Impact of climate change on tropospheric ozone and its global budgets
Texte intégral
Figure
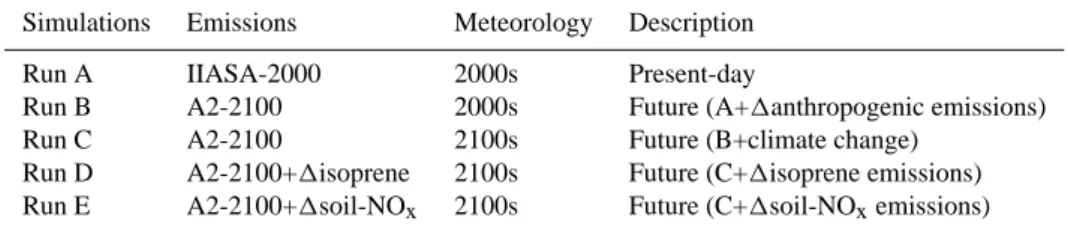
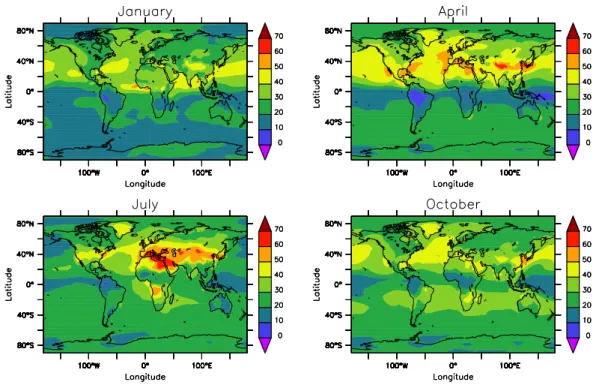


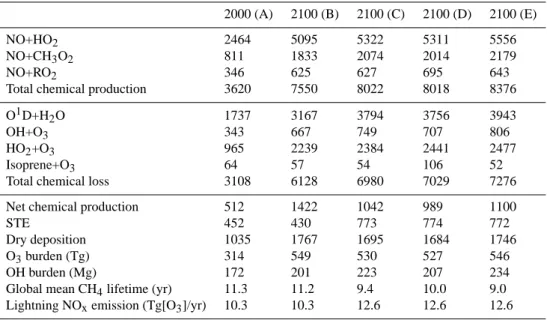
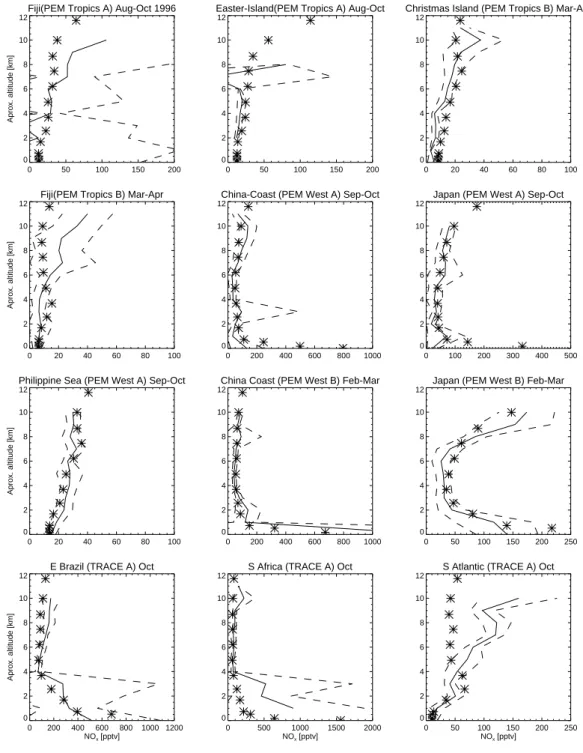
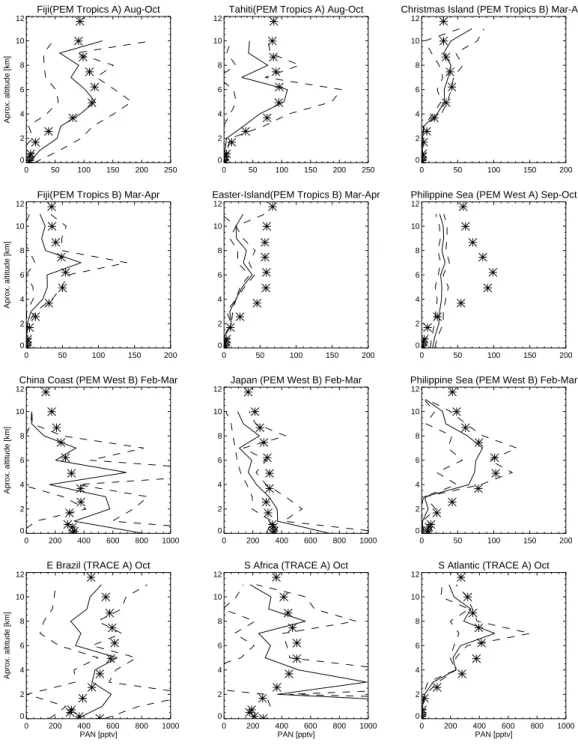
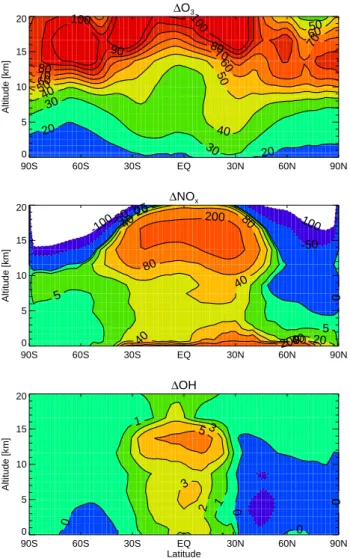

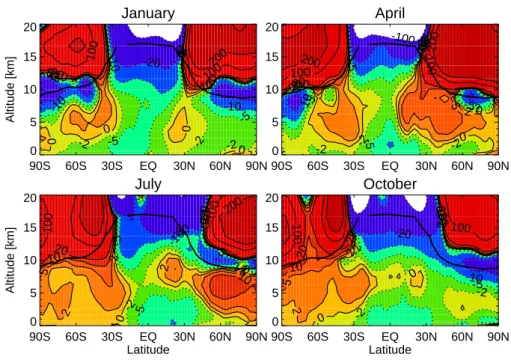
Documents relatifs
The choice of these projects was made with regard to the stormwater management devices implemented (detention and infiltration basins, swales, draining trenches,
B-measure for the Model described in Subsection ( 3.1 ) with initial temperature equal to 15 ◦ C for the minimal temperature and 22 ◦ C for maximum temperature and maturity time
The interplay between the mean flow, mesoscale stirring and bathymetry gen- erates complex and peculiar horizontal transport patterns in the Mediterranean that contributes to
Total new OH ( ΣOH new ) production and termination in reaction channels of the toluene system, calculated over the course of the 22/10/97 experiment.. Wagner et al. Time profiles
Cette relation entre hydrologie sous-glaciaire et dynamique glaciaire est la base de l’article présent dans ce chapitre (Article#4) qui propose une nouvelle lecture de la
Quantity and Quality of Dispersed Fine Particles after the Low-Energy Water-Dispersible Soil Test: Impact of the Initial Soil Matrix Potential... 1 Quantity and quality of
The main goal of this study is to evaluate the primary pro- ductivity of two contrasting years associated with La Ni˜na and El Ni˜no events in low and midland areas of
Worldwide distribution of the 5,565 islands with native monocot species and their classification according to four categories of vulnerability (low, moderate, high, very high)
