Influence des complexes protéiques sur la rétention de
copies de gène après une duplication de génome
Mémoire
Claudine Lamothe
Maîtrise en biochimie
Maître ès sciences (M. Sc.)
Québec, Canada
© Claudine Lamothe, 2018
Influence des complexes protéiques sur la rétention de
copies de gène après une duplication de génome
Mémoire
Claudine Lamothe
Sous la direction de :
iii
Résumé
Les duplications de gènes contribuent grandement à l'augmentation de la complexité des organismes en fournissant du nouveau matériel brut sur lequel agit la sélection naturelle. De ces duplications, c’est la duplication de génome qui a l’impact le plus important dû à la quantité de gènes impliqués. Plusieurs événements de duplication de génome ont eu lieu au fil de l'évolution de nombreuses lignées d'organismes. Une grande partie des gènes dupliqués créés lors de ces événements accumuleront des mutations délétères et seront inactivés ou disparaîtront du génome complètement, mais d'autres seront retenus. La rétention de certains gènes a été liée à divers facteurs comme le dosage génique et le niveau d'expression. Ce projet se concentre sur l’impact de la participation à des complexes protéiques sur la rétention des copies créés par des événements successifs de duplication de génome chez Paramecium tetraurelia. Nous avons d’abord prédit la composition de 885 complexes protéiques à travers les relations d'orthologie avec cinq espèces modèles. Ces complexes nous ont ensuite permis de déterminer que les gènes impliqués dans ces complexes avaient des niveaux d’expression plus élevés et plus corrélés, facteurs ayant déjà été associés avec un taux de rétention plus élevé. Nous avons également décelé une plus grande rétention d’un nombre pair de copies chez les gènes participant à des complexes protéiques, observation potentiellement reliée aux propriétés structurales des complexes. Parallèlement, nous avons noté un effet similaire à la participation à des complexes protéiques chez les gènes possédant des orthologues chez toutes les espèces modèles utilisées, démontrant que cet effet de conservation pouvait s’ajouter à celui de la participation à des complexes pour augmenter le niveau d’expression des gènes impliqués et le garder plus corrélé. Ensemble, ces facteurs présentent une image complexe de facteurs interreliés qui peuvent s’additionner pour influencer le sort des copies au fil de l’évolution.
iv
Abstract
Gene duplications contribute greatly to the increase in organismal complexity by providing new material for natural selection to act upon. Of these duplication events, whole-genome duplication has a major impact due to the sheer amount of gene copies produced. Several events of whole-genome duplication have occurred throughout the evolutionary history of many lineages. The greater part of the duplicated genes created during these events will accumulate deleterious mutations, become inactivated and will disappear completely from the genome, but some will be maintained over time. Several factors have been linked to the retention of certain genes such as gene dosage or the level of expression. This project focuses on the impact of participation in a protein complex on the retention of copies created during several successive events of whole-genome duplication in the ciliate
Paramecium tetraurelia. First, we predicted the composition of 885 protein complexes
through orthologous relationships with five model species. Those protein complexes then allowed us to determine that genes participating in those complexes had higher and more correlated expression, both factors previously linked in the literature with a higher retention rate. We also observed a greater retention of even numbers of copies for genes participating in protein complexes, observation which might be connected to structural properties of protein complexes. At the same time, we noted an effect similar to protein complex participation in genes with orthologs in all our model species used and determined that this might partially be caused by an overlap between the genes participating in protein complexes and those being conserved in all the model species. However, we also showed that the effect of widespread conservation was independent of that of complex participation. Together, those factors paint a complex picture of interconnected factors that can interact to influence the fate of copies through the course of evolution.
v
Table des matières
Résumé ... iii
Abstract ... iv
Table des matières ... v
Liste des tableaux ... vii
Liste des figures ... viii
Remerciements ... x
Avant-propos ... xi
1. Introduction générale ... 1
1.1. La duplication de gènes ... 1
1.2. Les types de duplication ... 1
1.3. Le sort des copies de gène ... 3
1.4. Les facteurs de rétention des copies ... 4
1.4.1. Contraintes liées à la stœchiométrie ... 4
1.4.2. Appartenance à certaines classes fonctionnelles ... 7
1.4.3. Distribution phylogénétique ... 7
1.4.4. Contenu en guanine et cytosine ... 7
1.4.5. Interactions ... 8
1.5. Paramecium tetraurelia ... 8
1.6. Problématique ... 9
1.7. Hypothèses et objectifs ... 9
1.8. Approche méthodologique ... 11
2. Protein complexes linked to greater copy retention after whole-genome duplication in Paramecium tetraurelia ... 12
2.1. Résumé ... 12
2.2. Abstract ... 13
2.3. Introduction ... 14
2.4. Results/Discussion ... 16
2.4.1. Finding orthologs between P. tetraurelia & model species ... 16
2.4.2. Inferring complexes in P. tetraurelia ... 16
2.4.3. Complex membership favors the retention of ohnologs ... 19
2.4.4. Highly conserved genes tend to retain ohnologs ... 21
2.4.5. Link between complexes and conservation ... 23
2.4.6. Complexes are enriched with ohnologons retaining an even number of ohnologs .. 24
2.4.7. Highly conserved ohnologons also retain preferentially even numbers of ohnologs 28 2.4.8. Understanding the bias for even numbers of ohnologs ... 28
2.5. Material and methods ... 31
2.5.1. Finding orthologs between P. tetraurelia & model species ... 31
2.5.2. Inferring complexes in P. tetraurelia ... 32
2.5.3. Computational analyses ... 34 2.5.4. Statistical analyses ... 35 Conclusion générale ... 36 Bibliographie ... 41 Appendix 1 ... 47 Appendix 2 ... 48 Appendix 3 ... 49 Appendix 4 ... 50
vii
Liste des tableaux
Table 1. Distribution of ohnologons according to the number of model species in which one or several orthologs could be found ... 16
viii
Liste des figures
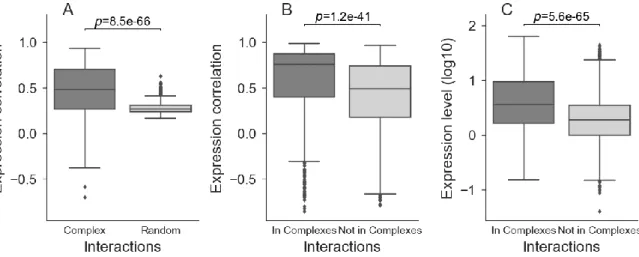
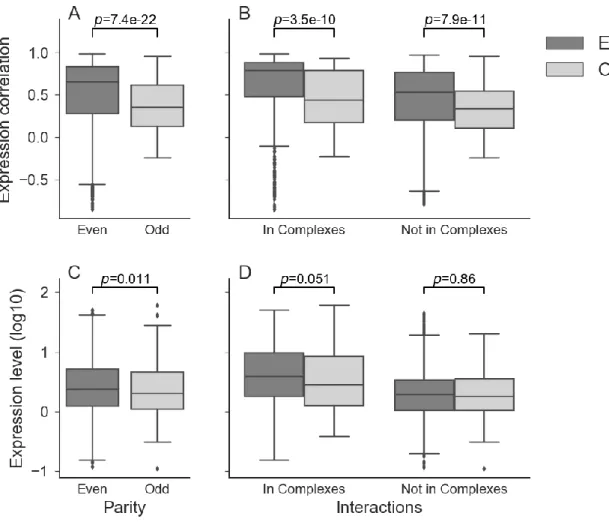
