of current drugs [61, 22]. This focus on "druggability" has led to
targets for which it may be relatively straightforward to develop a tight-binding inhibitor
without assessing the effectiveness with which the node can be used to control biological
functions and disease processes. Here we report the development of a network engineering
method to identify suitable drug targets based on their relative control over disease processes.
This approach is not only new, but it has the potential to lead to especially effective drugs,
2.1.1
The Biochemical Model
We develop and apply the method in the context of the epidermal growth factor (EGF) receptor signaling pathway. It is one of the most thoroughly studied biochemical signal transduction pathways with a wealth of experimental data supporting well established mod-els of network behavior 18, 83, 62, 90. The relatively detailed and well-calibrated nature of the model makes it a suitable candidate for our study, which is concerned with using detailed network-level understanding of biochemical processes to identify suitable places for interven-tion. The particular pathway version of the model that we used for this work was initially
developed by Schoeberl et al [94], later updated by Hornberg et al [50], and further modified
in our group by Apgar et al [6]. The pathway is modeled by a system of ordinary differential equations (ODEs) in which each ODE describes how a particular protein or protein complex in the pathway evolves over time in the presence of an EGF stimulus. The integration of the system of ODEs then gives the temporal dynamics for the concentration of all the proteins and protein complexes in the pathway as a function of time.
The pathway is shown schematically in Figure 2-1A. It is induced by the binding of
EGF ligand to the trans-membrane EGF receptor (EGFR) [11, 89]. For the purposes of
this work, we consider the EGF ligand to be the input of the system. Upon ligand binding
the receptor can dimerize and autophosphorylate [11, 89, 31]. This autophosphorylated
and activated EGF-EGFR dimer can recruit and activate a number of adaptor proteins by providing suitable binding sites. The sequential activation of the adaptor proteins leads to the activation of Ras [16] and of a canonical mitogen-activated protein kinase (MAPK) cascade composed of the proteins Raf, MEK, and ERK [78, 74]. ERK protein is the final protein as modeled, with ERK-pp representing a doubly phosphorylated and activated form, which is treated as the output of the pathway here. ERK-pp is itself an important signaling molecule; it is a kinase with a large number of substrates in both the cytoplasm and the nucleus, and can also act as a transcription factor to activate a number of growth and proliferation related genes [74, 17].
2.1.2
Model Variants
The model proposed by Schoeberl et al [94] describes normal pathway dynamics in the
presence of an EGF stimulus. Given that this pathway is deregulated (over-stimulated)
in a large number of human cancers of epithelial origin [91], we chose to study the ability
of inhibitors targeted to different nodes in the pathway to attenuate the over-stimulated
pathway response. To this end, we modeled three variants of the pathway that we refer
to as 'cancer variants'. These variants were modeled by introducing one of three common
mutations that are associated with over-stimulation of this pathway in various types of
cancers. These three mutations are: (1) EGFR over-expression together with a defect in
endocytosis [79, 31, 28, 9] (2) mutation of the Ras protein, which is a frequently mutated
oncogene [13, 9] and (3) mutation of Raf protein, which is again a common mutation in a
large number of cancers [13, 9]. Each of the three mutations leads to overstimulation of the
EGFR pathway represented by prolonged activation of ERK protein.
2.1.3
Format of Study
Here the question of target identification is formulated from a network perspective. The
goal is to find protein targets whose inhibition reduces network output most effectively. For
each candidate target protein or protein complex in the pathway, we augmented the models
to introduce and simulate a competitive inhibitor at a range of different concentrations and
evaluated the effect on pathway output (ERK-pp). This approach is shown systematically
in Figure 2-1B.
The relationship between the amount of inhibitor introduced in the model, its direct effect
on the target inhibited, and the effect on the pathway output was evaluated for 14 candidate
targets in the pathway. This comparative approach provided a quantitative evaluation of
relative target effectiveness and helped identify species in the network predicted to be most
effective in network attenuation.
