Evaluation du rôle du
microbiote intestinal dans
l'efficacité digestive du porc
Rapport de Stage Master 2 Biologie des Systèmes
1
Table des matières
1. Remerciements ... 2
2. Abstract ... 3
3. Introduction ... 4
4. Matériel et méthodes ... 6
4.1 Animaux, phénotypes et échantillons ... 6
4.2 Obtention des données de séquençage ... 7
4.3 Obtention de la table d'abondance décrivant les microbiotes intestinaux de porcs ... 8
4.3.1 Première méthode de création de la table d'abondances microbiennes: clusterisation par l'algorithme Esprit-tree ... 8
4.3.2 Seconde méthode de création de la table d'abondances microbiennes : répertorier les séquences Uniques ... 10
4.4 Analyse discriminante en composantes principales ... 10
4.5Analyse en corrélations canoniques régularisées(RCCA)... 12
5. Résultats et Discussion ... 14
5.1 Statistiques descriptives des données ... 14
5.1.1 Table d’abondance esprittree ... 14
5.1.2 Table d’abondance unique ... 14
5.1.3 Performances zootechniques ... 15
5.2 Transformation des tables d’abondance ... 17
5.3. Analyse discriminantes en composantes principales ... 19
5.3.1 La séparation des lignées sur la base du microbiote ... 19
5.3.2 Comparaison des temps ... 28
5.4 RCCA ; Analyse des corrélations entre table d’abondance et performances zootechniques ... 31
5.4.1 Comparaison des trois temps ... 31
6. CONCLUSION ... 34 7. Références ... 35 8. Annexes ... 36 8.1Annexe 1 ... 36 8.2 Annexe 2 ... 38 8.3 Annexe 3 ... 39
2
1. Remerciements
Lors des différentes étapes de mon stage de nombreuses personnes m’ont aidé. Je tiens donc à remercier l’INRA Toulouse plus précisément Genphyse pour m’avoir offert ma première expérience dans le monde du travail qui a été très enrichissante sur le plan professionnel et personnel.
Mes remerciements s’adressent plus précisément à Olivier ZEMB mon maître de stage pour ses conseils et son aide dans la compréhension et l’élaboration des différentes étapes de mon stage. Je souhaite tout autant remercier Hélène GILBERT mon autre encadrante pour son assistance sur la compréhension de la partie génétique animale de mon stage.
Je tiens aussi à remercier Laurent CAUQUIL qui m’a été d’une aide précieuse dans la partie statistiques et transformations des données dans mon stage.
Egalement je souhaite remercier Beatrice GABINAUD pour son explication sur l’approche biomoléculaire des échantillons. Enfin je souhaite également remercier toute l’équipe NED pour son accueil me permettant d’avoir des conditions optimales durant tout mon stage.
3
2. Abstract
Evaluation du rôle du microbiote intestinal dans l’efficacité alimentaire.
L’augmentation exponentielle de la population mondiale en corrélation avec la production et la consommation de viande dans le monde affecte plusieurs secteurs clé de notre société : l’économie, l’agriculture, l’environnement et la santé publique. Dans la filière porcine afin de limiter au maximum la compétition alimentaire entre alimentation animale et humaine une des pistes pour diminuer le coût lié à l’alimentation est l’amélioration de l’efficacité alimentaire. De nombreuses études ont montré qu’en plus de son rôle dans la digestion le microbiote intestinal était un acteur majeur dans la compréhension de certaines maladies et avait des liens dans certaines voies métaboliques. A partir de séquences 16S du microbiote intestinal de 23 porcs dont 12 appartenant à une lignée efficace et 11 à une lignée non-efficace, de performances zootechniques pour chacun des porcs étudiés les résultats ont montré que l’information basée sur les séquences 16S permet la distinction des deux lignées signifiant l’existence d’une différence de composition entre la flore intestinale d’une lignée efficace et non-efficace. D’autres résultats ont montré l’existence de lien de covariations entre les espèces bactériennes du microbiote et les caractères zootechniques.
Study of the role of the gut microbiota in feed efficiency.