Fig. 1. Expression evidence for inferred protein complexes. (A) Proteins within inferred protein complexes (n=839, complexes with missing expression values removed) are more correlated in expression than within random groups of ohnologons (n=853). The mean correlation value was used for each complex. Ohnologons participating in the inferred protein complexes also tend to have higher mean expression correlation between their ohnologs (B), as well as higher median expression levels (C) than those not participating in the inferred complexes. For the correlation of expression in ohnologons, only ohnologons with two or more ohnologs were considered (in complexes=980, not in complexes=1506). For the expression levels, all ohnologons with at least one ortholog were examined, regardless of the number of ohnologs (in complexes=1328, not in complexes=2420). All p-values calculated using Wilcoxon rank-sum test. ... 18
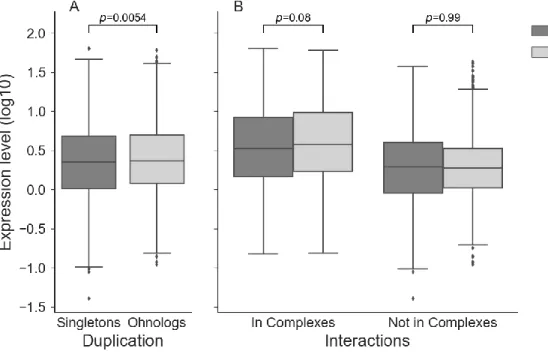
Fig. 2. Expression of singletons and ohnologs is correlated with complex membership. Singletons (n=1262) display slightly lower expression than ohnologs (n=2486) in general (A), but when controlling for complex membership (B), the difference in their expression levels is no longer significant, showing that the slightly higher expression level of the ohnologs was likely due to their enrichment within protein complexes. P-values calculated using Wilcoxon rank sum test. ... 20
Fig. 3. Ohnologons conserved in at least 5 model species (5sp) have more correlated and higher expression than those conserved in 4 or fewer species (4sp). Ohnologons with an ortholog in all 5 of our model species used (5sp) display expression that is both more correlated (A) and higher (C) than ohnologons with an ortholog in 4 or fewer of the model species. Participation in a protein complex amplified this effect, with highly conserved ohnologons participating in protein complexes having the most correlated (B) and highest (D) expression. All p-values calculated using Wilcoxon rank-sum test. ... 23
Fig. 4. Participation in protein complexes according to conservation in model species. The sudden jump in complex participation for ohnologons conserved in all 5 model species seems to indicate that highly conserved proteins are also much more likely to participate in protein complex. ... 24
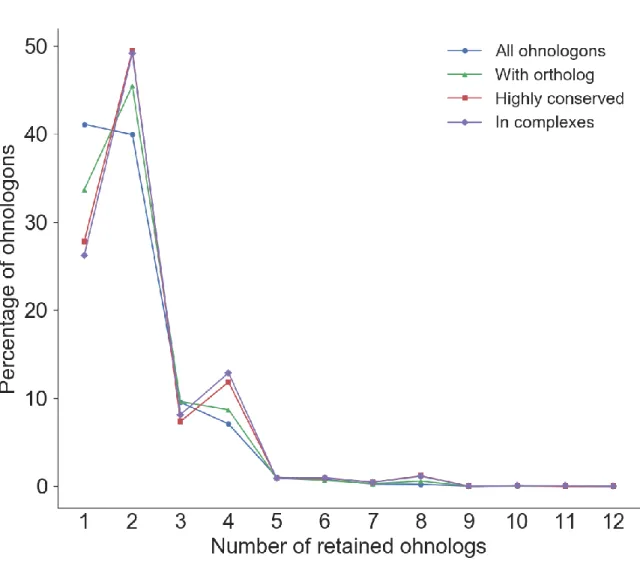
Fig. 5. Ohnologons participating in complexes and those with orthologs in all model species are enriched in even numbers of ohnologs. Blue circles: all the ohnologons found in the Aury et al. (2006) supplementary tables (n=18,496). Green triangles: ohnologons with an ortholog in at least one model species (n=3748). Red squares: highly conserved ohnologons with an ortholog in all the model species used in analyses (n=1090). Purple diamonds: ohnologons participating in protein complexes (n=1328). The latter two distributions show noticeable peaks at 2, 4 and 8 and a minor peak at 6 compared to the first two distributions. ... 25
Fig. 6. Parity influences significantly the expression correlation within ohnologons of 2 ohnologs and more, but barely impacts the expression level. (A) Mean expression correlation is higher within ohnologons with an even number of ohnologs (n= 2080) compared to those with an odd number (n=406). (B) Ohnologons with an even number of ohnologs participating in protein complexes have the highest correlation of expression while those with an odd number not
ix
participating in complexes have the lowest. (C) Expression is only marginally higher within ohnologons with an even number of ohnologs compared to those with an odd number. (D) When controlling for complex membership, expression levels between ohnologons with even and odd numbers of ohnologs are no longer significantly different. All p-values calculated using Wilcoxon rank-sum test. ... 27
Fig. S1 (A) Even-numbered ohnologons conserved in all 5 model species used in analysis (5sp) display higher expression correlation than those conserved in 4 species or fewer (4sp), or than odd-numbered ohnologons regardless of conservation status. (B) Parity does not impact the level of expression of ohnologons, regardless of conservation status. All p-values calculated using Wilcoxon rank-sum test. ... 47
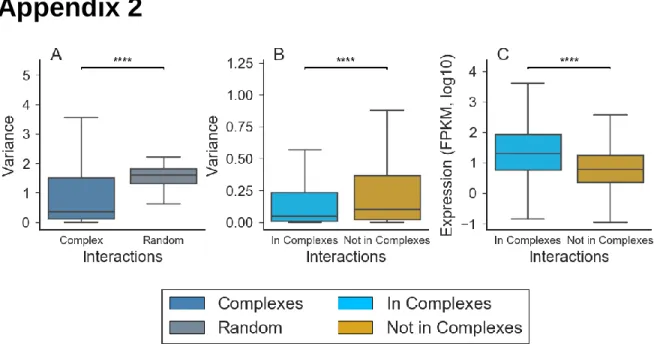
Fig. S2 RNA-Seq analysis confirms that expression is more similar and higher within protein complexes. (A) Expression variance is lower inside complexes (n=839) than random groupings (n=853). (B) Expression variance is lower within ohnologons participating in complexes than within those not participating in complexes. (C) Expression is higher in ohnologons participating in complexes than in those that do not. Outliers hidden in all variance figures for better visibility. All p-values calculated using Wilcoxon rank-sum test. ... 48
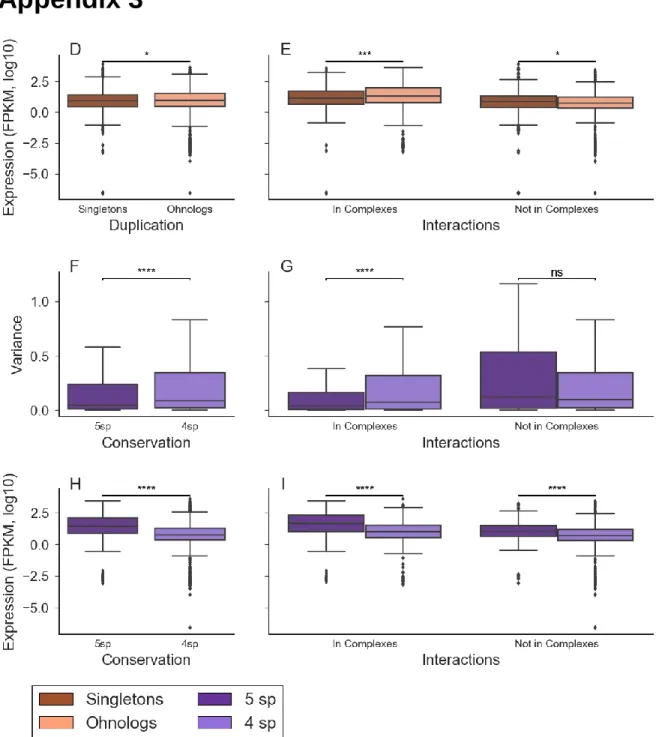
Fig. S3 (D) Expression levels of singletons (n= 1262) and ohnologs (n= 2486) estimated from RNA-Seq data. (E) Expression levels of singletons and ohnologs according to complex membership (F) Expression variance of ohnologons conserved in 5 model species (5sp) or 4 and fewer model species (4sp). (G) Expression variance of ohnologons conserved in 5 model species (5sp) or 4 and fewer model species (4sp) according to complex membership. (H) Expression level of ohnologons conserved in 5 model species (5sp) or 4 and fewer model species (4sp). (I) Expression level of ohnologons conserved in 5 model species (5sp) or 4 and fewer model species (4sp) according to complex membership. Outliers hidden in all variance figures for better visibility. All p-values calculated using Wilcoxon rank-sum test. ... 49
Fig. S4 (J) Expression variance for ohnologons according to parity. (K) Expression variance of ohnologons according to parity and complex membership. (L) Expression level of ohnologons according to parity. (M) Expression level of ohnologons according to parity and complex membership (N) Expression variance of ohnologons according to parity and phylogenetic distribution. (O) Expression level of ohnologons according to parity and phylogenetic distribution. Outliers hidden in all variance figures for better visibility. All p-values calculated using Wilcoxon rank-sum test. ... 50
x
Remerciements
Mon parcours académique n’a pas été facile et j’étais appréhensive à effectuer un retour aux études si tard dans ma vie. Il va sans dire que je dois beaucoup à mes professeurs qui m’ont épaulée tout au long de mon parcours. Merci au merveilleux Alan Anderson, directeur du programme de biologie au 1er cycle, qui a été le premier à me dire que
j’écrivais bien. Moi qui ai toujours refusé de faire une maîtrise dans mon ancienne vie par peur du mémoire, je me suis accrochée à ce compliment comme à une bouée de sauvetage durant toute ma rédaction et j’ai trouvé des mots en moi dont j’ignorais l’existence.
