HAL Id: hal-00303128
https://hal.archives-ouvertes.fr/hal-00303128
Submitted on 11 Oct 2007HAL is a multi-disciplinary open access
archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
The time evolution of aerosol composition over the
Mexico City plateau
L. I. Kleinman, S. R. Springston, P. H. Daum, Y.-N. Lee, L. J. Nunnermacker,
G. I. Senum, J. Wang, J. Weinstein-Lloyd, M. L. Alexander, J. Hubbe, et al.
To cite this version:
L. I. Kleinman, S. R. Springston, P. H. Daum, Y.-N. Lee, L. J. Nunnermacker, et al.. The time evolution of aerosol composition over the Mexico City plateau. Atmospheric Chemistry and Physics Discussions, European Geosciences Union, 2007, 7 (5), pp.14461-14509. �hal-00303128�
ACPD
7, 14461–14509, 2007
Time evolution of aerosol over the Mexico City plateau
L. I. Kleinman et al. Title Page Abstract Introduction Conclusions References Tables Figures ◭ ◮ ◭ ◮ Back Close Full Screen / Esc
Printer-friendly Version Interactive Discussion
EGU Atmos. Chem. Phys. Discuss., 7, 14461–14509, 2007
www.atmos-chem-phys-discuss.net/7/14461/2007/ © Author(s) 2007. This work is licensed
under a Creative Commons License.
Atmospheric Chemistry and Physics Discussions
The time evolution of aerosol composition
over the Mexico City plateau
L. I. Kleinman1, S. R. Springston1, P. H. Daum1, Y.-N. Lee1, L. J. Nunnermacker1, G. I. Senum1, J. Wang1, J. Weinstein-Lloyd2, M. L. Alexander3, J. Hubbe3,
J. Ortega3, M. R. Canagaratna4, and J. Jayne4
1
Brookhaven National Laboratory, Upton, NY, USA
2
SUNY, Old Westbury, NY, USA
3
Pacific Northwest National Laboratory, Richland, WA, USA
4
Aerodyne Research Inc., Billerica, MA, USA
Received: 12 September 2007 – Accepted: 20 September 2007 – Published: 11 October 2007 Correspondence to: L. I. Kleinman ([email protected])
ACPD
7, 14461–14509, 2007
Time evolution of aerosol over the Mexico City plateau
L. I. Kleinman et al. Title Page Abstract Introduction Conclusions References Tables Figures ◭ ◮ ◭ ◮ Back Close Full Screen / Esc
Printer-friendly Version Interactive Discussion
EGU Abstract
The time evolution of aerosol concentration and chemical composition in a megacity ur-ban plume was determined based on 8 flights of the DOE G-1 aircraft in and downwind of Mexico City during the March 2006 MILAGRO field campaign. A series of selec-tion criteria are imposed to eliminate data points with non-urban emission influences. 5
Biomass burning has urban and non-urban sources that are distinguished on the basis of CH3CN and CO. In order to account for dilution in the urban plume, aerosol con-centrations are normalized to CO which is taken as an inert tracer of urban emission, proportional to the emissions of aerosol precursors. Time evolution is determined with respect to photochemical age defined as –Log10 (NOx/NOy). The geographic distri-10
bution of photochemical age and CO is examined, confirming the picture that Mexico City is a source region and that pollutants become more dilute and aged as they are advected towards T1 and T2, surface sites that are located at the fringe of the City and 35 km to the NE, respectively. Organic aerosol (OA) per ppm CO is found to in-crease 7 fold over the range of photochemical ages studied, corresponding to a change 15
in NOx/NOy from nearly 100% to 10%. In the older samples the nitrate/CO ratio has leveled off suggesting that evaporation and formation of aerosol nitrate are in balance. In contrast, OA/CO increases with age in older samples, indicating that OA is still being formed. The amount of carbon equivalent to the deduced change in OA/CO with age is 56 ppbC per ppm CO. At an aerosol yield of 5% and 8% for low and high yield aromatic 20
compounds, it is estimated from surface hydrocarbon observations that only ∼9% of the OA formation can be accounted for. A comparison of OA/CO in Mexico City and the eastern U.S. gives no evidence that aerosol yields are higher in a more polluted environment.
ACPD
7, 14461–14509, 2007
Time evolution of aerosol over the Mexico City plateau
L. I. Kleinman et al. Title Page Abstract Introduction Conclusions References Tables Figures ◭ ◮ ◭ ◮ Back Close Full Screen / Esc
Printer-friendly Version Interactive Discussion
EGU 1 Introduction
Evidence is accumulating that the mass of organic aerosol (OA) can be an order of magnitude greater than can be explained on the basis of model calculations or mea-surements of precursor VOCs (volatile organic compounds) (Heald et al., 2005; de Gouw et al., 2005; Volkamer et al., 2006; Cubison et al., 2006; Johnson et al., 2006; 5
and Takegawa et al., 2006). At the same time other measurements and model calcu-lations yield reasonable agreement (e.g. Heald et al., 2006). Interest stimulated by this problem has yielded several plausible explanations and many new findings on aerosol – gas interactions (e.g., Jang et al., 2002; Kalberer et al., 2004; Goldstein and Galbally, 2007; Robinson et al., 2007).
10
The Mexico City MILAGRO field campaign conducted in March, 2006 offered an ex-cellent opportunity to investigate questions related to the production of OA in particular and the time evolution of aerosol properties in general. Eighteen million people re-side in Mexico City making it one of the world’s largest population centers. The region is characterized by high concentrations of gas phase and aerosol pollutants (Molina 15
and Molina, 2002). As an example of other megacities, whose numbers are predicted to grow in the coming decades, Mexico City is both a laboratory for understanding OA production in a polluted region and an example of a globally important source category. Volkamer et al. (2006) have extrapolated results on OA production observed in Mexico City to the global aerosol budget, finding that up to 1/3 of secondary organic aerosol 20
(SOA) could be due to anthropogenic VOCs – compared with a 6% contribution from current global models.
With these findings in mind, a focus of the MILAGRO campaign (http://www.eol.ucar. edu/projects/milagro/) and its DOE component (MAX-Mex; Megacity Aerosol Experi-ment in Mexico City (http://www.asp.bnl.gov/MAX-Mex.html)
25
was to examine the evolution of aerosols over time scales ranging from hours to days. This problem was attacked in several ways. Three surface sites were located on the Mexico City plateau in a configuration appropriate for Lagrangian sampling when
ACPD
7, 14461–14509, 2007
Time evolution of aerosol over the Mexico City plateau
L. I. Kleinman et al. Title Page Abstract Introduction Conclusions References Tables Figures ◭ ◮ ◭ ◮ Back Close Full Screen / Esc
Printer-friendly Version Interactive Discussion
EGU winds were from the S to SW (Doran et al., 2007). One site, T0, was located in a high
emission rate section of Mexico City, while the other two, T1 and T2, were located 30 and 63 km downwind. Aircraft measurements from the G-1, augmented by several C-130 flights, were made over T0 and other parts of the city as well as on E-W transects that went over T1 and T2. This allowed for capturing plumes from Mexico City that 5
went in a general northerly direction but happened to miss T1 and/or T2. Multi-day transport was investigated by the C-130 at locations where the Mexico City plume was predicted to be transported. A C-MET balloon was used as a Lagrangian marker. The NASA DC-8 investigated transport on still larger spatial scales, primarily over the Gulf of Mexico.
10
Previous studies have used a variety of sampling strategies to determine the time evolution of aerosol. Volkamer et al. (2006) relied primarily on observations in a source region, while de Gouw et al. (2005), Johnson et al. (2006), and Takegawa et al. (2006) primarily used observations from fixed receptor sites. Aircraft data from the 2004 NEAQS/ITCT campaign has been analyzed in terms of enhancements of water sol-15
uble OA and CO in individual plumes whose age is known by trajectory analysis or by ratios of reactive VOCs (Sullivan et al., 2006; Weber et al., 2007). The present study follows the approach used by Kleinman et al. (2007) in which measurements are assembled from multiple flights covering a range of photochemical ages between nearly fresh emissions and air masses that are approximately 1 day old. By using 20
photochemical age as a metric for atmospheric processing, we take advantage of the general layout of sampling sites but are not confined to the few instances where an air mass was intercepted at multiple downwind distances.
We are mainly concerned with urban emissions over the Mexico City plateau and restrict the G-1 data set by location and by trace gas composition, the latter set of 25
restrictions designed to minimize impacts from forest fires and industrial and utility emissions. In order to account for dilution of the urban plume with “background” air, CO is used as a conservative tracer of urban emissions and results are normalized to CO concentration. Photochemical age is marked by the oxidation of NOxand operationally
ACPD
7, 14461–14509, 2007
Time evolution of aerosol over the Mexico City plateau
L. I. Kleinman et al. Title Page Abstract Introduction Conclusions References Tables Figures ◭ ◮ ◭ ◮ Back Close Full Screen / Esc
Printer-friendly Version Interactive Discussion
EGU defined as – Log10(NOx/NOy).