The exponential increase of the world population correlated with the production and consumption of meat in the world affects key sectors of our society: economy, agriculture, environment and public health. In the pig production to minimize competition between animal food and human food, the improving of feed efficiency is one way to reduce the part of food in the production. Many studies have shown in addition to its role in digestion the gut microbiota had a function in the understanding of several diseases and had connections in metabolic pathways. From 16S sequences of the intestinal microbiota of 23 pigs including 12 belong to an effective line and 11 to a non-effective line and animal performance for each pig, the results showed with information based on 16S sequences we can see distinction between the two lines. This mean the existence of a difference in composition between the gut microbiota of an effective and non-effective pig. Other results showed the existence of a covariance between bacterial species in the gut microbiota and animal characters.
4
3. Introduction
Aujourd’hui la population mondiale et la production et la consommation de viande dans le monde ne cessent d’augmenter depuis déjà plusieurs décennies ayant pour cause d’affecter plusieurs secteurs clé du monde : l’économie, l’agriculture, l’environnement et la santé publique. En quarante ans la production mondiale de viande a triplé, où on observe dans la dernière décennie une augmentation de 20%. Dans un contexte où on observe une forte augmentation de la demande pour l’alimentation animale, l’agriculture et l’élevage des animaux doivent être orientés afin de permettre une réduction de l’impact environnemental et limiter une compétition entre alimentation humaine et animale. Cela signifie que les animaux dans le futur devront être plus efficaces dans la transformation et l’utilisation de l’aliment en réduisant leur prise alimentaire tout en maintenant une performance de production assez haute, en réduisant leurs rejets dans l’environnement et en acceptant une large gamme d’aliments (protéines ou fibres). Par exemple en France le coût de production lié à l’alimentation correspond à environ 60% du cout de production du porc. Une des pistes pour diminuer le coût lié à l’alimentation est l’amélioration de l’efficacité alimentaire, c’est un enjeu majeur dans la filière porcine afin de limiter au maximum la compétition alimentaire qu’entraine la sélection.
L’efficacité alimentaire est l’efficacité d’utilisation des aliments par les animaux en croissance, elle est classiquement évaluée par le rapport entre le gain de poids et la consommation alimentaire pendant une période donnée. Il s’agit d’un indicateur de rendement, compris entre 0 et 1. Il traduit l’efficacité de conversion de l’aliment en gain de poids, et est appelé indice de consommation (IC).L’IC est influencé par la quantité et la qualité d’aliment disponible, le type d’animal et l’environnement d’élevage. La quantité d’énergie disponible dans la ration est le facteur le plus déterminant pour l’IC.
L’efficacité alimentaire dépend probablement de plusieurs facteurs : Par exemple, le phénotypage pour rechercher plus précisément des marqueurs biologiques spécifiques, comme l’élevage d’animaux en modifiant leurs ressources alimentaires pour étudier l’efficacité digestive selon l’aliment et essayer de l’améliorer. Un autre exemple est l’étude du microbiote intestinal pour l’efficacité alimentaire car la flore intestinale joue un rôle important dans la digestion des fibres de l’aliment. Cela est important vu que les produits destinés à l’alimentation animale seront de plus en plus fibreux pour tenter de restreindre la compétition entre alimentation animale et humaine. Au jour d’aujourd’hui aucune recherche n’a été entreprise sur l’existence d’un effet du microbiote intestinal sur l’efficacité alimentaire de manière approfondie chez le porc. (Gilbert, 2015)
5
Le microbiote intestinal est l’ensemble des bactéries du tube digestif. On estime le nombre de ces bactéries à 100 000 milliards chez l’humain soit 10 à 100 fois plus que le nombre de cellules dont est constitué le corps humain et leur diversité est importante vu qu’un millier d’espèces bactériennes ont pu être distinguées. Ces différentes espèces bactériennes étaient définies comme ayant seulement un rôle dans la digestion des aliments mais diverses études ont montré que le microbiote intestinal était un acteur majeur dans la compréhension de certaines maladies comme la maladie de Crohn (Erickson et al., 2012), l’obésité (Meng et al., 2014) ou le diabète de type 2 (Qin et al., 2012). Des liens ont été aussi montrés entre les bactéries du microbiote et le cerveau ainsi que le système immunitaire. On parle dans ces études du métagénome correspondant à l’étude de l’ensemble des gènes de ces bactéries. Après ces différents travaux sur les effets du microbiote intestinal, d'autres scientifiques se sont intéressés à l’existence d’un lien entre le microbiote et l’efficacité alimentaire chez les animaux.