Un autre remerciement bien senti va à mon directeur de recherche, Christian Landry, aux encouragements et à l’optimisme apparemment inépuisables. Avec sa capacité remarquable à communiquer les concepts les plus complexes, il a le don de nous faire sentir plus intelligent juste en étant à proximité, dommage que l’effet s’estompe avec la distance…
Merci aux membres de mon comité d’encadrement, Louis Bernatchez et Arnaud Droit (qui se prend une deuxième dose de remerciements pour être sur mon jury d’évaluation), ainsi qu’à Patrick Lagüe qui a eu la gentillesse d’accepter d’évaluer mon mémoire à la dernière minute sans avoir entendu parler de moi auparavant. Merci à tous ceux du labo Landry, mes adorables voisins de bureau Anna et Angel avec qui j’ai eu tant de plaisir à jouer à roche-papier-ciseau, édition éternuement multilingue, à Véro qui a le cœur sur la main (et il est tellement gros, je ne sais pas comment il tient), à Chris mon grand frère d’Allemagne et Mani ma petite sœur d’Inde et à tous ceux qui vont me manquer terriblement : Axelle, Matteo, Mathieu, Caro, Simon, Éléonore, Marie, Carla, Yacine, Gil, Lou, Phil, Clara, Hélène, Souhir, Alex, Isabelle, François, Guillaume, Ugo, Pauline et Andrée-Ève.
Merci aux gens fantastiques du laboratoire Ogura qui m’ont accueillie en plein milieu de la semi-campagne japonaise et avec qui j’ai passé trois merveilleux mois : Atsushi Ogura, Ryuuhei Minei et Yuki Ueda.
Et finalement, les derniers mais non les moindres, merci à ma famille, ma mère Lucie qui a toujours cru en moi, mon père Daniel qui m’a donné un amour pour la science et ma cousine Laurence qui vit l’agonie de son mémoire en même temps que moi.
xi
Avant-propos
Ma thèse englobe l'ensemble de mes recherches sur les particularités de la rétention de copies dans un organisme ayant subi plusieurs cycles de duplication du génome entier,
Paramecium tetraurelia. Ce manuscrit contient une introduction au sujet de la duplication
de gènes en général et de la duplication du génome entier en particulier, suivi du chapitre principal de mon mémoire, écrit en anglais sous forme d’article et suivant l'ordre des sections utilisé par la revue PLoS Computational Biology, où nous espérons pouvoir le publier. Cet article est suivi d'une conclusion générale passant en revue les objectifs de mon projet, la façon dont ils ont été atteints, les lacunes de mon projet et ce qui pourrait être fait pour les corriger, ainsi que des pistes potentielles pour de futures analyses.
Je suis auteure principale de ma thèse et de l'article inclus. J'ai acquis les données nécessaires aux analyses, effectué les analyses, créé toutes les figures et rédigé l'article sous la supervision de mon directeur de recherche Christian Landry, professeur titulaire au département de biologie ainsi qu’au département de biochimie, microbiologie et bio-informatique. En tant que coauteur, Christian Landry m’a également aidé à orienter mes analyses ainsi qu’à interpréter mes résultats. Mon autre coauteur Angel Cisneros a écrit une partie du code utilisé pour obtenir les orthologues entre P. tetraurelia et les espèces modèles et a fourni de précieux conseils et suggestions au cours des analyses. L'article est présentement en préparation pour publication.
1
1. Introduction générale
1.1. La duplication de gènes
La duplication de gènes est un événement inévitable au cours de l'évolution. Son occurrence est reconnue comme un facteur majeur dans l'apparition d'innovations phénotypiques, ainsi que dans l'évolution de la complexité morphologique et des réseaux d'interactions (1–3). Son occurrence est si fréquente dans l’évolution qu’il est encore possible de trouver une grande fraction de copies de gènes, parfois même jusqu’à 65%, dans le génome de la plupart des espèces (4). Elle est généralement reconnue pour augmenter également la robustesse du génome en diminuant l'impact de mutations délétères (5–8), mais paradoxalement, elle peut également engendrer la fragilité lorsque les deux copies deviennent dépendantes l’une de l’autre pour leur bon fonctionnement au sein d’un réseau d’interactions (9). Certaines familles de gènes qui ont vu leur nombre de copies augmenter au cours de l’évolution des vertébrés sont même associées à certains cancers et maladies génétiques (10). D’autres familles ont eu une expansion en relation avec le mode de vie, comme les récepteurs olfactifs chez les eucaryotes multicellulaires (11).
1.2. Les types de duplication
L'étendue de cette duplication peut aller du gène unique au génome entier. La duplication en tandem ou à petite échelle, ou small-scale duplication (SSD), couvre un seul gène ou segment de chromosome, par exemple suite à une recombinaison inégale durant la méiose (1) ou une rétroposition (4). Il peut également s'agir d'une duplication d'un chromosome entier (aneuploïdie). Un exemple de duplication en tandem bien connu est celui de la duplication chez les singes de l’Ancien Monde d’un gène sur le chromosome X codant pour un pigment de la rétine (12). Les deux copies ont par la suite divergé, devenant sensibles à des longueurs d’onde différentes et permettant l’émergence de la vision trichromatique chez les primates.
À l'autre extrémité de l'échelle, il y a la duplication de génome entier ou whole-genome
duplication (WGD) où tous les chromosomes sont dupliqués, causant une polyploïdie (1).
Ce genre d’événement est rare chez les vertébrés car généralement fatal, mais relativement plus commun chez les plantes (13–15). Ce genre d’événement est à l’origine
2
du blé cultivé (Triticum aestivum), dont le génome hexaploïde s’est formé au fil d’événements successifs d’hybridation entre trois espèces différentes. Cette propriété des plantes de pouvoir former des hybrides fertiles après des duplications de génome a été exploitée afin de produire de nouvelles variétés, comme le triticale, un hybride de blé et de seigle qui contient les génomes des deux espèces parentes (16). Il semblerait donc que la polyploïdie soit un facteur important dans l'augmentation de la complexité d'un organisme (3,5,17,18). En fournissant de nouveaux allèles comme matériel brut à la sélection naturelle, la WGD agit comme source d'innovations fonctionnelles (5). De fait, des événements de WGD ont été répertoriés dans de nombreuses familles, entre autres chez la levure (19), les angiospermes (15,20) et même dans la lignée des vertébrés (1,21), précédant généralement la radiation de nombreuses espèces (1,22–24). Les événements de WGD sont certainement reliés à des événements de spéciation, car le changement de ploïdie engendre généralement un isolement reproducteur avec la ou les espèces parentes (10).