This study presents results on the photochemical age dependence of SOA; organic aerosol mass peaks amu/z = 44 and 57 (M44 and M57) which are surrogates for oxy-genated organic aerosol (OOA) and hydrocarbon-like organic aerosol (HOA); and other non-refractory aerosol constituents (NO−3, SO2−4 , NH+4, and Cl−) all of which are mea-5
sured with an Aerodyne C-ToF-AMS. Preliminary to determining the age dependence of aerosol constituents, a set of selection criteria are described with particular attention to the use of CH3CN and CO in identifying and removing from our data set air masses that are impacted by non-urban biomass burning. Ambient aerosol concentrations and concentrations normalized to CO are provided as a function of photochemical age. The 10
increase with age of OA per ppm CO is compared with a calculated value based on urban measurements of aromatic hydrocarbons and literature values of aerosol yields. Comparisons are made with measurements in the eastern U.S.
2 Experimental
The G-1 was equipped with instruments to measure chemical and microphysical prop-15
erties of aerosol particles as well as gases that are either aerosol precursors or tracers of emission sources. Flight time was concentrated on characterizing fresh emissions over Mexico City and determining their evolution over time durations of order 1 day or less and spatial scales of order 100 km or less.
Data used in this study are 10 s average values. Trace gas concentrations are ex-20
pressed as mixing ratios by volume. Aerosol properties are reported at standard condi-tions of 20◦C and 1 atmosphere. Local times are used in this study. Data are archived at ftp://ftp.asd.bnl.gov/pub/ASP%20Field%20Programs/2006MAXMex/. In this study flights are referred to by month (m), day (dd), and an “a” or “b” for the 1st or 2nd flight of a day, i.e. 318a for the first flight on 18 March 2006. The format for a flight identifier 25
ACPD
7, 14461–14509, 2007
Time evolution of aerosol over the Mexico City plateau
L. I. Kleinman et al. Title Page Abstract Introduction Conclusions References Tables Figures ◭ ◮ ◭ ◮ Back Close Full Screen / Esc
Printer-friendly Version Interactive Discussion
EGU 2.1 Flights
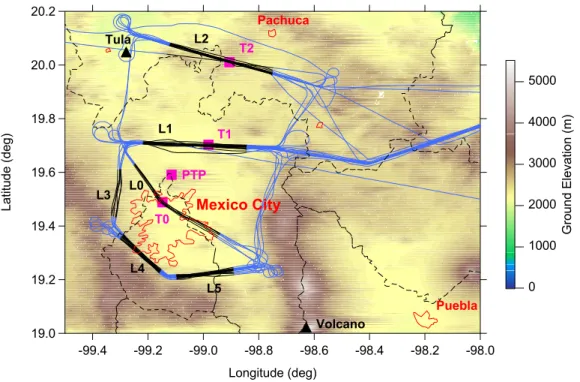
Measurements discussed in this paper are confined to the Mexico City plateau, west of 98◦ Longitude. Figure 1 shows the sampling area, surface measurement sites, and the G-1 ground track. There were 15 G-1 flights between 3 March and 27 March 2006, of which 8 had the requisite combination of measurements to examine the time 5
evolution of aerosol properties using CO as a conservative tracer and NOx/NOy to determine photochemical age. Date, time and the number of transects over Legs L0 to L5 (defined in Fig. 1) are given in Table 1. Often a back and forth pattern was flown above the T1 and T2 surface sites. Altitudes above ground level (a.g.l.) were 500 to 2500 m and 200 to 2400 m for transects above the T1 and T2 surface sites, 10
respectively. Urban crossings over T0 were mainly at an altitude of ∼550 m a.g.l. The multi-stranded appearance along the L0, L1, and L2 legs is due respectively to 6, 18, and 13 overflights. As indicated in Table 1 sampling over the Mexico City plateau was distributed between mid-morning and late afternoon.
According to emission maps shown by Lei et al. (2007), T0 is located near the area 15
of peak emission density. T1 is at the fringe of the Mexico City urban area, and T2 is 35 km outside. Figure 1 shows additional urban areas as well as the locations of the Tula power plant refinery complex, and the Popacatapetl volcano. Biomass burning on the plateau and adjacent mountains was visually observed from the G-1. Satellite observations of fires in adjacent areas are given by Yokelson et al. (2007).
20
2.2 Instruments
Table 2 provides a list of instruments used in this study. CO, NOx, NOy, and SO2 measurements have been described by Springston et al. (2003). Acetonitrile (CH3CN), toluene, and terpenes (and other species not used in this study) were quantified by a proton transfer mass spectrometer (PTR-MS) with a measurement cycle that was 25
typically 15 s (Lindinger et al., 1998).
ACPD
7, 14461–14509, 2007
Time evolution of aerosol over the Mexico City plateau
L. I. Kleinman et al. Title Page Abstract Introduction Conclusions References Tables Figures ◭ ◮ ◭ ◮ Back Close Full Screen / Esc
Printer-friendly Version Interactive Discussion
EGU factor for the instrumentation used in this study. Particle size between 0.1 and 3µm
was measured using a PCASP-100X (Particle Measuring Systems, Inc., Boulder, CO) with SPP-200 electronics (Droplet Measurement Technologies, Boulder, CO) mounted on an external pylon. The number distribution of particles over the size range 16 to 444 nm was determined using an SMPS consisting of a cylindrical Differential Mobility 5
Analyzer (TSI Inc., model 3081) and a condensation particle counter (TSI Inc., model 3010). SMPS data were analyzed using the inversion procedure described by Collins et al. (2002). Aerosol size distributions are obtained at a relative humidity below 25%. For the PCASP this was achieved by using the de-icing heater (Haller et al., 2006). Air flow to the DMA passes through a Nafion dryer. In general, the atmosphere over 10
the Mexico City plateau was very dry (average relative humidity =27%) and one would expect that particles would have little associated water even without active drying.
Non-refractory aerosol mass, composition, and size distributions were determined with an Aerodyne C-ToF AMS (Drewnick et al., 2005). A measurement cycle for ac-quiring a mass spectrum was typically 12 s. In this study we make use of AMS derived 15
concentrations of organics, NO−3, SO2−4 , NH+4, Cl−, and the 44 and 57 amu/z peaks which are primarily CO+2 and C4H+9, respectively. These later peaks denoted here as M44 and M57 have been used as markers for oxidized and un-oxidized hydrocarbons, respectively (Zhang et al., 2005).
The AMS collection efficiency (CE) is the fraction of particles with diameters within 20
the acceptance range of the aerodynamic lens that actually contribute to the mass spectrometer signal. Many field studies have established that the CE of the AMS is about 0.5 (e.g. Canagaratna et al. 2006 and reference contained therein). We have evaluated CE by comparing aerosol volumes deduced from AMS measurements with that determined from the PCASP and DMA number distributions. The following prelim-25
inary steps were taken: 1) AMS concentrations were converted into volumes using the measured non-refractory speciation and densities of 1.2 and 1.77 g cm−3 for organic and inorganic constituents, respectively. 2) PCASP size bins which were based on an assumed refractive index of 1.55 were adjusted according to the refractive index
deter-ACPD
7, 14461–14509, 2007
Time evolution of aerosol over the Mexico City plateau
L. I. Kleinman et al. Title Page Abstract Introduction Conclusions References Tables Figures ◭ ◮ ◭ ◮ Back Close Full Screen / Esc
Printer-friendly Version Interactive Discussion
EGU mined from the AMS speciation using Mie scattering results from Liu and Daum (2000).
As it turned out these corrections were small, typically changing the PCASP volumes by less than 10%. 3) The DMA volume for particles smaller than 100 nm was added to the PCASP volumes. These summed volumes will be referred to as PCASP volumes. The sizes of particles detected by the AMS depends on the transmission characteris-5
tics of the aerodynamic lens. Laboratory results have shown a 100% transmission for particles with a vacuum aerodynamic (va) diameter between ∼60 and 600 nm (Jayne et al., 2000). At a typical aerosol density of 1.5 g cm−3, the equivalent geometric diam-eter range of a spherical particle is 40 to 400 nm. Effects of non-spherical particles on sizing have been discussed in the literature (DeCarlo et al., 2004; Slowik et al., 2004). 10
Particles smaller than 40 nm are not a concern in this study because their mass is neg-ligible. Larger particles, up to 1000 nm are detected, albeit with decreased efficiency.