L’objectif est donc d’évaluer le rôle du microbiote intestinal dans l’efficacité alimentaire chez le porc. Pour cela j’ai à ma disposition des données concernant 23 porcs sélectionnés sur le critère de la consommation moyenne journalière résiduelle (CMJR), qui est un paramètre de l’efficacité alimentaire, quantifiant l’ingéré non utilisé pour la production (Koch et al. 1963). Ce paramètre est un critère de sélection possible pour explorer de nouvelles pistes de réduction de l’ingéré des porcs. Il se définit comme étant la différence entre la consommation observée et une consommation d’aliment « théorique » en fonction des besoins estimés de l’animal pour l’entretien et la croissance. En restriction alimentaire ou avec une alimentation ad libitum il s’agit de sélectionner les animaux gaspillant le moins d’énergie dans leur entretien, dans leur métabolisme, dans leur activité et aussi dans le dépôt dans les tissus (Drouilhet et al., 2013)On cherche à savoir s’il existe des différences significatives dans le microbiote intestinal selon l’efficacité alimentaire en comparant porcs efficaces et porcs peu efficaces. Si l’hypothèse suivante : le microbiote intestinal possède un effet dans l’efficacité alimentaire chez le porc est accréditée alors le microbiote intestinal pourra être pris en compte dans le processus de sélection de l’efficacité alimentaire et conduire à une meilleure précision de celle-ci. Mon stage vise donc à analyser des données précédemment acquises au laboratoire sur les performances zootechniques et le microbiote intestinal de porcs de deux lignées, efficace et non-efficace. Plus précisément j’ai cherché à savoir si ces deux groupes avaient des microbiotes différents et si oui quelles espèces bactériennes les caractérisent. Pour cela une clusterisation des différentes séquences de bactéries des microbiotes a été réalisée pour obtenir une table d’abondance. J’ai ensuite réalisé différents traitements statistiques pour confirmer ou infirmer l’hypothèse d’une séparation des lignées à partir de l’information basée sur les séquences 16S.
6
4. Matériel et méthodes
4.1 Animaux, phénotypes et échantillons
Pour étudier les différences de microbiote entre lignées efficace et moins efficace, des données ont été produites en 2011 sur 23 porcs qui sont issus de lignées sélectionnées de façon divergente pendant 8 générations selon un des paramètres de l’efficacité alimentaire la CMJR. Sur ces 23 porcs 12 de la lignée ‘m’ sont des porcs consommant moins d’aliment par rapport à leur poids vif et leur composition corporelle en moyenne, 11 de ces 23 porcs sont quant à eux issus de la lignée moins efficaces (‘p’) c’est-à-dire des porcs qui ont une mauvaise utilisation de l’aliment entraînant une prise alimentaire excessive par rapport à leurs performances. Pour mener à bien cette étude il a été décidé avant mon stage d’étudier le microbiote sur trois temps différents comme l’indique le Tableau 1 pour les deux lignées pour en voir aussi l’évolution car on sait qu’à la naissance des porcs leur microbiote était totalement vierge et que la composition de ce microbiote a été donc influencé par les conditions d’environnement et par l’alimentation, en plus de leur génotype. Des prélèvements de fèces ont donc été réalisés à chaque temps et pour chaque porc pour connaître la composition du microbiote de chaque porc et son évolution à l’aide du séquençage.
T1 11 semaines T2 17 semaines T3 20 semaines TOTAL Lignée m 12 12 12 36 Lignée p 11 11 11 33 Total 23 23 23 69 ECH
Tableau1. Distribution des échantillons de fèces des porcs aux trois temps.
Des performances zootechniques ont été mesurées pour chaque porc pour permettre de les comparer selon la composition de leur microbiote fécal. Ces données zootechniques correspondent à différentes mesures réalisées entre la naissance du porc et le temps trois à vingt semaines: le poids du porc aux trois temps, le poids à la naissance, l’indice brut correspondant à la CMJR, l’indice de consommation, l’épaisseur de lard mesurant la composition corporelle , la consommation journalière entre 35kg et 95kg, qui quantifie la prise d’aliment par porc, le
7
gain moyen quotidien entre 35 et 95 kg pour chaque porc correspondant à la vitesse moyenne de croissance.