La distinction entre les deux types de duplication est cruciale, car elle a un impact majeur sur le sort des copies de gène générées par l'événement. Cet impact se manifeste notamment dans la classe fonctionnelle des copies perdues ou conservées durant les générations successives. En effet, les deux types de duplication produisent souvent des patrons de rétention contraires (17,25–27). Par exemple, les gènes associés à la transcription et à la transduction du signal ont tendance à être retenus après un événement de WGD, mais sont plus souvent perdus après une duplication en tandem, alors que les gènes associés à la régulation de l'activité enzymatique sont sur-représentés après une duplication en tandem mais sous-représentés dans le cas de la WGD (5,17,26). Cet effet est souvent attribué au dosage génique, car un changement de stœchiométrie peut avoir des effets délétères sur l'organisme (28). Il y aurait donc une pression sélective pour éliminer ce genre de déséquilibre, par exemple celui causé par la duplication en tandem d'un seul gène codant pour une sous-unité à l'intérieur d'un complexe protéique (29). Cependant, comme une duplication du génome entier préserve les proportions respectives des composantes du complexe, les effets délétères associés à une duplication en tandem ne se manifestent pas et la pression sélective est inverse, tendant à préserver les copies de toutes les composantes du complexe pour éviter un débalancement des sous-unités et garder le complexe fonctionnel (29–31).
3
1.3. Le sort des copies de gène
Après une duplication, les copies de gène ainsi créées peuvent évoluer de quatre façons générales (32–34):
Maintien de la fonction originale
Les deux copies restent similaires, fournissant plus de robustesse au génome ou un niveau plus élevé d’une certaine protéine. La similarité de séquence entre les deux copies peut être préservée à travers des processus comme la conversion génique ou la sélection purificatrice (35,36). Selon le type de duplication, l’abondance de la protéine résultante peut être maintenue au même niveau que la protéine ancestrale ou altérée, par exemple afin de respecter les contraintes stœchiométriques de ses partenaires d’interaction ou afin d’augmenter l’abondance d’une protéine constituant l’étape limitante d’une voie métabolique. Le gène de l’amylase (AMY1) est un exemple souvent cité de duplication en tandem où certaines populations humaines présentent un nombre plus ou moins élevé de copies en lien avec la proportion d’amidon dans leur diète (37). Les copies gardent la même fonction, mais le niveau plus élevé d’amylase permet de traiter plus rapidement l’amidon.
Néofonctionnalisation
Une mutation dans une des deux copies va lui conférer une nouvelle fonction. Si cette fonction s’avère bénéfique à l’organisme, la mutation sera fixée par sélection directionnelle (38). La nouvelle fonction peut prendre plusieurs formes, par exemple une nouvelle activité enzymatique permettant de synthétiser un nouveau produit métabolique (39). Ses interactions pourraient également être altérées, la rendant insensible à un inhibiteur (par exemple, chez l’humain, GLUD1 est inhibée par la GTP alors que sa copie GLUD2 y est résistante) ou lui donnant de nouveaux partenaires d’interaction (40,41).
Subfonctionnalisation
Parfois, les deux copies vont accumuler des mutations délétères complémentaires qui vont subdiviser la fonction de la copie ancestrale en deux sous-fonctions. Les deux copies sont donc requises pour effectuer la fonction ancestrale. Au sein d’un réseau d’interactions, ce type d’évolution pourrait se traduire par une répartition des partenaires
4
d’interaction de la protéine ancestrale entre ses deux copies filles (38). Cette subdivision des tâches pourrait s’accomplir en changeant le patron d’expression des deux copies de sorte qu’elles sont exprimées dans des tissus différents (32,39)
Nonfonctionnalisation (pseudogénisation)
Comme la majorité des mutations sont délétères pour le fitness de l’organisme (32), la présence d’une deuxième copie permet l’accumulation de ces mutations sur une des copies, en autant qu’il n’existe pas une forte pression sélective qui s’exerce de manière égale sur les deux copies. Le sort le plus fréquent d’une des copies est donc la pseudogénisation, où la copie devient non-fonctionnelle par accumulation de mutations de perte de fonction. La vaste majorité des copies suivront le chemin de la pseudogénisation et seront éventuellement éliminées du génome (19,30,32,42). Le taux de perte des copies est le plus élevé immédiatement après l'événement de duplication et diminue graduellement au fil des générations (30,42,43). Cependant, certaines catégories de gènes vont systématiquement perdre leur copie à chaque événement de duplication. Ces gènes sont généralement considérés ‘résistant à la duplication’ (duplication-resistant
genes) et sont associées à des catégories fonctionnelles hautement conservées telles que
la réparation de l’ADN, la recombinaison et la réponse aux dommages de l’ADN. Ce phénomène a été observé suite à des événements de duplication indépendants dans plusieurs taxons (44,45). En revanche, ce qui nous intéresse ici, ce sont plutôt les facteurs qui aident à conserver les copies actives au sein du génome, peu importe leur divergence fonctionnelle. Autrement dit, pourquoi une certaine paire de copies va réussir à conserver les deux copies au fil du temps?
1.4. Les facteurs de rétention des copies
1.4.1. Contraintes liées à la stœchiométrie
Les réactions chimiques exigent des quantités particulières des réactants et les protéines n’y font pas exception. Cet impact peut se manifester sous plusieurs aspects, qu’il s’agisse du dosage génique au sein de l’organisme entier, du niveau d’expression des gènes suivant une duplication ou de l’impact de l’appartenance à des complexes ou à des voies métaboliques.
5
L'importance du dosage génique sur le phénotype a été constatée dès les années 1920 par des scientifiques comme Albert Blakeslee et Calvin Bridges sur des organismes aussi différents que les plantes et les drosophiles (46,47). Leurs expériences ont montré non seulement une influence du gène proportionnelle au nombre de copies, mais ont fait une distinction majeure entre la duplication d'un seul gène et celle du génome entier. Dans le cas d'une duplication affectant un seul gène ou chromosome, les individus étaient plus faibles ou stériles alors que les individus polyploïdes étaient viables et fertiles (48). Lors d'une duplication de génome entier, les gènes sont dupliqués en même temps que leurs éléments régulateurs, éliminant l'instabilité causée par la duplication du gène seul (1). Cependant, lorsqu'un seul gène ou chromosome est dupliqué, les protéines concernées verront leur concentration augmenter indépendamment de celles avec lesquelles elles pourraient interagir à l'intérieur d'un sentier métabolique ou d'un complexe. Ce genre de déséquilibre dans la concentration des sous-unités d'un complexe va altérer la stœchiométrie du complexe, résultant en la formation de complexes incomplets et inactifs (28,29,31).
Ce concept peut être également appliqué à la perte d'une copie à la suite d’une WGD, résultant cette fois-ci en une baisse d'expression d'une des sous-unités. Qu’il s’agisse d’une duplication en tandem ou d’une perte de copie suite à une WGD, un gain ou une perte affectant un seul gène au lieu du génome complet et causant un déséquilibre dans la stœchiométrie occasionnera une diminution de la valeur adaptative (fitness) et sera contre-sélectionné (29). Cet effet de dosage est présumé être un des facteurs majeurs influençant la rétention des paralogues après un événement de WGD (30,43,49–51). Cependant, les gènes concernés doivent être sensibles au dosage pour que cette pression sélective s'effectue sur eux.