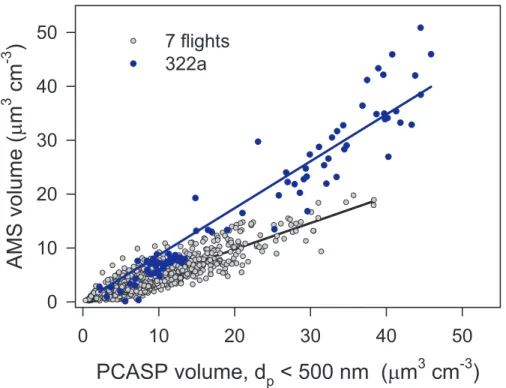
For each flight we have done 3 linear regression of AMS volume versus 1) PCASP volume with Dp<400 nm, 2) PCASP volume with Dp<500 nm, and, 3) DMA volume
with Dp<444 nm. These sharp size cutoffs only approximate the AMS response
func-15
tion but in view of the decreasing amount of mass above Dp(va)=600 nm, these re-gressions should capture most of the information on CE. Figure 2 shows the relation between AMS volume and that from the PCASP with Dp<500 nm. Data are restricted
to urban air masses as described in a following section. Similar results, with slightly shifted scales are obtained using PCASP Dp<400 nm and DMA volumes (figures not
20
shown). According to Fig. 2, the first 7 flights show a compact relation between AMS and PCASP volume with a slope of ∼0.5. Higher aerosol concentrations were observed on the 8th flight (322a) and the ratio of AMS to PCASP volume was distinctly greater than found on any of the other flights. High values of CE, above, 0.5, have been ob-served for acidic aerosols (Quinn et al, 2006; Kleinman et al., 2007) and CE should 25
approach 1 for aerosol with a high proportion of NH4NO3as that compound is used for calibration. Neither condition, however, applies on the 322a flight.
Consistent with results summarized in Table 3, AMS concentrations were calculated with a CE of 0.5 except for flight 322a in which CE = 0.85. Without 322a, the flight to
ACPD
7, 14461–14509, 2007
Time evolution of aerosol over the Mexico City plateau
L. I. Kleinman et al. Title Page Abstract Introduction Conclusions References Tables Figures ◭ ◮ ◭ ◮ Back Close Full Screen / Esc
Printer-friendly Version Interactive Discussion
EGU flight standard deviation of CE is ∼0.08, possibly due to shifts in calibration or changes
in aerosol-type encountered on different days.
3 Methods and supporting data
3.1 Selection criteria
Measurements made during the G-1 flights over the Mexico City plateau are affected 5
primarily by Mexico City emissions with secondary contributions from utility and in-dustrial sources, biomass burning, and volcanic emissions. In order to focus on the time evolution of urban emissions, data selection criteria are imposed as indicated in Table 4. These criteria have three purposes. First to restrict attention to the bound-ary layer over the Mexico City plateau. Second, to minimize sources of non-urban 10
aerosols, with the exception of sulfate formed from SO2point sources and possibly vol-canic emissions. Third, to eliminate those instances in which fresh NOxemissions from a downwind non-urban source resets the photochemical age clock to a younger age than is appropriate for the urban mixture of pollutants. Of the aerosol that is derived from biomass burning, a fraction is from urban sources such as cooking, heating and 15
trash incineration. To a first approximation these sources are co-located with urban CO and NOx sources and are properly treated as part of the urban emissions whose time dependence we are interested in. The remaining biomass fraction is from fires outside the city and therefore can cause distortions in the aerosol – age relations if incorrectly attributed to urban emissions. Selection criteria should discriminate against non-urban 20
biomass burning, hereinafter referred to as “forest fire emissions”.
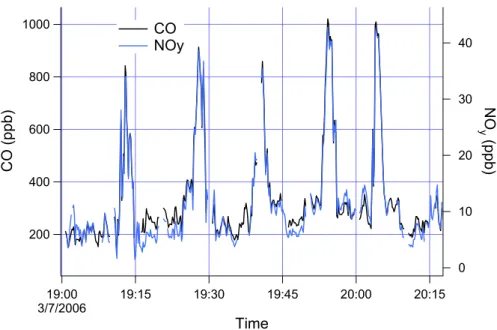
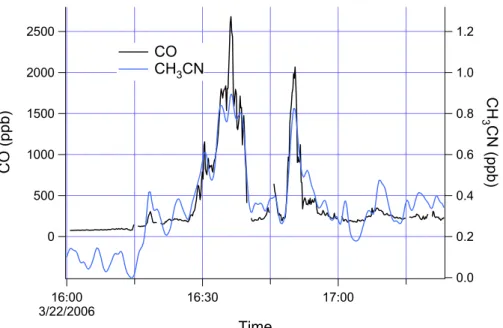
Once the geographic constraints and boundary layer constraints are satisfied, the primary criteria for data inclusion is that the CO/NOy ratio be close to that observed in very polluted air masses from Mexico City, in which the pollution origin is clearly urban. As an example, Fig. 3 shows a time series from the 307a flight in which the 25
ACPD
7, 14461–14509, 2007
Time evolution of aerosol over the Mexico City plateau
L. I. Kleinman et al. Title Page Abstract Introduction Conclusions References Tables Figures ◭ ◮ ◭ ◮ Back Close Full Screen / Esc
Printer-friendly Version Interactive Discussion
EGU That the plumes in Fig. 3 represent near-by urban emissions is substantiated by the
NOx to NOy ratio which was approximately 60%. On other flights, earlier in the day, even less processed emissions are observed with NOx sometimes exceeding 90% of NOy. A linear regression of the data in Fig. 3 yields CO=98 + 20.6 NOy (r2=0.98). Similar results are obtained on other flights. The criteria that:
5
15<(CO − 100)/NOy<25 (1) is based on these measurements. The 15 to 25 range in Eq. (1) is greater than the spread in the urban CO to NOyratio but allows for uncertainties in background concen-trations and for decreases in NOy due to dry deposition of HNO3. At concentrations approaching an ill-defined background, Eq. (1) is of limited utility so it is not used as a 10
screening tool unless CO>170 ppb.
Industrial and utility point sources are identified as having high concentrations of SO2 and/or a low ratio of CO to NOy. An inspection of CO, NOy, and SO2time series data (graphs not shown) indicates that the criteria in Table 4 eliminate short duration high concentration spikes which are due to near-source plume penetrations. A majority of 15
these events are due to the Tula industrial complex. In order to obtain a representa-tive picture of sulfate concentrations on the plateau, the SO2 threshold is not set low enough to discriminate against all SO2plumes. What is important for this study is that the SO2plumes that are left in the data set are not otherwise different from urban pol-lution, in particular that the SO2 is not associated with extra (i.e. non-urban) NOx and 20
CO which would affect our determination of photochemical age or dilution.
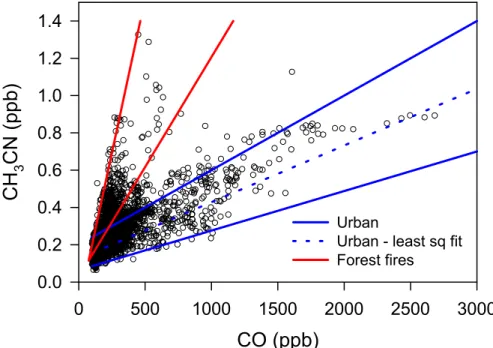
Air masses affected by forest fire emissions are identified by an elevated CH3CN to CO ratio. Figure 4 shows the relation between CH3CN and CO after all of the selection criteria in Table 4 have been applied except for the limits on CH3CN and CO/NOy. Data in Fig. 4 fall in 2 categories. There is a branch with a high CH3CN/CO ratio due 25
primarily to forest fires and one with a low ratio due primarily to anthropogenic sources. De Gouw et al. (2006) and Warneke et al. (2006) give examples of forest fire plumes and anthropogenic plumes which, for the most part, can be clearly differentiated on
ACPD
7, 14461–14509, 2007
Time evolution of aerosol over the Mexico City plateau
L. I. Kleinman et al. Title Page Abstract Introduction Conclusions References Tables Figures ◭ ◮ ◭ ◮ Back Close Full Screen / Esc
Printer-friendly Version Interactive Discussion
EGU the basis of CH3CN/CO. Red lines in Fig. 4 indicate the range of ∆CH3CN/∆CO from
Alaskan and Canadian forest fires observed by de Gouw et al. (2006) near the east coast of the U.S. The area between these lines is seen to encompass much of the forest fire data in the G-1 data set. A pair of blue lines in Fig. 4 delineates a second data branch with a lower CH3CN/CO ratio. As discussed below the CH3CN in the low 5
ratio branch appears to have an urban origin The upper bound of the urban branch is given by
CH3CN(ppb) = 0.2 + 0.4 × 10−3CO(ppb) (2)
An air mass is considered to have a minimal forest fire influence if it has a CH3CN concentration below the value given in Eq. (2)
10
Time series data for flight 322a presented in Fig. 5 shows that CH3CN concentrations up to 1 ppb can occur at the high CO concentrations encountered over Mexico City. On that flight a fresh urban plume (NOy is 77% NOx) was encountered on the L3-L4 and L0 legs between 10:00 and 11:00. CO concentrations exceed 2 ppm and the CO/NOy ratio (see figure caption) is nearly identical to other urban transects. Hydrocarbon spe-15
ciation (not shown) is also as expected for an urban source. Location, concentration, and speciation all support the contention that these plumes are composed of anthro-pogenic pollutants. Over most of the urban plume CO and CH3CN concentrations are proportional, strongly suggesting a co-located source. In Fig. 5 the scales for CO and CH3CN are related by Eq. (2) with slope and intercept qualitatively determined so as to 20
describe the proportionality between CO and CH3CN in these urban plumes. Having identified CO and CH3CN as arising from common sources, and having identified the CO plumes as urban emissions, this establishes that CH3CN concentrations up to the limit given by Eq. (2) can arise primarily from urban sources.