4.2 Obtention des données de séquençage
Après que les fèces ont été prélevées chez les porcs (au laboratoire avant mon stage) le but a été comparer les microbiotes de porcs efficaces et non-efficaces grâce au séquençage de la parties variable V3 du gène de la petite sous-unité ribosomique comme marqueur (i.e. gène ARN 16S). La première étape a été de congeler les fèces pour les conserver, ensuite l’analyse technique a été faite une fois que tous les échantillons de fèces ont été récupérés pour diminuer au maximum le bruit qui peut être lié à l’extraction de l’ADN ou à l’environnement du laboratoire. Ensuite il a fallu extraire et purifier l’ADN de chaque échantillon à l’aide de différentes lyses puis cet ADN a dû être amplifié à l’aide de PCR pour permettre sa quantification et son séquençage. Pour la PCR les amorces qui ont été choisis pour l’expérimentation sont les régions hypervariables V3/V4 de l’ARN 16S car presque toutes les bactéries sont identifiables sur la base de ces régions et aussi que des banques de données publiques telles que GreenGenes ou RDP répertorient ces séquences pour permettre une comparaison avec les résultats du séquençage obtenu.La RDP (Ribosomal Database Project II) a été développée afin de répondre à la demande croissante d'analyse à haut débit des séquences d'ARNr, que ce soit en écologie microbienne ou en médecine et a été utilisé pour l’affiliation taxonomique des différentes bactéries composant le microbiote des lignées efficaces et non-efficaces.
Suite aux PCR les échantillons ont été séquencés à la plateforme de séquençage puis renvoyés sous forme de fichiers FASTQ. Ce dernier est un format texte qui permet de stocker une séquence biologique (généralement une séquence nucléotidique) et ses scores de qualité correspondants. Le séquençage de l’ARN 16S est très fréquemment utilisé pour l'identification au niveau spécifique car il s'agit de molécules ubiquitaires possédant une structure conservée et abondantes dans les cellules. Les séquences des différents échantillons sont ensuite mis sous le format fasta qui permet de représenter une ou plusieurs séquences où la première ligne commence par le symbole ‘>’ suivi d’un identifiant correspondant à la séquence se situant sur la deuxième ligne, FLORALIM_1.1P1134572.clean.fasta (exemple d’un échantillon). In fine j’ai eu à ma disposition 69 fichiers multifasta correspondant au séquençage de l’ARN 16S des 69 échantillons avec un total de séquences de 1178918 séquences. Ces dernières aux sorties du séquençage ont subi un prétraitement suivant la taille, ensuite suivant les N cela correspond à
8
enlever les erreurs de nucléotide c’est-à-dire enlever tous les nucléotides qui ne sont ni A, T, C ou G. Comme autre prétraitement réalisé, la suppression des homopolymères (stretch de A d’environ 9 nucléotides), ces derniers sont une grosse source d’erreur du séquençage 454, technologie utilisée dans cette expérimentation.
4.3 Obtention de la table d'abondance décrivant les microbiotes
intestinaux de porcs
4.3.1 Première méthode de création de la table d'abondances
microbiennes: clusterisation par l'algorithme Esprit-tree
J’ai utilisé un workflow en shell bash du laboratoire que mon maître de stage a créé que j’ai dû adapter aux données que l’on a mis à ma disposition pour permettre la conversion des séquences 16S en table d’abondance d’espèce bactériennes. La clusterisation des séquences sorties du séquençage au format fasta a été faite à l’aide d’Esprit-tree qui est un des algorithmes existants pour l’analyse des communautés microbiennes(Sun et al., 2012). Esprit-tree est une mise en œuvre rapide de l'algorithme de classification hiérarchique pour l’analyse des données de séquençage. Il atteint une complexité de calcul quasi-linéaire et maintient une grande précision comparable à la clusterisation hiérarchique. Il est basé sur l'apprentissage en traitant simultanément l'espace et les problèmes de calcul de travail préalable. L'idée de base est de partitionner un espace de séquence dans un ensemble de sous-espaces en utilisant un arbre de partition construit en utilisant un espace métrique, puis de manière récursive d’affiner une structure de regroupement dans ces sous-espaces. La technique repose sur de nouvelles méthodes de clustering, d'insertion de séquence et de suppression de nœuds de l'arbre de partition. Pour éviter les calculs exhaustifs des distances entre les clusters, chaque cluster de séquences est représenté en tant que séquence probabiliste, et l’algorithme définit un ensemble d'opérations pour aligner les séquences probabilistes et calculer les distances génétiques entre elles (Cai et al 2011). Esprit-tree a été implémenté sur le serveur GenoToul qui est une plateforme génomique créé en 2000 afin d’étudier les métadonnées produites par les nouvelles techniques de séquençage (NGS).