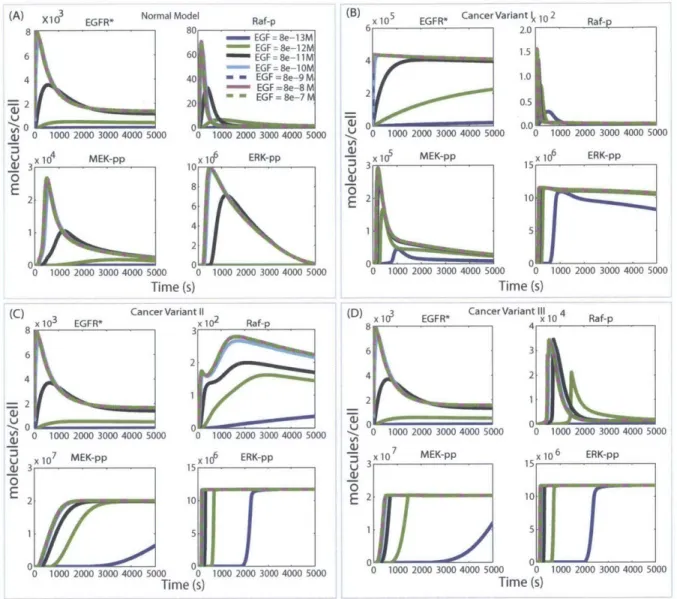
attenuation. The 'Target Effect' is a direct measure of the fraction of target inhibited whereas the 'Output Effect' is quantified as the reduction in ERK-pp signal measured as either the integral under the curve of the concentration trajectory or the peak height for the trajectory [50, 8].
2.1.4
Summary of Findings
The results of the study show a very wide range of effectiveness across the panel of poten-tial targets examined, with more effective targets found downstream, close to the output. These observations are not strongly sensitive to which pathway model of EGFR signaling signaling was used or the particular parameters used in simulating models. Furthermore, we demonstrate that network dissection and detailed analysis of signaling dynamics of the pathway can provide important insights that can be used to understand the basis for the target behaviors observed.
2.2
Methods
2.2.1
The Normal Model
The ODE pathway model for signaling downstream from EGFR utilized in the current study evolved from the original model by Schoeberl et al [94], as modified by Hornberg et al [50]
and further updated by Apgar et al [6]. Here our normal model is the Apgar et al [6]
version. The term "normal" refers to the published model of the pathway to distinguish it from perturbed versions containing cancer-associated changes that lead to exaggerated responsiveness. The model has 13 unique proteins that comprise 101 unique chemical species through the formation of complexes and catalytic modification. Model dynamics are driven
by 163 elementary chemical reactions that are described using mass-action kinetics. A feature
of mass-action kinetic formulations is that they contain only zeroth-, first-, and second-order reactions; all higher-second-order abstracted reactions are written as a series of these more
elementary ones. Parameters of the model include 107 distinct rate constants and 101 initial concentrations; in addition, there is 1 input (EGF).
2.2.2
Cancer Variant Models
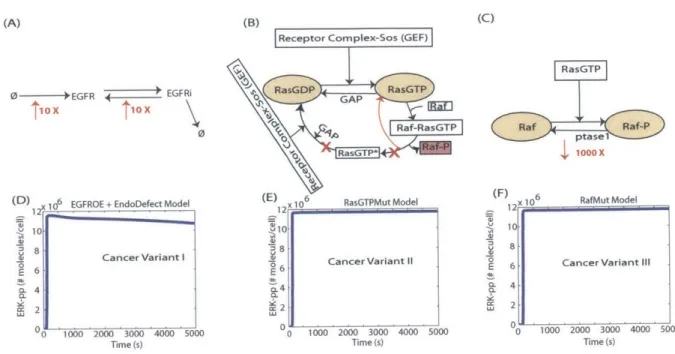
Three variants of the normal model were constructed as plausible mechanisms of deregulation that might represent processes operating in cancer cells. Variant I was obtained by increasing the rate of production of EGFR protein by 10 fold while also increasing its recycling rate from endosomes to the plasma membrane by 10 fold (Figure 2-2A) [79, 1]. Whereas the
unstimulated normal model has a steady-state receptor number of 8.28 x 10' cell-1, for
the Variant I model this value was increased 53 fold to 4.38 x 105 cell- fold. Variant II
was obtained by short-circuiting the activation-deactivation--reactivation process of Ras to reflect compromised GTPase activity that arises from point mutations of the same class as G12V, which we model as preventing GTP hydrolysis thus leading to prolonged Ras-GTP activity (Figure 2-2B). In the published model, upon activation of one molecule of Raf protein, Ras-GTP is hydrolyzed back to Ras-GDP to start the next round of the activation cycle. This hydrolysis step was removed in the Variant II model here, keeping Ras-GTP in an activated form longer [95]. Variant III was obtained by decreasing the association rate constant for p binding the phosphatase for the dephosphorylation step of activated
Raf-p Raf-protein to Raf by 1000 fold. This mimics the Raf-presence of a constitutively activated form
of the protein in the model (Figure 2-2C) and acts similarly to a common mutation V600E
[30]. While different investigators might have chosen different implementation details, the
processes represented here are directly drawn from common mutational alterations known to affect tumor cells.