Urban plume points from other flights can have CH3CN/CO slopes about a factor of 25
two lower than that given by Eq. (2) as indicated from the bottom envelope of points in Fig. 4. This variability deserves closer attention as it may provide insights into sources of urban CH3CN. Also shown in Fig. 4 is a least squares fit to the anthropogenic branch
ACPD
7, 14461–14509, 2007
Time evolution of aerosol over the Mexico City plateau
L. I. Kleinman et al. Title Page Abstract Introduction Conclusions References Tables Figures ◭ ◮ ◭ ◮ Back Close Full Screen / Esc
Printer-friendly Version Interactive Discussion
EGU which shows a CH3CN enhancement of 0.3×10−3 ppb CH3CN/ppb CO. The CH3CN
enhancement observed in Mexico City is comparable to that measured in New York City (0.25×10−3 ppb CH
3CN/ppb CO) by de Gouw et al. (2006).
Figure 5 includes several segments in which data is excluded from analysis because CH3CN exceeds the value given by Eq. (2). Some of the excluded data falls within 5
the forest fire branch defined by the de Gouw et al. (2006) data set but some of it is not clearly from anthropogenic sources or forest fires. This situation is typical of other flights. The demarcation between branches in Fig. 4 is fuzzy because there is mixing of urban and forest fire influenced air masses. Such mixing could be responsible for the relatively high concentrations of CO and CH3CN found in less polluted ”background” 10
air masses.
In the construction of an urban-only data set, biomass burning is of most concern because models indicate that it is the dominant global source of OA (Intergovernmental Panel on Climate Change, 2001). An extrapolation of fire plume measurements made in the mountains surrounding Mexico City by Yokelson et al. (2007) indicates significant 15
impacts on regional concentrations of CO and aerosol. In addition to the data selection criteria, forest fire data is discriminated against by determining OA/CO ratios from linear regressions which yield aerosol per CO over and above background concentrations. 3.2 Photochemical age
Exposure of pollutants to atmospheric processing is quantified using the ratio of NOx 20
to NOy as a photochemical age (Olszyna et al., 1994; Kleinman et al., 2007). NOx is emitted largely as NO which rapidly reacts with O3 to form a steady state mixture of NO and NO2. Subsequent oxidization reactions form HNO3, PAN and organic nitrates. The sum total of NOx and its oxidation products is denoted as NOy and to a good approximation these compounds, including fine particle aerosol NO−3, are detected with 25
near 100% efficiency. On the time scales of interest here, hours to 1 day, NOyis almost conservative. Aside from rain events which were avoided, the primary NOy removal
ACPD
7, 14461–14509, 2007
Time evolution of aerosol over the Mexico City plateau
L. I. Kleinman et al. Title Page Abstract Introduction Conclusions References Tables Figures ◭ ◮ ◭ ◮ Back Close Full Screen / Esc
Printer-friendly Version Interactive Discussion
EGU mechanism is by dry deposition, which is limited to a maximum loss of ∼25% by the
CO/NOyconstraint in Eq. (1).
We define photochemical age as –Log10 (NOx/NOy), so that it has a value of 0 for fresh emissions (NOy = NOx) and a value of 1 when 90% of NOx has been converted into oxidation products. In the daytime NOxis primarily NO2and the dominant oxidation 5
reaction is OH + NO2→ HNO3. In that case Z
k[OH]dt = −2.303 Log10(NOx/NOy) (3)
This relation allows us to assign a time scale to a photochemical age given an assumed OH concentration. For OH = 107molec cm−3, a photochemical age of 1 is reached in approximately 8 h. The average OH concentration is generally not known and there 10
are other reactions that convert NOx into oxidation products. Thus, Eq. (3) can only qualitatively translate a NOx/NOyratio into an actual time. Assigning 10
7
OH cm−3 as a peak day time value, we a set a time scale of about 1 day for the transition between a photochemical age of 0 and 1.
There is an extensive literature on the use of photochemical age techniques, much 15
of which points out ambiguities and biases in age that are inherent in atmospheric samples which contain emissions from mixtures of sources (Kleinman et al., 2003; Parrish et al., 2007, and references contained therein). While not quantitative, the NOx/NOyclock (in common with VOC clocks) should be monotonic if significant down-wind emissions sources are avoided as we attempt to do by screening out non-urban 20
NOx plumes. It is therefore a useful metric for ordering air masses according to atmo-spheric exposure.
Because Mexico City is the major source of NOx on the plateau it is expected that photochemical age will be low over the city and increase towards the T1 and T2 down-wind sites. That is shown in Fig. 6. Although flights were preferentially scheduled for 25
days when transport would be from the city to the L2 leg (Doran et al., 2007), the gen-eral increase in age with distance away from the city is not necessarily due to direct
ACPD
7, 14461–14509, 2007
Time evolution of aerosol over the Mexico City plateau
L. I. Kleinman et al. Title Page Abstract Introduction Conclusions References Tables Figures ◭ ◮ ◭ ◮ Back Close Full Screen / Esc
Printer-friendly Version Interactive Discussion
EGU transport. As long as there are flows in the basin mixing material outward from the
source region, the behavior shown in Fig. 6 is expected. 3.3 Dilution
As Mexico City emissions are transported away from their source region they become dilute due to a mixing-in of cleaner air from elsewhere. We account for dilution in the 5
determination of age-related changes to aerosol properties by normalizing results to CO, which, on the time scales of interest is a conservative tracer. Because of the selection criteria in Table 4, CO concentrations above background are mainly from urban emissions. It is assumed that emission sources for aerosols and their precursors are co-located with CO sources either because these compounds are emitted from a 10
common source type (i.e. diesel vehicles) or because they are emitted in proportion to a common factor such as population density. On this basis,
EPOA=αECO
[POA − OAB]=α[CO − COB] (4)
where E’s are emission rates, POA is primary organic aerosol, square brackets indi-15
cate concentration, subscript B indicates a background value, andα is a proportionality
constant. Before any SOA is formed, OA is identical to POA. SOA production is evi-denced from the ratio [OA–OAB ]/[CO–COB] increasing aboveα to, say α′. Note that
α′can be evaluated without knowing background concentrations by
α′= d[OA]i/d[CO]
i (5)
20
where subscript “i” identifies a data sub set with a particular photochemical age. Similar consideration apply to other aerosol components.
Dilution of pollutants on the Mexico City plateau is illustrated in Fig. 7. As expected highest concentrations of CO are located over the city and concentrations are progres-sively lower over L1 and L2, which as shown in Fig. 6 tend to be where older air masses 25
ACPD
7, 14461–14509, 2007
Time evolution of aerosol over the Mexico City plateau
L. I. Kleinman et al. Title Page Abstract Introduction Conclusions References Tables Figures ◭ ◮ ◭ ◮ Back Close Full Screen / Esc
Printer-friendly Version Interactive Discussion
EGU reflects flight to flight differences in plume location and ventilation. By almost any
mea-sure a non-city CO concentration of 200 to 300+ ppb such as observed on L2 is very high. In this case, peak concentrations over the city are sufficiently high that a 300 ppb downwind concentration can be consistent with the good ventilation that is predicted by particle dispersion calculations (Fast and Zhong, 1998; de Foy et al., 2006)
5
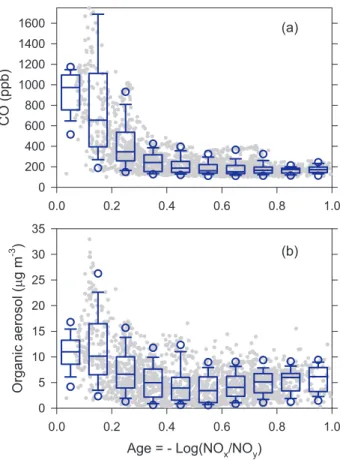
A decrease in CO concentration as a function of photochemical age is shown in Fig. 8a. Peak CO concentrations reach 2.5 ppm. For ages greater than 0.5, CO con-centration vary between 100 and more than 300 ppb, with an average of ∼180 ppb which does not decrease significantly with age. (There were a few CO measurements below 100 ppb which are most likely not boundary layer air and are removed from our 10
data set according to the selection criteria in Table 4.) An analogous dilution graph for organic aerosol is shown in Fig. 8b. The age dependence of organic aerosol is clearly different than CO indicating that the ratio OA/CO increases with age. This finding is put on a firmer footing in the next section.
