The water–amorphous calcium carbonate interface and its interactions with amino acids
Texte intégral
Figure
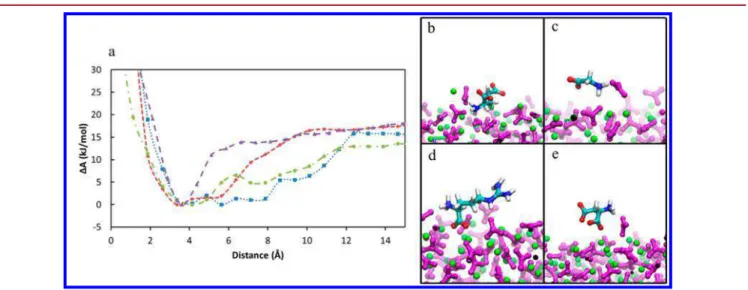
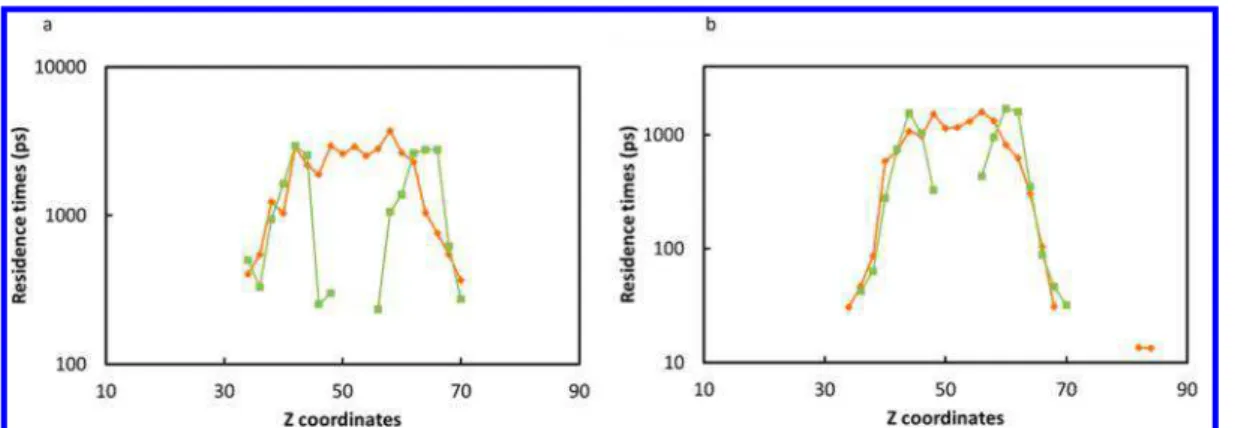
Documents relatifs
Here is presented a detailed analysis of the photochemical and photophysical properties of all the possible chemical forms of oxyluciferin and their analogues in explicit water
The effect of the calcium source, level and the particle size on calcium retention, eggshell quality and the overall calcium requirement in laying hens... For Peer
The gammaproteobacterium Achromatium forms intracellular amorphous calcium carbonate and not (crys- talline)
Oral appliances and maxillomandibular advancement surgery: an alternative treatment protocol for the obstructive sleep apnea-hypopnea syndrome. Maxillomandibular advancement
All the Baseline and Filler-2 trials plus 3 Filler-1 trials were presented before the Critical trials in order to test whether in the second half of the task
Some discrepancies have been detected in the intensifica- tion rates of the pressure at the centre of the cyclones and also in their position at the moment of maximum development
[r]
The World Health Organization (WHO) assembled a diverse group of nutrition, medical, epidemiological and other scientific experts and water technologists at the Pan American