Le niveau d'expression des gènes
La stœchiométrie semble être reliée au niveau d'expression des gènes dupliqués, que ce soit avant ou après l'événement de duplication. Les gènes les plus fortement exprimés ont une plus grande tendance à être retenus, même à travers plusieurs événements de duplication (5,30,38,52). La perte d'un de ces paralogues survient lorsqu'un fort déséquilibre se développe entre les copies, où une copie va être graduellement surexprimée pour compenser la perte d'expression de l'autre copie et préserver l'équilibre stœchiométrique (30). La perte de la copie avec la plus faible expression devient alors
6
effectivement neutre d'un point de vue évolutif (51). Parmi les gènes qui sont les plus affectés par ce type de pression sélective, on peut retrouver notamment des gènes codant pour des protéines ribosomiques et des gènes métaboliques (3,26,29,52,53).
L’appartenance à la même voie métabolique ou au même complexe
Le lien entre la rétention des gènes et l'appartenance à la même voie métabolique ou complexe protéique a déjà été postulé auparavant (30). Cependant, les voies métaboliques et les complexes protéiques démontrent un certain degré de robustesse face au changement d'expression d'une de leurs composantes et la pression sélective pour retenir une seule copie devrait être minime, surtout lorsqu'elle provient d'une duplication en tandem(28,53,54). C'est pourtant le contraire qui est observé et l'explication se trouve très probablement dans le type de duplication impliqué. Lorsqu'une voie métabolique est dupliquée au complet par WGD, la perte de plusieurs copies aurait une influence plus globale sur la voie et son flux. La pression sélective aurait donc tendance à s'effectuer sur la voie au complet et non pas seulement sur ses composantes individuelles. Ce modèle explique le fait que la perte d'une seule copie provenant d'une duplication en tandem au sein d'une voie métabolique est relativement neutre, alors qu'une plus grande pression sélective s'exerce pour retenir plus de copies lorsqu'elles sont générées par une WGD (53). De plus, considérant que la duplication du sentier métabolique entier va augmenter son flux, et par conséquent le taux de métabolisme de l'organisme, ce genre de modification globale serait favorisé par la sélection naturelle et un retour à la copie simple serait contre-sélectionné (55).
De la même façon, les gènes dupliqués codant pour des sous-unités de complexes protéiques auront tendance à être retenus. Sinon, leur perte devra être compensée par la surexpression de l'autre copie ou par la perte de toutes les autres copies des partenaires dans le complexe (30).
Un autre élément qui vient brouiller les cartes vient du fait que selon les travaux de Gout et al. (2009), les gènes métaboliques sont « significativement plus exprimés que d'autres types de gènes ». Cependant, en corrigeant pour les niveaux d'expression, la différence de rétention entre les gènes métaboliques et non-métaboliques n'est plus significative, démontrant que le niveau d'expression a une influence majeure sur la rétention, surpassant l'appartenance à un sentier métabolique (53).
7
1.4.2. Appartenance à certaines classes fonctionnelles
Certaines classes fonctionnelles de gènes sont sur-représentées parmi les copies retenues, indiquant un avantage sélectif pour certains gènes à être retenus en plus grand nombre de copies. Tel que mentionné plus haut, les protéines ribosomales sont très fréquemment conservées à l'état dupliqué après un événement de WGD (3,26,29,30,52). Dans les catégories plus générales, notons les gènes reliés à la transduction du signal et à la régulation tels que les kinases et phosphatases ainsi que les facteurs de transcriptions (3,5,17,25,52). Comme précédemment, certaines de ces catégories comportent toutefois de nombreux gènes fortement exprimés (30,52). Cependant, à niveau d'expression égal, l'impact de la classe fonctionnelle sur la rétention reste notable et serait donc indépendant du niveau d'expression (49). La rétention de ces catégories spécifiques pourrait indiquer que la WGD est un facteur majeur dans le développement de la complexité des réseaux de régulation, ainsi qu'un moyen puissant de diversification fonctionnelle et d'adaptation (3,17).
1.4.3. Distribution phylogénétique
Gout et al. (2010) ont rapporté que chez Paramecium tetraurelia, les gènes conservés chez un grand nombre d’eucaryotes, donc ayant une plus grande distribution phylogénétique, avaient une plus grande tendance à retenir des copies à la suite d’événements de WGD que ceux qui étaient spécifiques au genre Paramecium. Cependant, aucune explication n’a été avancée pour cette particularité au-delà du fait que les gènes plus conservés subissaient des pressions sélectives différentes que ceux spécifiques à certaines lignées (56,57). En effet, les gènes plus anciens, plus conservés ont tendance à évoluer plus lentement que ceux plus jeunes restreints à une lignée spécifique.
1.4.4. Contenu en guanine et cytosine
Un lien possible entre le contenu d'un gène en guanine et cytosine (GC) a été noté par McGrath et al. (2014) (43), corrélation qui restait bien visible même en corrigeant pour le niveau d'expression. Cependant, aucune explication formelle pour cet effet n'a encore été démontrée. McGrath et al. proposent un lien hypothétique entre une forte sélection de purification sur la séquence codante des gènes riches en GC et une pression sélective pour leur rétention après une WGD.
8
1.4.5. Interactions
Les protéines qui interagissent avec d'autres protéines sont plus souvent conservées après une WGD, en lien avec l'hypothèse de l'équilibre des gènes et du dosage (3,27,29). Cependant, Hakes et al. (2007) présentent un portrait différent, n'ayant pas observé que les copies retenues étaient en général plus connectées que celles non retenues. Ils ont toutefois noté que les copies provenant d'une WGD partageaient un plus grand nombre de partenaires d'interaction que celles provenant d'une duplication en tandem, indiquant que les copies avaient conservé des fonctions similaires. Un cas spécial d'interaction est celui où la protéine interagit avec elle-même pour former des homodimères. Pereira et al. (2007) ont remarqué que lorsque ce type de protéine était dupliqué, il y avait souvent formation de complexes de paralogues, puisque les interactions étaient préservées. Leurs observations mènent à l'hypothèse que de nombreux complexes proviendraient de la duplication d'une telle protéine, suivie par la divergence des paralogues et l'accrétion de nouvelles sous-unités (58). Ce modèle ne concerne pas spécifiquement la WGD, mais est compatible en tant que facteur de rétention des paralogues et démontre encore l'importance de la WGD pour l'évolution de la complexité.
1.5. Paramecium tetraurelia
Paramecium tetraurelia est un organisme unicellulaire de l’embranchement des ciliés qui
commence à s’imposer comme organisme de choix pour l’étude de la duplication des gènes à cause de son génome très particulier. Elle aurait subi au cours de son évolution au moins trois événements connus de WGD en plus d’un autre théorique plus ancien, les traces desquels sont encore très visibles grâce à un nombre limité de réarrangements du génome (30). C'est cette conservation remarquable de la disposition des gènes (synténie) qui a permis à Aury et al. (2006) de distinguer très clairement les trois événements les plus récents chez P. tetraurelia en alignant les blocs de copies conservés. De plus, son taux de rétention de copies est exceptionnellement élevé. En effet, contrairement à d'autres espèces comme la levure Saccharomyces cerevisiae qui a également subi un événement de WGD dans un ancêtre lointain, mais qui n'a conservé que 12.9% de ses gènes pré-WGD sous forme de paires de copies (52), P. tetraurelia en a conservé 51% (30). En plus du grand nombre de copies à étudier, l'occurrence relativement récente (~230 millions d'années (50)) de la dernière WGD permet d'étudier les processus de perte et rétention en action (53).