4 Age dependence of normalized aerosol concentration 15
At this point we have limited the G-1 data set to urban emissions and have described the use of CO as a conservative tracer and the oxidation of NOxas a way of determining photochemical age. In a previous study (Kleinman et al., 2007) we constructed ratios such as OA/CO by subtracting background values for OA and CO. In Mexico City, aged air masses can have high and variable concentrations of trace gas and aerosols, as 20
illustrated in Fig. 8. It is problematic to define background conditions.
Instead we have divided the data set into 10 subsets, each spanning 0.1 units of age. Subsets have over 100 data points except for the youngest (39) and oldest (50). For each subset a linear least squares regressions was performed with total AMS aerosol concentration or organic aerosol concentration as the dependent variables (Y) and CO 25
as the independent variable (X). Regression slopes are determined from the reduced major axis approach which, according to Isobe et al. (1990), is appropriate when “the
ACPD
7, 14461–14509, 2007
Time evolution of aerosol over the Mexico City plateau
L. I. Kleinman et al. Title Page Abstract Introduction Conclusions References Tables Figures ◭ ◮ ◭ ◮ Back Close Full Screen / Esc
Printer-friendly Version Interactive Discussion
EGU objects under study have an intrinsic scatter that is much larger than the uncertainties
due to the measurement process”. In this approach the slope is the geometric mean of the ordinary least squares regression slope of Y vs. X and one over the slope of X vs. Y. As it is assumed that CO is a conservative tracer of the urban plume which is also the source of aerosols and aerosol precursors, these slopes represent urban aerosol 5
impacts normalized to account for dilution.
The age dependence of aerosol components after accounting for dilution is deter-mined from
d[X]/d[CO]i= d[Aerosol]/d[CO]i× Average([X]/[Aerosol])i (6) where X can be organics, nitrate, sulfate, ammonium, or chloride and i is an index for 10
photochemical age. One gets slightly different results with this approach compared to a regression of individual species versus CO (as is done for organics). Equation (6), however, has the advantage that the 5 components are guaranteed to add up to the to-tal aerosol concentration. The age dependence of ambient aerosol (without accounting for dilution) is simply given by [X]i.
15
In order to summarize the time evolution of aerosol we define an age growth factor, G, which is the fractional change in concentration between the youngest and oldest age bins. This factor is defined with respect to the value of a linear least squares fit, i.e. if
d[X]/d[CO]=a + b × Age
20
then G is given by
G=(a + bAge10)/(a + bAge1) (7)
where Age1and Age10 are average ages in the first and last age bins, 0.083 and 0.94, respectively. Changes in aerosol between zero age and 0.083 are not included in G because there are too few measurements to support an extrapolation of the least 25
ACPD
7, 14461–14509, 2007
Time evolution of aerosol over the Mexico City plateau
L. I. Kleinman et al. Title Page Abstract Introduction Conclusions References Tables Figures ◭ ◮ ◭ ◮ Back Close Full Screen / Esc
Printer-friendly Version Interactive Discussion
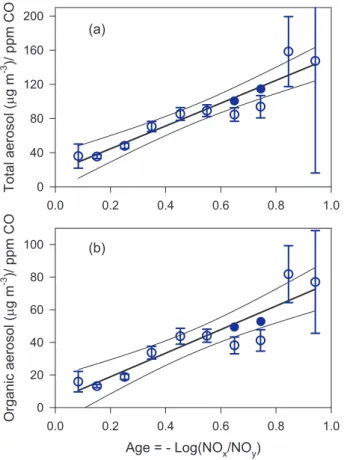
EGU Figure 9 shows the time evolution of total non-refractory aerosol and organic aerosol,
normalized to CO, based on the regression analysis described above. It was not possi-ble to determine a priori the errors in aerosol concentration and CO since the variability in Aerosol/CO has a not-easily-quantified component from atmospheric heterogene-ity. Instead error bounds are calculated fromχ2 according to the method in Press et 5
al. (1986). Error bars are largest for the youngest and two oldest data subsets, which have a smaller number of samples and/or a poor correlation compared to other data subsets.
A linear least squares fit has been added to Fig. 9 as a description of the increase in these quantities as functions of photochemical age. According to Eq. (7), there is 10
a factor of 5.0 and 6.9 increase in dilution-corrected total aerosol and organic aerosol due to photochemical aging.
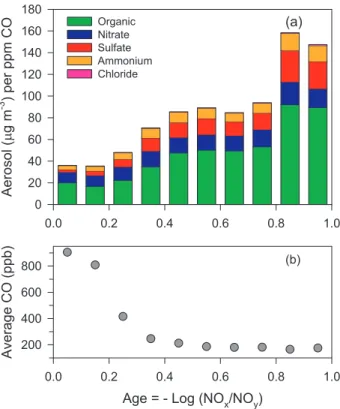
Normalized aerosol composition calculated as a function of photochemical age ac-cording to Eq. (6) is shown in Fig. 10a. There is a more or less uniform increase in total aerosol concentration with age, except for a dip in the 2 age bins between 0.6 15
and 0.8, which is discussed below. Compared to young air masses, aged air masses tend to have a higher proportion of organics and sulfate and a lower proportion of ni-trates. Ammonium concentrations vary with nitrate and sulfate so as to yield a nearly age-independent neutralization of 88–99%. Aerosol age growth factors based on the data in Figs. 9a, 9b, and 10a are given in Table 5.
20
Figure 11 presents average ambient aerosol concentrations for 10 age bins. Al-though aerosol/CO increases several-fold with age, dilution causes actual aerosol centrations to be lowest in older air masses. Ambient levels of the 5 aerosol con-stituents can be reconstructed by multiplying the “per CO” concentrations in Fig. 10a by the average CO concentrations in Fig. 10b, taking care to subtract background CO. 25
Background CO is actually a difficult quantity to determine directly from the data which is why the ”per CO” concentrations were derived from a regression analysis. Figure 11 includes re-constructed total aerosol concentrations determined from
ACPD
7, 14461–14509, 2007
Time evolution of aerosol over the Mexico City plateau
L. I. Kleinman et al. Title Page Abstract Introduction Conclusions References Tables Figures ◭ ◮ ◭ ◮ Back Close Full Screen / Esc
Printer-friendly Version Interactive Discussion
EGU where Aerosol/CO is the data in Fig. 10a, CO concentrations are from Fig. 10b, and
background CO is set at 100 ppb. There is good agreement between reconstructed and measured aerosol. This agreement implies that in the clean conditions represented by a 100 ppb CO concentration, background aerosol concentrations are near zero. An inspection of the CO data, however, shows that the low concentration points cluster 5
near 130 ppb. If we take this value as background and evaluate Eq. (8) we get a second reconstructed aerosol concentration that is on average 3µg m−3lower than previously obtained. Except for the 1st age bin, the reconstructed aerosol is 2 to 4.5µg m−3 lower than the observed ambient, which we can assign as background aerosol. As seen by these 2 reconstructions, background aerosol is sensitive to assumptions on 10
background CO. It is encouraging that over a reasonable range of CO concentrations, reasonable predictions for background aerosol result.
Age trends in M44/Org and M57/Org are shown in Fig. 12. With atmospheric aging the proportion of M44 is seen to increase and the proportion of M57 to decrease. This is consistent with two identifications made by Zhang et al. (2005): first that M44 and 15
M57 are approximate surrogates for OOA and HOA, and second that that OOA and HOA have the properties of SOA and POA. The increase in M44 and the decrease in M57 then follows when the addition of organic mass with age is accomplished by adding SOA to pre-existing aerosol.