Une des limites de cette méthode c’est qu’il faut qu’avant l’étape de clusterisation on ait au maximum 500 000 séquences. A ma disposition je possède 69 échantillons ce qui signifie que
9
par échantillon le maximum de séquences que l’on peut avoir est d’environ 7 246 séquences. Le workflow a été exécuté à l’aide de Putty et de WinSCP et son détail est en annexe 1.
La première étape du workflow est une normalisation permettant de comparer les différents échantillons, elle se fait en prenant comme nombre maximal de séquences le nombre minimal de séquences retrouvé dans un échantillon. C’est une normalisation par le bas. La deuxième étape a été de trouver les chimères et de les supprimer par l'algorithme UCHIME (Edgar et al., 2011).
Ces chimères surviennent suite à l’amplification PCR et au séquençage, ce sont des séquences artéfactuelles produites par le protocole expérimental. Elles représentent un problème important car elles suggèrent la présence d’organismes non importants qui n’existent pas en réalité dans les échantillons. Cette étape est très importante afin d’obtenir un résultat de clustering le plus précis possible. A la sortie de cette étape j’ai récupéré un fichier par échantillon dans lesquels les séquences chimériques ont été supprimées, c’est à partir de ce moment que j’ai pu vérifier que le nombre de séquences totales sans chimères était largement inférieur à 500 000 séquences permettant de passer à l’étape de clusterisation d’Esprit-tree. Il initialise son algorithme de clusterisation en regroupant les séquences par similitude. L’avantage d’Esprit-tree c’est qu’il génère ici des fichiers par cluster ou OTU (unité taxonomique opérationnelle), c’est-à-dire un fichier dans lequel toutes les séquences appartenant à l’OTU sont répertoriées. La répartition de chaque séquence de chaque échantillon a été faite à un seuil 3% de clusterisation c’est-à-dire que chaque cluster regroupe des séquences ayant un pourcentage de similitude supérieur ou égal à 97%. Ce seuil a été établi dans la publication Esprit-tree pour précisément les régions V3 et V4 de l’ARN 16S. Suite à la génération des différents clusters, on a compté le nombre d’apparitions de chaque séquence d’un échantillon dans chaque OTU pour en faire une table d’abondance. Après pour chaque OTU, une assignation taxonomique a été donné en prenant compte comme séquence de référence le centroïde du cluster correspondant à la séquence moyenne, c’est-à-dire la séquence ayant une distance similaire entre elle et les différentes séquences du cluster. Cette assignation est faite grâce à la base de données RDP. A la fin du workflow, une table d’abondance est obtenue contenant 26 824 OTUs où l’on retrouve le nombre de fois où une séquence d’un échantillon est présente dans un cluster. L’assignation taxonomique de la séquence du centroïde de chaque OTU, son séquençage et son identifiant sont également présents dans la table d’abondance.
10
4.3.2 Seconde méthode de création de la table d'abondances
microbiennes : répertorier les séquences Uniques
Pour observer et comparer la performance d’Esprit-tree, à partir des fichiers sans les chimères crées pour les 69 échantillons une autre méthode pour rassembler les séquences a été réalisée q ui ne prend pas en compte de seuil de clusterisation de 0.03.