2.2.3
Drug Intervention Models
A series of modified versions of the normal model and of cancer variants I, II, and III
competitive inhibitor that specifically targeted one of 14 plausible chemical species in each
model; 14 modified versions of the normal and of each of the three variants were constructed
to represent targeting each plausible species in the system. In the work reported here each
target bound inhibitor in a second-order reaction to form a complex that was completely
inactive. This inhibitor-target complex was either allowed to dissociate back to the target
and the inhibitor or degrade at the rate of degradation of the target protein. In other
work we treated the inhibitor-target complex as inhibiting only some of the activities of
the target or as being a non-competitive inhibitor of target. The models used here had the
inhibitor act in a non-depleting manner to simulate the effect of a large volume of drug
present in cell culture or in circulation that replenished drug that bound to target. Two
new parameters were introduced in each model variant for the kon and koff for the inhibitor
binding to target. Values of 1.66 x 10-6 cell molecule-1 s-1 and 1 x 10- 3 S-1 were used for second-order association rate and first-order dissociation rate, respectively. These values
are equivalent to 1 x 106 M- 1 S-1 for the association and 1 x 10-3 S-1 for the dissociation
rate constants using typical dimensions of a mammalian cell (1 x 10-l L) [94], giving a unit
nanomolar equilibrium dissociation constant.
In simulations the pathway was equilibrated in the presence of the inhibitor before
stim-ulation with the EGF growth signal. For each target of interest, inhibitor was introduced at
100 different logarithmically spaced concentrations, between 6 and 6 x 108 molecules cell- 1, which corresponded to a maximum concentration of 1 mM using typical dimensions for a
mammalian cell.
For each level of inhibitor concentration introduced, output signatures of interest were
measured and compared with the case in which the intervention was not present in the
pathway. A schematic of this process is shown in Figure 2-1B, where stage (i) represents
the model with no EGF stimulus and no inhibitor (intervention), stage (ii) represents the
pathway behavior when the model has been equilibrated in the presence of the inhibitor but
stimulus without any intervention, and stage (iv) represents the system with intervention in
the presence of EGF stimulus. Simply stated, we equilibrated at (i) and used that as the
starting state for a type (iii) simulation; likewise, we equilibrated at (ii) as a starting point
for a type (iv) simulation.
(A) (B V ---- Extracellular Cytoplasm Endosome .RAF RAF 'L 6(D) 6 (E)
106 Normal Model x10 Cancer Model 1.
8 .210 110.8 6 00.8 4 0. 2 0.2 C0 LU0 0.C0 -0 1000 2000 3000 4000 5000 0 1000 2000 3000 4000 5000 0.0 Time (s) Time (s) 00i Wi No EGF No Inhib M C Cii 0.2 0.4 0.6 0.8 1.0 Target Effect m -n n C (iv U aj (F) 0.8
0.61 /
0.2 0. r 0 90 99 99.9 99.99 %Target InhibitionFigure 2-1: Overview of target evaluation strategy. (A) Schematic representation of the EGFR signaling pathway studied here. (B) The strategy compares network behavior in the presence and absence of candidate inhibitors. (C) Dynamics of pathway output (ERK-pp) upon stimulation with step increase of 8 nM EGF at time zero, for the normal model. (D) Idealized representation of overstimulated ERK-pp output dynamics in the presence of activating mutations in the pathway. This is the phenotype typical of variants of the pathway with activating mutations. (E) Illustrative behaviors expected for different types of targets, depending on the relationship between the target and the output. The black line represents the case with a linear relationship between the target and output. The non-linear nature of signal transduction means the actual trend can deviate from this linear behavior either in a sub-linear (green line) or super-linear (red line) manner. (F) Expected trends of (E) on a semi-log scale, as this is the scale used in presenting the simulation results.
)
2.2.4
Target and Output Effect Metrics
The focus of this study is to quantify the relationship between target binding and output
attenutation. Thus, metrics were chosen to quantify each of these system perturbations.