5 Discussion
20
5.1 Non-uniform sources
Compared to the linear fits, the data points in Figs. 9 and 10 show some bumps and dips, the largest being low values in the age bins 0.6–0.7 and 0.7–0.8. This feature is due to a collection of points from flight 320b, located just to the east of the L2 leg. At this location, 18 km of a high CO plume was sampled, with almost identical results at 3 25
ACPD
7, 14461–14509, 2007
Time evolution of aerosol over the Mexico City plateau
L. I. Kleinman et al. Title Page Abstract Introduction Conclusions References Tables Figures ◭ ◮ ◭ ◮ Back Close Full Screen / Esc
Printer-friendly Version Interactive Discussion
EGU detached from the more numerous lower concentration points at the same
photochem-ical age. According to the selection criteria in Table 4 and other measurements, there is nothing else unusual about this plume. Yet, something is different and the high CO concentrations yield low values for the dilution corrected quantities in Figs. 9 and 10. Effects of removing this plume are shown in Fig. 9. The premise of a uniform mixture 5
of emissions is a good approximation but there are exceptions. 5.2 Aerosol composition
Most of the non-refractory mass measured by the AMS is of secondary origin, as shown in the previous section for organics. For sulfate, nitrate, and ammonium ions, gas to particle conversion, augmented by aqueous phase chemistry in the case of SO2−4 , 10
are known to be the major pathway for adding these materials to the aerosol phase. The addition of secondary material to the aerosol phase will cause concentrations, normalized to CO, to increase. Sulfate is non-volatile, so additions to the aerosol phase are not reversible. NH+4 and NO¯3, are volatile. If aerosols are advected into regions with low partial pressures of NH3 and HNO3, these substances can evaporate. Although 15
the age dependence of dilution-adjusted aerosol concentrations are affected by many processes besides volatility, the behavior of nitrate suggests that partitioning between gas and aerosol phases is important (Lee et al., 2006). Aerosol nitrate concentrations (Fig. 10a) are almost constant above a photochemical age of 0.4 causing the age growth factor to be the lowest in Table 5. On average, a point has been reached where 20
aerosol nitrate growth balances loss. Presumably, individual samples gain or loose of nitrate depending on conditions. Organic aerosol concentration (Fig. 10a), in contrast, continues to increase over the entire age range. Although it is possible that the average includes some samples in which OA is evaporating, the overall trend suggests that this is not a dominant process.
ACPD
7, 14461–14509, 2007
Time evolution of aerosol over the Mexico City plateau
L. I. Kleinman et al. Title Page Abstract Introduction Conclusions References Tables Figures ◭ ◮ ◭ ◮ Back Close Full Screen / Esc
Printer-friendly Version Interactive Discussion
EGU 5.3 Carbon mass balance
In this section we estimate the gas phase hydrocarbon precursor concentrations re-quired to account for the 6.9 fold increase in organic aerosol that occurs during aging. The increase in aerosol concentration, normalized to CO, is given by
∆(OA/CO)=(OA/CO)LSQ(10)−(OA/CO)LSQ(1) (9)
5
where the value of OA/CO is evaluated at the 2 endpoints of the linear least squares fit shown in Fig. 9b. Equation (9) yields ∆(OA/CO) = 62 µg m−3/ppm CO. It is assumed that the organic mass added during aging is OOA with a carbon content, OC, given by OOA/OC = 2.2 µg/µg C (Zhang et al., 2005). The carbon added to the aerosol phase during aging is then ∆(OC/CO) = 28 µg C m−3/ppm CO, equivalent to 56 ppbC per ppm 10
CO.
The next step is to determine the amount of organic aerosol that would be produced from gas phase precursors. Our calculations are based on Mexico City surface urban aromatic hydrocarbon (HC) measurements reported by Velasco et al. (2007) in their Table 4. Velasco et al. 2007 do not report CO measurements, so conversion into 15
ppb HC/ppm CO is accomplished by using toluene as a transfer standard. PTR-MS observations coincident with the aerosol data set used in this study yield the following relation:
[toluene(ppb)]=−0.48+4.2 [CO(ppm)], r2=0.85 (10)
which is equivalent to 29 ppbC per ppm CO. Thus, the inferred increase in OA is sto-20
ichiometrically equivalent to 193% of ambient toluene. Hydrocarbon/CO ratios are obtained by combining Eq. (10) with the surface HC observations, yielding
HC (ppb)/CO (ppm)=(HC/toluene)Surface × (4.2 ppb toluene/ppm CO)G−1 (11) Smog chamber data for low and high yield aromatic OA precursors, presented by Odum et al. (Fig. 1, 1997) give an aerosol yield of 5% and 8%, respectively, at an OA con-25
ACPD
7, 14461–14509, 2007
Time evolution of aerosol over the Mexico City plateau
L. I. Kleinman et al. Title Page Abstract Introduction Conclusions References Tables Figures ◭ ◮ ◭ ◮ Back Close Full Screen / Esc
Printer-friendly Version Interactive Discussion
EGU (Pankow, 1994) and the data of Odum et al. 1997, aerosol yields increase as the
or-ganic aerosol phase available for partitioning is made larger. Yields used here are likely to be overestimates for the G-1 data set as ambient organic aerosol concentrations were considerably lower than 100µg m−3 (see Fig. 11). Organic aerosol production is calculated from
5
∆(OA ppbC/ppm CO)Predicted =X(ppbC HCi/ppm CO) × Yi (12) where Yi is the aerosol yield of the ith compound and the sum includes 13 aromatic compounds measured by Velasco et al. (2007). The predicted OA formation is 4.8 ppbC per ppm CO, 2.4 ppbC of which is from toluene. Equation (12) accounts for only 9% of the observed 56 ppbC per ppm CO added to the aerosol phase in aging.
10
The obvious question is: Where does the additional 51 ppbC/ppm CO accounting for 91% of the organic aerosol mass added during aging come from? Other anthro-pogenic precursors with significant organic aerosol yields include longer chain alkanes and alkenes (Seinfeld and Pandis, 1998). In a simulation of aerosol formation in Mexico City these compounds added only another 25% to the aerosol yields from aromatics 15
(Volkamer et al., 2006). Biogenic VOCs have high aerosol yields (Griffin et al., 1999) but the G-1 PTR-MS measurements of terpenes were typically less than 0.2 ppb. An-other pathway for organic aerosol production is partitioning of hydrophilic VOCs into the aerosol aqueous phase (Aumont et al., 2000). However, in most models (e.g. Griffin et al., 2002) this mechanism is of lesser importance than the addition of hydrophobic 20
compounds typified by oxidation products of aromatics. Enhanced yields from acid cat-alyzed reactions seem unlikely because Mexico City aerosols are nearly neutralized. Furthermore, recent studies have not found evidence that aerosol yields increase with acidity under the range of conditions encountered in the ambient atmosphere (Peltier et al., 2007; Zhang et al., 2007). It is possible that extra sources of organic aerosol 25
are to be found among high molecular weight, low and intermediate volatility VOCs (Goldstein and Galbally, 2007; Robinson et al., 2007). Such compounds could include oxidation products of VOCs that are evaporated from POA during the rapid dilution that
ACPD
7, 14461–14509, 2007
Time evolution of aerosol over the Mexico City plateau
L. I. Kleinman et al. Title Page Abstract Introduction Conclusions References Tables Figures ◭ ◮ ◭ ◮ Back Close Full Screen / Esc
Printer-friendly Version Interactive Discussion
EGU occurs shortly after emission (Robinson et al., 2007). Aerosol phase polymerization
reactions that yield non-volatile products are another possibility (Kalberer et al., 2004). Such a mechanism would be consistent with the apparent lack of an evaporative loss of OA (in fact a continued growth) as an air mass ages and gas phase concentrations decrease.
5
Organic aerosol mass in excess of model predictions has been observed in several locations using different methodologies. Of most relevance to the present study are the observations of Volkamer et al. (2006) at the CENICA urban site in Mexico City during April, 2003. Model calculations that were based on measured oxidants and VOCs accounted for less than 1/8th of the OOA increase at CENICA between sunrise 10
and mid-afternoon. De Gouw et al. (2005) reach a similar conclusion based on obser-vations up to 2 days downwind of the northeastern U.S. coast, namely that measured anthropogenic aerosol precursors could only account for 7% of the observed SOA, with little likelihood that biogenic compounds could make up much of the difference.