9
1.6. Problématique
Les duplications de génome entraînent la création d’un nombre énorme de copies, désignées sous le nom d’ohnologues (59) en l’honneur de Susumu Ohno, pionnier de la recherche sur les duplications. Ces gènes qui descendent d’un même ancêtre sont regroupées au sein d’un ohnologon (49). Comme nous savons que P. tetraurelia a subi trois duplications de génome connues en plus d’une duplication théorique, nous devrions nous attendre à retrouver 24 ohnologues au sein de chaque ohnologon, soit 16
ohnologues. Cependant le nombre d’ohnologues retrouvés dans les 18 496 ohnologons identifiés par Aury et al. (2006) varie entre 1 et 12, un écart considérable. Le fait que certains gènes soient des singletons, c’est-à-dire systématiquement retournés à l’état simple après chaque événement de WGD est tout aussi étrange que le fait que certains gènes aient conservé presque tous leur ohnologues. Tels que présentés plus haut, les facteurs gouvernant la rétention des ohnologues suivant une duplication de génome sont multiples et souvent interconnectés. Il devient difficile de cerner le rôle exact de chacun de ces facteurs dans la rétention des copies et il est très probable que ces facteurs agissent en concert. Comment alors déterminer quels facteurs sont responsables de ces deux extrêmes?
1.7. Hypothèses et objectifs
Notre projet s’intéresse à deux facteurs influençant la rétention : la participation à des complexes protéiques et la distribution phylogénétique. La sur-représentation des membres de complexes protéiques parmi les protéines retenues après un événement de WGD avait déjà été notée (3,27,29) et cet effet est généralement attribué au dosage génique. Il est également reconnu que les protéines qui participent dans des complexes protéiques ont une expression plus corrélée (60). Cette co-expression pourrait garder le niveau d'expression des deux copies suffisamment élevé pour éviter la perte d’une d’entre elles. En effet, tel que mentionné plus haut, la perte d’un ohnologue peut être neutre d’un point de vue évolutif si l’autre ohnologue au sein du même ohnologon peut compenser pour la diminution de son niveau d’expression (51). Suivant un événement de WGD, les deux ohnologues participeront au même complexe protéique durant un certain temps et auront le même niveau d’expression avant de diverger. La participation à des complexes protéiques pourrait assurer que ces deux ohnologues conservent un niveau d’expression
10
relativement corrélé et élevé, évitant qu’un des ohnologues ne soit perdu par dérive génétique lorsque son niveau d’expression tombe sous un certain seuil. De plus, la participation à des complexes protéiques pourrait possiblement expliquer le fait que les gènes ayant une plus grande distribution phylogénétique (ceux qui sont conservés chez un plus grand nombre d’espèces distantes) semblent avoir un taux de rétention plus élevé. Comme les complexes protéiques tendent à être conservés à travers les espèces, nous devrions observer un chevauchement important entre les gènes les plus conservés et ceux participant à des complexes protéiques. La rétention des ohnologues pour ces gènes serait donc reliée aux mêmes facteurs qui favorisent la rétention des ohnologues au sein des complexes, soit le dosage génique et l’expression.
Notre hypothèse comporte donc trois éléments : que les complexes protéiques augmentent la rétention des ohnologues, que les ohnologons présents dans les complexes protéiques ont un niveau d’expression plus élevé et plus corrélé que ceux qui en sont absents, et que les ohnologons les plus conservés présentent ces mêmes traits grâce à leur participation dans des complexes protéiques. Afin de vérifier cette hypothèse, voici nos objectifs spécifiques :
1. Déterminer si la rétention d’ohnologues est plus fréquente au sein des complexes protéiques. Autrement dit, valider que le ratio de singletons à ohnologons contenant toujours des ohnologues est plus faible à l’intérieur qu’à l’extérieur des complexes.
2. Vérifier que l’expression des ohnologues est plus corrélée et plus élevée au sein des ohnologons qui participent à des complexes protéiques.
3. Comparer l’impact de la distribution phylogénétique à celui de la participation à des complexes protéiques au niveau du ratio singletons/ohnologues et de l’expression au sein des ohnologons.
4. Établir l’étendue du chevauchement entre les ohnologons participant à des complexes protéiques et ceux qui sont le plus conservés afin de voir si la participation à des complexes protéiques peut expliquer à elle seule l’impact de la distribution phylogénétique.
11
1.8. Approche méthodologique
Présentement, P. tetraurelia est encore peu utilisée en recherche comme organisme modèle. Comme il n'y a pas de données d'interactions publiées pour P. tetraurelia, celles de cinq espèces modèles ont été utilisées à la place. Les espèces modèles utilisées sont
Drosophila melanogaster, Homo sapiens, Mus musculus, Saccharomyces cerevisiae et Schizosaccharomyces pombe. Les relations d’orthologie entre P. tetraurelia et les cinq
espèces modèles ont été obtenues à partir de tableaux de correspondance disponibles sur la base de données InParanoid (61,62). Les complexes protéiques ont par la suite été inférés à partir de ces données en utilisant les relations d'orthologie entre les protéines des espèces modèles et celles de P. tetraurelia. 885 complexes protéiques ont pu être inférés chez P. tetraurelia de cette manière. Le choix des espèces modèles a été basé sur la disponibilité de données sur leurs complexes protéiques ainsi que sur la grande distance phylogénétique qui les sépare. Les ohnologons de P. tetraurelia qui ont des orthologues chez les cinq espèces modèles ont été considérés comme étant hautement conservés pour les besoins de notre comparaison entre la participation à des complexes protéiques et la distribution phylogénétique.
L’impact des complexes protéiques inférés et de la distribution phylogénétique sur l’expression au sein des ohnologons a été analysé grâce à des données microarray générées par Arnaiz et al. (2010) et disponibles publiquement sur le Gene Expression
Omnibus (63). Les conclusions ont été vérifiées avec un ensemble de données RNA-Seq
12
2. Protein complexes linked to greater copy
retention after whole-genome duplication in
Paramecium tetraurelia
2.1. Résumé
Plusieurs événements de duplications du génome entier (WGD) ont fourni une grande quantité de copies de gènes à Paramecium tetraurelia, dont la plupart ont été perdus avec le temps. Plusieurs facteurs ont déjà été proposés comme influençant la rétention de copies. Dans cette analyse, nous avons constaté que la participation à des complexes protéiques augmentait la rétention des copies créées par WGD et était reliée à une expression plus élevée et plus corrélée au sein des groupes de copies provenant du même gène ancestral. Nous proposons également que la participation à des complexes protéiques pourrait partiellement expliquer la rétention accrue des copies observée pour les gènes conservés à travers plusieurs espèces eucaryotes. Enfin, nous avons noté que le taux de rétention accru dans les complexes était largement biaisé vers un nombre pair de copies et nous suggérons que cela pourrait être lié à certaines propriétés structurales des complexes protéiques.
13
2.2. Abstract
Several events of whole-genome duplications (WGD) have shaped Paramecium
tetraurelia’s evolutionary history and provided it with a large amount of gene copies, of
which the greater part has been lost. Several factors have already been suggested to explain the patterns of loss and retention found in P. tetraurelia. In this study we found that participation in protein complexes increases the retention of copies created through WGD and was linked to higher and more correlated expression within groups of copies descended from the same ancestral gene. We also propose that protein complex participation might partially explain the increased retention of copies previously observed for genes conserved through several eukaryotic species. Finally, we observed that the increased rate of retention found within complexes was largely skewed toward an even rather than odd number of copies and suggest that this might be related to certain structural properties of protein complexes.
14
2.3. Introduction
Gene duplication is a major factor shaping the evolution of organisms, linked to the appearance of certain phenotypic innovations, as well as to the rise of morphological complexity and interaction networks (1–3,5,17,18). Gene duplication can augment genome robustness by alleviating the impact of deleterious mutations (5–8), although the opposite effect has also been observed (9). Duplication events vary in scale from small-scale duplication (SSD), where only one gene or a limited region of the genome is duplicated, to whole-genome duplication (WGD) where the entire genome is duplicated. Each type arises from different mechanisms and affects the genome in radically different ways, influencing the fate of the copies created through the duplication event. However, regardless of the type of duplication, the fate usually awaiting one of the copies is pseudogenization through accumulation of non-functionalizing mutations (19,30,32,42). The copies that survive this pruning process are therefore the exception rather than the rule and warrant closer examination. This is especially true in the case of WGD where a large number of copies are created at the same time and are gradually lost over time, with the highest rate of loss being right after the event and then slowing down. Given that WGD events preceded many major radiation of species, from unicellular organisms (1,19) to vertebrates (1,21), clarifying the mechanisms determining which genes retain multiple copies amounts to better understanding the evolutionary trajectory of organisms at the molecular and organismal levels.