In each case we chose a fraction (or percentage) metric. The Target Effect is defined as
the fraction of available target that is bound by inhibitor and thus inactivated; the Output
Effect is the fractional associated decrease in output signal. For each model (normal and the
variants) these fractional changes were calculated with respect to the signal strength in the
absence of any intervention in the model.
Target Effect (2.1)
[1 ] + Ki
Where, [I] = inhibitor concentration, Ki = inhibitor binding affinity.
Output Effect = (unperturbed output - perturbed output) (2.2)
unperturbed output
Where, "unperturbed" represents model without inhibitor and "perturbed" represents model
with inhibitor.
2.2.5
Signal Transduction between MEK and ERK
In order to understand why MEK went from being super-linear in normal model to sub-linear
in overstimulated variant models, the signal transduction dynamics between MEK and ERK
was analyzed further by perturbing the normal model and measuring the signals at MEK-pp
and ERK-pp levels. More specifically, we varied a parameter in the model ('k42'- one that
affects the binding of phosphorylated-Raf to its phosphotase) by three orders of magnitude
on either side of its nominal value in the normal model. This parameter span was sampled
at 100 different values in a log scale. The normal model was simulated for each of these 100
values for 'k42'. This resulted in 100 different signals at MEK-pp and ERK-pp levels (the
resulting ERK-pp values were plotted against MEK-pp values to evaluate the relationship
between these two species in the network. Further, we evaluated the MEK-pp and ERK-pp
signals for normal and the three variant models. These four data points were overlayed on
the curve describing the relationship between MEK-pp and ERK-pp values obtained from
'k42' variation.
2.2.6
Parameter Variability Study
Each parameter in the unperturbed normal model was sampled using Latin Hyper-cube
Sampling (LHS) method. We used a log-normal distribution with mean values of the normal
model and the standard deviation of 0.5. This meant that 95% (2o-) of the parameters
sampled were within 10 fold of the nominal parameter values (i.e. the 95% of the parameter
range was from 0.1 x nominal values to 10 x nominal values). 10,000 parameter sets and
hence 10,000 models were generated from this parameter space. Each model was first run to
steady state before applying an EGF stimulus of 8-nM.
Only the models with parameter sets that were able to stimulate the pathway were chosen
for further analysis. The criterion used in selecting model for further analysis was that the
model should produce at least half the output (ERK-pp area for 5000s) of the unperturbed
normal model. For each of the chosen models, the target behavior was evaluated by inhibiting
the model at each of the 14 nodes in the network at 100 different concentrations of inhibitor.
Resulting target behavior for each target was classified as sub-linear, super-linear, and
in-between (ambiguous) behaviors using a three point classifier. The three point classifier
compared the output effect of the inhibition to that of linear effect at three different values
of target effect. If all the three point showed output effect less than that expected from a
linear response then that particular target behavior of the model under question was classified
as sub-linear. Similarly, if the output effect was greater than what would be expected from
linear response at all the three target effect levels the behavior was classified as super-linear.
![Figure 1-1: Schematic biochemical pathway of EGF induced EGFR system [6].](https://thumb-eu.123doks.com/thumbv2/123doknet/14431273.515222/19.918.201.684.129.417/figure-schematic-biochemical-pathway-egf-induced-egfr.webp)
![Figure 2-4: Comparison between the simulation results and the results reported in Chen et al [20] for a cell-based assay with inhibition of equivalent targets](https://thumb-eu.123doks.com/thumbv2/123doknet/14431273.515222/39.918.104.788.124.710/figure-comparison-simulation-results-results-reported-inhibition-equivalent.webp)
![Figure 2-7: Parameter Variability and Target Behaviors: The parameters (kinetic rate constants and initial protein concentrations) were sampled from log-normal distribution with the mean value set to the value published in [94],](https://thumb-eu.123doks.com/thumbv2/123doknet/14431273.515222/46.918.110.790.177.801/parameter-variability-behaviors-parameters-constants-concentrations-distribution-published.webp)
![Table 2.1: A sample of intervention strategies that are currently available (or under development) for down regulation of EGFR pathway [25, 70].](https://thumb-eu.123doks.com/thumbv2/123doknet/14431273.515222/50.918.127.758.160.445/table-intervention-strategies-currently-available-development-regulation-pathway.webp)