5.4 Comparison with the Eastern U.S. 15
Mexico City has a more concentrated set of emission sources than found in most, if not all, areas of the U.S. resulting in very high concentrations of gas phase pollutants (Figs. 3 and 5; Molina and Molina, 2002). Our emphasis in this study, however, has been on intensive properties, in particular on OA production per unit CO. It is of interest to determine whether this intensive property depends on absolute concentration. If it 20
does, then the growth of megacities would lead to a different set of impacts compared to the situation where the same number of people and the same amount of industrial activity are spread out over several smaller population centers. An expectation that ∆(OA/CO) might be greater in regions with high emissions can be justified on the basis of the absorptive/ partitioning model, in which aerosol yields increase when there is 25
a large amount of organics in the aerosol phase into which low volatility VOCs can partition.
correspond-ACPD
7, 14461–14509, 2007
Time evolution of aerosol over the Mexico City plateau
L. I. Kleinman et al. Title Page Abstract Introduction Conclusions References Tables Figures ◭ ◮ ◭ ◮ Back Close Full Screen / Esc
Printer-friendly Version Interactive Discussion
EGU ing quantity calculated from the 2002 NEAQS (Kleinman et al., 2007) and 2004
NEAQS/ITCT (Sullivan et al., 2007; Weber et al., 2007) field campaigns. The 2002 and 2004 NEAQS studies used data from the northeastern U.S. and in 2004 also from northern GA. As a qualitative indicator of pollution levels, CO concentrations in Mexico City reached 2.5 ppm, while those measured in the U.S., which included plumes from 5
NYC and Atlanta, had CO below 325 ppb. On average, CO concentrations in Mexico City were twice that observed in the 2002 NEAQS campaign.
The amount of OA produced during aging in Mexico City is 62µg m−3per ppm CO, as compared to 66 and 63µg m−3per ppm CO for the 2002 and 2004 NEAQS studies. This comparison offers no evidence that OA production per CO in Mexico City is any 10
greater than in the eastern U.S.
There are a few caveats to the comparison: 1) In all 3 studies OA is normalized to CO as a tracer of urban emissions. The utility of CO rests on the assumption that CO is emitted in proportion to compounds that are responsible for OA production. While this assumption may hold within a region, emission sources in Mexico City and the eastern 15
U.S. do differ. Further work is needed to determine how per-CO numbers translate from one region to another. 2) Related to item 1 are differences in biogenic emissions which are sources of SOA that are not expected to be correlated with CO. 3) Com-parisons should be performed over the same time interval. The one day aging time for Mexico City estimated from NOx/NOy is less accurate than back trajectory ages 20
determined for most of the 2004 NEAQS data. If the older Mexico City samples have undergone less aging than the older NEAQS samples this would make the Mexico City ∆(OA/CO) appear to be low. 4) The 2004 results should be considered a lower bound as organic aerosol measurements are of the water soluble component – operationally defined as the aerosols that grow into a collectible size when exposed to supersatu-25
rated conditions within the PILS. 5) There were few near-source measurements in the 2002 NEAQS campaign and the inferred value of OA/CO at low age is relatively un-certain. 6) Even though very high gas phase pollutant concentrations are encountered in Mexico City, a comparison of Fig. 8b and 11 with Fig. 9 of Kleinman et al. (2007)
ACPD
7, 14461–14509, 2007
Time evolution of aerosol over the Mexico City plateau
L. I. Kleinman et al. Title Page Abstract Introduction Conclusions References Tables Figures ◭ ◮ ◭ ◮ Back Close Full Screen / Esc
Printer-friendly Version Interactive Discussion
EGU indicates comparable OA concentrations in both regions. We defer a comparison of
absolute concentrations except to note that OA in the NEAQS region was uncharac-teristically high compared with long term monitoring data from the IMPROVE network (Malm et al., 2004).
Our results leave open the possibility that more “efficient” OA production would have 5
been observed in Mexico City if there were stagnation events such that pollutants aged under more concentrated conditions.
6 Conclusions
Changes in aerosol concentration and speciation have been determined as a function of photochemical age based on 8 flights of the DOE G-1 aircraft in and downwind of 10
Mexico City. Over the City we find young concentrated plumes, with CO concentra-tions up to 2.5 ppm and NOx/NOy ratios that can exceed 90%. Successively lower CO concentrations and older air masses are found over the T1 and T2 surface sites. The measurements considered here are confined to the Mexico City plateau and yield al-most an order of magnitude spread in photochemical age defined as –Log10(NOx/NOy). 15
A qualitative estimate of age since emission is less than 1 h to about 1 day.
Our interest is in the evolution of urban plumes and towards that end a set of selec-tion criteria were imposed to eliminate air masses significantly impacted by non-urban biomass burning and utility and industrial sources. Forest fire plumes are known to have elevated CH3CN/CO ratios (de Gouw et al., 2006; Warneke et al., 2006). An in-20
spection of data taken in well defined urban plumes supports a designation that pollu-tants were primarily urban if CH3CN(ppb)<0.2+0.4×10−3CO (ppb). Because CO con-centrations could reach more than 2 ppm, the urban source of CH3CN could contribute over 1 ppb CH3CN, presumably from embedded burning such as cooking, heating, and waste incineration.
25
As an air massed aged from the youngest to oldest category the fraction of aerosol that was organic increased from ∼50 to 60%, nitrate decreased from ∼25 to 10%,
ACPD
7, 14461–14509, 2007
Time evolution of aerosol over the Mexico City plateau
L. I. Kleinman et al. Title Page Abstract Introduction Conclusions References Tables Figures ◭ ◮ ◭ ◮ Back Close Full Screen / Esc
Printer-friendly Version Interactive Discussion
EGU and sulfate increased from ∼7 to 17%. At all ages, ammonium concentrations were
sufficient to neutralize ∼90 to 100% of the aerosol acidity from sulfate and nitrate. With increasing age M44/OA increases and M57/OA decreases, consistent with the added OA mass being OOA.
The urban data set was split into 10 subsets and for each the amount of aerosol 5
per ppm CO was determined as the slope of a linear regression. This procedure has the advantage that one does not have to specify background concentrations which are difficult to estimate. Between the youngest and oldest data subset, total non-refractory aerosol measured by an AMS increased by a factor of 5, while organics showed a 7 fold increase. OA/CO shows a steady increase with no sign that OA is evaporating 10
in older air masses, in which lower gas phase VOC concentrations might be expected to cause re-partitioning of semi-volatile organic compounds back into the gas phase. Nitrate/CO, in contrast, has reached a plateau suggesting that condensation and evap-oration of NH3 and HNO3 are in balance. Upon multiplying the per CO aerosol con-centrations by an average CO above background, one recovers reasonable conditions, 15
i.e. background aerosol = 2–4.5 µg m−3 at CO = 130 ppb.
The change in OA/CO between the youngest and oldest age bins is 62µg m−3 per ppm CO. At an OC/OA ratio characteristic of OOA (Zhang et al., 2005) this is equivalent to 56 ppbC per ppm CO. OA production calculated from surface HC measurements (Velasco et al., 2007) and smog chamber data (Odum et al., 1997) can only explain 20
9% of the inferred increase in OA during aging, a fraction similar to what others have found (de Gouw et al., 2005; Volkamer et al., 2006). Continued OA formation over 1 day is consistent with mechanisms in which low volatility VOC are continuously produced and/or aerosol-phase polymerization reactions yield low volatility products such that OA does not re-partition to the gas phase in cleaner air.
25
OA production was compared with observations made in the eastern U.S. during the 2002 NEAQS (Kleinman et al., 2007) and 2004 NEAQS/ITCT (Sullivan et al., 2006; Weber et al., 2007) field campaigns. In approximately 1 days aging, similar amounts of OA were produced per unit CO. We find no evidence for a megacity effect, whereby
ACPD
7, 14461–14509, 2007
Time evolution of aerosol over the Mexico City plateau
L. I. Kleinman et al. Title Page Abstract Introduction Conclusions References Tables Figures ◭ ◮ ◭ ◮ Back Close Full Screen / Esc
Printer-friendly Version Interactive Discussion
EGU conversion of OA precursors to OA proceeds to a greater extent in a region with very
high emission rates.
Acknowledgements. We thank chief pilot B. Hannigan and the flight crew from PNNL for a
job well done. We gratefully acknowledge the Atmospheric Science Program within the Office of Biological and Environmental Research of DOE for supporting field and analysis activities
5
and for providing the G-1 aircraft. Use of a PTR-MS provided by EMSL is appreciated. The MILAGRO campaign owes its success to many people. We would like to single out L. Molina (MIT and MCE2), S. Madronich (NCAR) and J. Gaffney (Univ. Arkansas) for organizational efforts and scientific guidance, and J. Fast (PNNL) and colleagues for weather and pollution forecasts. We thank D. Sueper (Aerodyne and Univ. CO) for her assistance in reducing the
10
AMS data. This research was performed under sponsorship of the U.S. DOE under contracts DE-AC02-98CH10886.
References
Aumont, B., Madronich, S., Bey, I., and Tyndall, G. S.: Contribution of secondary VOC to the composition of aqueous atmospheric particles: A modeling approach, J. Atmos. Chem.
15
Phys., 35, 59–75, 2000,http://www.atmos-chem-phys.net/35/59/2000/.