As with many complex evolutionary processes, there are many factors governing not only which genes will lose their copy, but the rate at which this process occurs. Here, the type of duplication that yielded the copies is paramount as it has been shown to affect in a major way the fate of copies. SSD and WGD often produce opposite retention patterns (17,25–27) depending on certain factors like the functional category of the genes involved (5,17,26), their level of expression (5,30,38,49,52,53), gene dosage (28,29,31,46,47), participation in complexes or metabolic pathways (28,30,53). Those factors have all been linked to the retention rate and it is likely that there is not one single factor governing which copies are preferentially retained, but rather a large number of interconnected factors, each nudging the fate of a copy towards loss or retention.
From these important factors, it is difficult to delineate how much of an impact each individual factor contributes to the final result. Here we aim to understand the manner in
15
which participation in a protein complex can affect the retention of copies following WGD, as well as measure the impact of other connected factors such as gene dosage and conservation throughout several eukaryotic taxa.
To this end, we used Paramecium tetraurelia, a ciliate that is quickly growing in popularity as the organism of choice to study WGD. At least three different WGD events in its history have been confirmed through synteny analysis of its genome, with traces of an even more ancient fourth event also being detected (30). Even more remarkable is the degree of conservation of the copies generated through those multiple events, with roughly 51% of the genes present before the last WGD event having retained their copy (30), in stark contrast to the 13% found in a more well-studied organism having also undergone WGD like Saccharomyces cerevisiae (52). Unfortunately, while P. tetraurelia is a useful organism for the study of WGD, there is still relatively little data available for it, notably concerning its protein interactions. Our study was therefore divided into two steps: inferring protein complexes in P. tetraurelia through orthology using protein complex data for widely-used model species and combining them with expression and conservation data to draw conclusions about their respective impact on gene copy retention after several rounds of WGD.
16
2.4. Results/Discussion
2.4.1. Finding orthologs between P. tetraurelia & model species
Copies descended from the same ancestral gene through WGD have been dubbed ‘ohnologs’, in honor of Susumu Ohno, pioneer in WGD research. Those ohnologs are grouped together in what is called an ohnologon. Our study looked at the 18,496 ohnologons identified in P. tetraurelia by Aury et al. (2006) to which were added 402 ORFs that also had orthologs in at least one model species and are presumed to be singletons, for a total of 18,898 ohnologous groups. For the sake of clarity, we will refer to all these groups as ‘ohnologons’ even when they are singletons, under the understanding that these singletons were once two ohnologs of which one was lost. These ohnologons contain between 1 and 12 copies generated through the three known WGD events and a further hypothesized ancient fourth event. Of these ohnologons, 3748 (19.8%) had an ortholog in at least one of the model species used (Table 1). Therefore, complexes could only be inferred through orthology for about 20% of P. tetraurelia’s ohnologons. While this is a limited set, we assume that it is large enough to reflect important factors associated with protein complexes that may contribute to the maintenance of ohnologs after WGD.
Table 1. Distribution of ohnologons according to the maximum number of model species in which one or several orthologs could be found
Number of model species Percentage of ohnologons
0 80.17 1 4.76 2 3.87 3 3.46 4 1.98 5 5.77
2.4.2. Inferring complexes in P. tetraurelia
After obtaining orthologs in P. tetraurelia for each of the model species, we inferred the orthologous complexes and merged together those that had the same subunits after mapping, yielding a total of 885 different complexes (see Methods). Of those, 8.2% (n=71) were present in 2 model species or more. There is a certain amount of uncertainty
17
concerning this complex list. First of all, it was inferred through orthology based on the assumption that if a protein is conserved in other species, so are its interactions. This assumption may not hold true in all cases and some ohnologons may have changed interaction partners during the long timespan that separates P. tetraurelia from the other model species. Similarly, all descendants from an ancestral gene in P. tetraurelia are treated as one single unit interacting with the same partners as the ancestral gene. While this has to be true just after the WGD event, this most likely erodes as the ohnologs diverge. Finally, the complexes were inferred from a limited list of known complexes that is not comprehensive. This inferred list is therefore far from exhaustive and absence of an ohnologon from the inferred complexes does not preclude its membership in a complex without orthologs in the available model species.
The validity of the inferred complexes was tested using data that is independent from protein sequence and orthology comparison. Using data such as gene ontology would not be independent since P. tetraurelia’s annotation would be largely based on orthology to model species, whose annotation partly derive from protein complexes or related functions information or vice-versa. We used co-expression analyses among members of complexes on the basis that subunits in a complex tend to have correlated expression, which often serves to predict protein-protein interactions (60,65). We used independent microarray data and RNASeq data to measure the similarity of expression among member subunits. For the microarray data, all probes matching to more than one locus were eliminated to avoid spurious correlation due to cross-hybridization. Because all members of an ohnologon are assigned to a complex by homology, we used the median expression of all members to summarize the expression level of the ohnologon. We found that the mean expression correlation inside complexes was higher compared to random groupings of ohnologons that respects the size distribution (Fig. 1A). It is worth noting that the mean expression correlation within the randomly-created groups is not centered on 0 but slightly higher because the random groups were created from random ohnologons rather than random proteins. Therefore, there is a residual correlation of expression from within the ohnologons themselves.
18
Fig. 1. Expression evidence for inferred protein complexes. (A) Proteins within inferred protein complexes (n=839, complexes with missing expression values removed) are more correlated in expression than within random groups of ohnologons (n=853). The mean correlation value was used for each complex. Ohnologons participating in the inferred protein complexes also tend to have higher mean expression correlation between their ohnologs (B), as well as higher median expression levels (C) than those not participating in the inferred complexes. For the correlation of expression in ohnologons, only ohnologons with two or more ohnologs were considered (in complexes=980, not in complexes=1506). For the expression levels, all ohnologons with at least one ortholog were examined, regardless of the number of ohnologs (in complexes=1328, not in
complexes=2420). All p-values calculated using Wilcoxon rank-sum test.
These results were still valid when creating random complexes using strictly ohnologons participating in complexes as opposed to all ohnologons with an ortholog in at least one model species. The median correlation of expression was 0.48 for the real complexes and 0.40 for the random ones (p-value=1.38e-09, Wilcoxon rank-sum test). The same conclusion was reached when looking at expression variance among the genes within a complex as a metric for expression dissimilarity using the RNA-Seq data (see Methods), with the inferred complexes having generally lower expression variance compared to the equivalent random groupings (median values 0.36 versus 1.6, Fig. S2A). This shows that at least part of the co-expression between the subunits of the inferred complexes came from membership in these inferred complexes and was not simply due to higher correlation within ohnologons themselves, or due to the mere property of participating in a protein complex.
As the previous observations suggest, we found that expression was highly correlated within ohnologons of two and more ohnologs, particularly those involved in the inferred complexes. The mean correlation coefficient for pairs of ohnologs inside ohnologons found within complexes was higher than that of those not found in the inferred complexes (median values 0.76 vs 0.49, Fig. 1B). We observe the same results using RNASeq data (Fig. S2B). Ohnologons in complexes showed lower within-ohnologon expression
19
variance, and therefore more similar expression (median values 0.045 vs 0.0980, p-value=4.62e-11, Wilcoxon rank-sum test). As ohnologs within an ohnologon descend from the same ancestral gene, it is expected that their expression would be correlated to some degree, depending on their degree of divergence. This would indicate a cumulative effect of both being part of an ohnologon and said ohnologon participating in a complex. Finally, not only were ohnologons in complexes more co-expressed, they also had higher expression levels (median values 0.55 vs 0.28, Fig. 1C). This result was once more confirmed with RNASeq data (Fig. S2C, median values 1.28 vs 0.78, p-value=3.48e-75, Wilcoxon rank-sum test). This latter observation is especially relevant as higher expression has already been shown to result in a higher retention rate in P. tetraurelia (49).