Brechtel, F. J.: Description and Assessment of a New Aerosol Inlet for the DOE G-1 Research Aircraft. Final Technical Report of work performed by BMI under contract #0000058843 to Brookhaven National Laboratory, Aug., 2003.
Canagaratna, M. R., Jayne, J. T., Jimenez, J. L., et al.: Chemical and microphysical
character-20
ization of ambient aerosol with the Aerodyne Aerosol Mass Spectrometer, Mass Spectrom. Rev., 26, 185–222, doi:10.1002/mas.20115, 2007.
Collins, D. R., Flagan, R. C., and Seinfeld, J. H.: Improved inversion of scanning DMA data, Aerosol Sci. Technol., 36(1), 1–9, 2002.
Cubison, M. J., Alfarra, M. R., Allan, J., Bower, K. N., Coe, H., McFiggans, G. B., Whitehead, J.
25
D., Willimams, P. I., Zhang, Q., Jimenez, J. L., Hopkins, J., and Lee, J.: The characterisation of pollution aerosol in a changing photochemical environment, Atmos. Chem. Phys., 6, 5573– 5588, 2006,http://www.atmos-chem-phys.net/6/5573/2006/.
DeCarlo, P., Slowik, J. G., Worsnop, D. R., Davidovits, P., and Jimenez, J. L.: Par-ticle morphology and density characterization by combined mobility and aerodynamic
ACPD
7, 14461–14509, 2007
Time evolution of aerosol over the Mexico City plateau
L. I. Kleinman et al. Title Page Abstract Introduction Conclusions References Tables Figures ◭ ◮ ◭ ◮ Back Close Full Screen / Esc
Printer-friendly Version Interactive Discussion
EGU
diameter measurements. Part 1: Theory, Aerosol Sci. Technol., 38, 1185–1205, doi:10.1080/027868290903907, 2004.
de Foy, B., Varela, J. R., Molina, L. T., and Molina, M. J.: Rapid ventilation of the Mexico City basin and regional fate of the urban plume, Atmos. Chem. Phys., 6, 2321–2335, 2006,
http://www.atmos-chem-phys.net/6/2321/2006/.
5
de Gouw, J. A., Middlebrook, A. M., Warneke, C., Goldan, P. D., Kuster, W. C., Roberts, J. M., Fehsenfeld, F. C., Worsnop, D. R., Canagaratna, M. R., Pszenny, A. A. P., Keene, W. C., Marchewka, M., Bertman, S. B., and Bates, T. S.: Budget of organic carbon in a polluted atmosphere: Results from the New England Air Quality Study in 2002, J. Geophys. Res., 110, D16305, doi:10.1029/2004JD005623, 2005.
10
de Gouw, J. A., Warneke, C., Stohl, A., et al.: Volatile organic compounds composition of merged and aged forest fire plumes from Alaska and western Canada, J. Geophys. Res., 111 D10303, doi:10.1029/2005JD006175, 2006.
Doran, J. C., Barnard, J. C., Arnott, W. P., et al.: The T1-T2 study: evolution of aerosol proper-ties downwind of Mexico City, Atmos. Chem. Phys., 7, 1585–1598, 2007
15
Drewnick, F., Hings, S. S., DeCarlo, P. F., Jayne, J. T., Gonin, M., Fuhrer, K., Weimer, S., Jimenex, J. L., Demerjian, K. L., Borrmann, S., and Worsnop, D. R.: A new Time-of-Flight Aerosol Mass Spectrometer (ToF-AMS) – Instrument description and first field deployment, Aerosol. Sci. Technol., 39, 637–658, 2005.
Fast, J. D. and Zhong, S.: Meteorological factors associated with inhomogeneous ozone
con-20
centrations within the Mexico City basin, J. Geophys. Res., 103, D15, 18 927–18 946, 1998. Goldstein, A. H. and Galbally, I. E.: Known and unexplored organic constituents in the Earth’s
atmosphere, Environ. Sci. Technol., 41(5), 1514–1521, 2007.
Griffin, R. J., Cocker III, D. R., Flagan, R. C., and Seinfeld, J. H.: Organic aerosol formation from the oxidation of biogenic hydrocarbons, J. Geophys. Res., 104(D3), 3555–3567, 1999.
25
Griffin, R. J., Daddub, D., Kleeman, M. J., Fraser, M. P., Cass, G. R., and Seinfeld, J. H.: Secondary organic aerosol 3. Urban/regional scale model of size-and composition-resolved aerosols, J. Geophys. Res., 107(D17), doi:10.1029/2001JD000544, 2002.
Haller, A. G., Strawa, A. W., Schmid, B., Andrews, E., Ogren, J., Sheridan, P., Ferrare, R., Covert, D., Elleman, R., Jonsson, H., Bokarius, K., and Luu, A.: Atmospheric Radiation
30
Measurements Aerosol Intensive Operating Period: Comparison of aerosol scattering during coordinated flights, J. Geophys. Res., 111, D05S09, doi:10.1029/2005JD006250, 2006. Heald, C. L., Jacob, D. J., Park, R. J., Russell, L. M., Huebert, B. J., Seinfeld, J. H., Liao, H., and
ACPD
7, 14461–14509, 2007
Time evolution of aerosol over the Mexico City plateau
L. I. Kleinman et al. Title Page Abstract Introduction Conclusions References Tables Figures ◭ ◮ ◭ ◮ Back Close Full Screen / Esc
Printer-friendly Version Interactive Discussion
EGU
Weber, R. J.: A large organic aerosol source in the free troposphere missing from current models, Geophys. Res. Lett., 32, L18809, doi:10.1029/2005GL023831, 2005.
Heald, C. L., Jacob, D. J., Turquety, S., et al.: Concentrations and sources of organic carbon compounds in the free troposphere over North America, J. Geophys. Res., 111, D23S47, doi:10.1029/2006JD007705, 2006.
5
Intergovernmental Panel on Climate Change: Climate Change 2001 – The Scientific Basis, edited by: J. T. Houghton et al., Cambridge Univ. Press, 2001.
Isobe, T., Feigelson, E. D., Akritas, M. G., and Babu, G. J.: Linear regression in astronomy, I., The Astrophysical Journal, 364, 104–113, 1990.
Jang, M., Czoschke, N. M., Lee, S., and Kamens, R. M.: Heterogeneous atmospheric aerosol
10
production by acid-catalyzed particle-phase reactions, Science, 298, 814–817, 2002. Jayne, J. T., Leard, D. C., Zhang, X., Davidovits, P., Smith, K. A., Kolb, C. E., and Worsnop,
D. R.: Development of an aerosol mass spectrometer for size and composition analysis of submicron particles, Aerosol Sci. Technol., 33, 49–70, 2000.
Johnson, D., Utembe, S. R., Jenkin, M. E., Derwent, R. G., Hayman, G. D., Alfarra, M. R.,
15
Coe, H., and McFiggans, G.: Simulating regional scale secondary organic aerosol formation during the TORCH 2003 campaign in the southern UK, Atmos. Chem. Phys., 6, 403–418, 2006,http://www.atmos-chem-phys.net/6/403/2006/.
Kalberer, M., Paulsen, D., Sax, M., Steinbacher, M., Dommen, J., Prevot, A. S. H., Fisseha, R., Weingartner, E., Frankevich, V., Zenobi, R., and Baltensperger, U.: Identification of polymers
20
as major constituents of atmospheric organic aerosols, Science, 303, 1659–1662, 2004. Kleinman, L. I., Daum, P. H., Lee, Y.-N., Nunnermacker, L. J., Springston, S. R.,
Weinstein-Lloyd, J., Hyde, P., Doskey, P., Rudolph, J., Fast, J., and Berkowitz, C.: Photochemical age determinations in the Phoenix metropolitan area, J. Geophys. Res., 108(D3), 4096, doi:10.1029/2002JD002621, 2003.
25
Kleinman, L. I., Daum, P. H., Lee, Y.-N., Senum, G. I., Springston, S. R., Wang, J., Berkowitz, C., Hubbe, J., Zaveri, R. A., Brechtel, F. J., Jayne, J., Onasch, T. B., and Worsnop, D.: Aircraft observations of aerosol composition and ageing in New England and Mid-Atlantic States during the summer 2002 New England Air Quality Study field campaign, J. Geophys. Res., 112, D09310, doi:10.1029/2006JD007786, 2007.
30
Lee, Y.-N., Jayne, J., Alexander, L., Canagaratna, M., Springston, S. R., Senum, G. I., Hubbe, J., Daum, P. H., and Kleinman, L.: Aerosol composition determined on board the DOE G-1 aircraft during MAX-Mex in March 2006, First MILAGRO Science Meeting, Boulder, CO, 23