Overall, these analyses support the existence of the inferred complexes in P. tetraurelia. We believe that the difference between the ohnologons inferred to participate in protein complexes and the rest to be significant enough to clearly set those two groups apart and allow us to see the impact of protein complex membership in the rest of the analyses. Furthermore, since we restricted our analyses to the set of ohnologons with an ortholog in at least one model species, we can see that the higher correlation of expression and higher expression level of ohnologons inferred to be in complexes is independent from the impact of being conserved in other species. Participating in complexes therefore might affect how ohnologs will diverge after gene duplication through higher and more correlated expression, factors already linked to higher retention rate of ohnologs in previous literature (49,51).
2.4.3. Complex membership favors the retention of ohnologs
All genes are duplicated during a WGD duplication event and they generally slowly return to singleton state, albeit at different rates. 22% (8003/35905) of genes in the ohnologons used in our analysis are in a singleton state. These genes that remain singletons despite several rounds of duplication have been called duplication-resistant genes (44,45). We examined whether complex membership could be a factor in these genes returning to singleton state. For all ohnologons conserved in at least one model species, those with ohnologs far outnumbered singletons almost two to one (2486 versus 1262), suggesting an association between conservation across species and after duplication.
20
We first looked at the distribution of singletons versus ohnologs inside and outside inferred protein complexes. While ohnologs were more numerous than singletons regardless of complex membership, there was a significant difference in their respective ratios. For ohnologons participating in complexes, 26.20% (348/1328) were singletons while the proportion for ohnologons not participating in complexes was 37.77% (914/2420), showing that ohnologons participating in complexes had a greater tendency to retain ohnologs (p-value=5.78e-13, Fisher's exact test).
We were also interested in seeing if singletons and ohnologs differed at the level of expression. In general, ohnologs have slightly higher expression than singletons (median values 0.36 vs 0.35, Fig. 2A), but this higher expression is likely due to enrichment of ohnologs in protein complexes (which show higher expression), as the difference between singletons and ohnologs is no longer significant when controlling for complex membership (Fig. 2B).
Fig. 2. Expression of singletons and ohnologs is correlated with complex membership. Singletons (n=1262) display slightly lower expression than ohnologs (n=2486) in general (A), but when controlling for complex membership (B), the difference in their expression levels is no longer significant, showing that the slightly higher expression level of the ohnologs was likely due to their enrichment within protein complexes. P-values calculated using Wilcoxon rank sum test.
Both groups displayed higher expression levels when participating in complexes, but they seemed to trend in different directions. Ohnologs participating in complexes seemed to have slightly higher expression levels than singletons and the reverse was true for
21
ohnologs not participating in complexes. However, neither of those differences were statistically significant. RNA-Seq analyses showed the same general trends, but with some level of significance when controlling for complex membership (Fig. S3D and S3E). Notably, for both sets of expression data the gap between ohnologs and singletons is larger for those in complexes, showing that complex membership seems to affect ohnologs to a greater degree than singletons at the expression level. The higher retention of ohnologs in protein complexes cannot be conclusively linked to higher expression as the difference with singletons was not significant. However, as observed in Fig. 1B, ohnologons in protein complexes had significantly higher expression than those not in protein complexes. The singletons present in protein complexes could be the remnants of ohnologons who failed to maintain a high enough correlation of expression across both ohnologs to prevent the loss of one of them, and their lower proportion in complexes could be explained by the influence of complexes on higher expression correlation within ohnologons (60). Another explanation could be that those singletons are associated with functional categories under strong selective pressure to return to single state, such as DNA repair and metabolism (17).
2.4.4. Highly conserved genes tend to retain ohnologs
Another factor we wanted to look at was phylogenetic distribution, that is the degree to which a gene is conserved throughout the phylogenetic tree, as it had previously been linked to a higher ohnolog retention rate (49). The mechanism for this association is not yet clear, however we suspect that highly conserved ohnologons also tend to participate in protein complexes. We divided our ohnologons according to whether they had an ortholog in all five of our model species, or four species or fewer. We found that this factor had an impact on retention analogous yet independent to that shown by membership in a protein complex. For example, the proportions of singletons to ohnologs were very similar to those found in complex membership. Singletons were proportionally fewer in ohnologons conserved in all model species, forming 27.80% (303/1090) of the set, compared to 36.08% (959/2658) in ohnologons conserved in 4 model species or fewer. (p-value=9.04e-07, Fisher’s exact test). Compare with singletons forming 26.20% (348/1328) of ohnologons in protein complexes and 37.77% (914/2420) ohnologons not in complexes. Genes conserved in all our model species therefore also display a similar bias towards retention of multiple ohnologs.
22
Larger phylogenetic distribution was also associated with more correlated and higher expression, an effect that was further compounded by complex participation. Ohnologons conserved in all the model species had more correlated expression than those conserved in four or fewer species (Fig. 3A, Fig. S3F) and their expression level as higher as well (Fig. 3C, Fig. S3H). This relationship between phylogenetic distribution and expression was modulated by participation in protein complexes, with ohnologons conserved in 5 model species and participating in protein complex having significantly more correlated expression (Fig. 3B, Fig. S3G), while for those not participating in complexes the degree of phylogenetic distribution did not seem to impact significantly the correlation of expression. On the other hand, ohnologons conserved in 5 model species had higher expression regardless of complex participation (Fig. 3D, Fig. S3I), but the latter factor resulted in even higher expression for each category of conservation, which would indicate that the impacts of phylogenetic distribution and complex membership can add up in driving up expression, potentially influencing the retention of ohnologs. The increased expression of ohnologons that are both highly conserved and participating in protein complexes likely points to those ohnologs containing essential genes, as it is acknowledged that essential genes are highly expressed (66). The higher correlation of expression found within the highly conserved ohnologons probably also reflects lower sequence divergence between the ohnologs, a crucial element that would prevent pseudogenization.
23
Fig. 3. Ohnologons conserved in at least 5 model species (5sp) have more correlated and higher expression than those conserved in 4 or fewer species (4sp). Ohnologons with an ortholog in all 5 of our model species used (5sp) display expression that is both more correlated (A) and higher (C) than ohnologons with an ortholog in 4 or fewer of the model species. Participation in a protein complex amplified this effect, with highly conserved ohnologons participating in protein complexes having the most correlated (B) and highest (D) expression. All p-values calculated using Wilcoxon rank-sum test.
2.4.5. Link between complexes and conservation
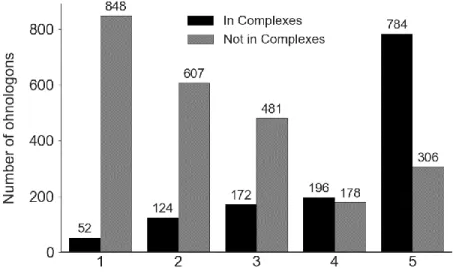
The similarities between the impacts of complex membership and phylogenetic distribution might be explained by the fact that we used orthology to infer complexes. Since complexes tend to be conserved across species, there might be a large overlap between ohnologons found in complexes and those with orthologs in all six model species. In effect, the link between complexes and conservation appears to be significant. We find that ohnologons conserved in all model species are vastly over-represented in the complexes. 59.04% (784/1328) of ohnologons involved in complexes are also conserved in all the model species, versus only 12.64% (306/2420) for ohnologons not in complexes (p-value=4.93e-194, Fisher's exact test).