Cette méthode est simple : elle consiste à rassembler dans un fichier toutes les séquences des 6 9 fichiers fasta sans les chimères en un seul fichier. Ensuite dans ce fichier le principe est de c
ompter le nombre de fois que l’on observe une séquence unique et on regroupe ces séquences dans un groupe que l’on appelle cluster. Avec cette méthode qu’on appellera
‘Unique’ une table d’abondance contenant 10 4812 OTUs est obtenue avec les mêmes caractéristiques que la table d’abondance esprittree. La table d’abondance sera appelé unique,
il faut noter que pour cette table la taxonomie n’est pas présente pour les différents groupes créés.
A partir de ces deux tables d’abondances obtenues, un traitement statistique a été réalisé à l’aide du logiciel R pour étudier les différences de microbiote qu’il peut exister entre lignée efficace et non-efficace.
Avant toute manipulation de données une étape de normalisation pour toutes les data frames que l’on a analysé avec la DAPC a été réalisé sur l’abondance afin de nous donner la possibilité de comparer les clusters crées entre eux.
4.4 Analyse discriminante en composantes principales
L'analyse discriminante en composantes principales (DAPC) est une approche méthodologique qui conserve tous les avantages de l’analyse discriminante en en optimisant la perpendicularité des axes. La DAPC repose sur la transformation de données en utilisant l'analyse en composantes principales (PCA) comme une étape préalable à l’analyse discriminante, ce qui garantit que les variables ne soit pas corrélées entre elles et que leur nombre est inférieur à celui des échantillons analysés. Comme la PCA, la DAPC peut être appliquée à de très grands ensembles de données, comme les données de séquençage générées par les nouvelles techniques de séquençage. Avec l'affectation des séquences des échantillons aux clusters, la DAPC fournit
11
une bonne visualisation des structures génétiques entre-populations permettant une meilleure visualisation des données. La DAPC est une méthode qui a pour but d’identifier et de décrire des clusters génétiques. On s’intéresse peu à la diversité qui peut exister intra-groupes mais entre groupes d’individus. Cette fonction a été mise en place au vu des différents problèmes des méthodes traditionnelles comme les algorithmes basés sur du clustering bayésien qui ne sont pas capables de faire face à des quantités de données trop importantes ou comme les méthodes d’analyses multivariées qui ne sont pas suffisantes à l’étude de la structure génétique des communautés microbiennes.
VAR(X) = B(X) + W(X)
où B(X)=variance entre groupe et W(X)= variance dans groupe et X représente la population étudiée.
La DAPC optimise B(X) et minimise W(X) permet de montrer les meilleures différences possibles entre groupe sans se soucier des différences intra-groupe. C’est la principale différence avec les autres méthodes d’analyses en composantes principales qui cherchent à étudier la diversité globale en se focalisant sur la variance globale.
Dans le cas des données de ce rapport la comparaison des porcs efficaces et non-efficaces, la DAPC est le meilleur moyen pour savoir si ces deux lignées peuvent être ou non séparées significativement.(Jombart et al, 2010). Sous R la DAPC a été implémenté dans le package ‘adegenet’ qui est dédié à l'analyse exploratoire de données génétiques. Il met en œuvre un ensemble d'outils allant de méthodes multivariées à la génétique spatiale et aux analyses de mutations ponctuelles (Jombart et al, 2011).
La DAPC est une analyse statistique qui comprend un test de significativité à 5%. Pour faire cette analyse deux hypothèses ont été posée : l’hypothèse nulle (H0) qui explique que la séparation des deux groupes efficaces et non-efficaces est due au hasard et l’hypothèse alternative (H1) où la séparation des groupes est une réalité biologique différente d’une distribution au hasard. La DAPC prend plusieurs paramètres en entrée comme le nombre de composantes principales, un fichier d’assignation de chaque échantillon à la lignée, la table d’abondance que l’on souhaite analyser et un nombre de simulations.
Cette analyse permet de calculer les probabilités des séparations des deux lignées à l’aide d’un paramètre appelé le a-score qui calcule le biais entre une observation biologique et une randomisation des groupes sur 500 simulations pour savoir si on peut mieux distinguer les deux lignées que le hasard à partir des OTUs. La DAPC en sortie donne deux probabilités
12
correspondant à la lignée efficace ‘m’ et la lignée non-efficace ‘p’. Ces probabilités sont calculées en comparant l’appartenance des échantillons aux lignées selon les composantes principales et les 500 simulations où l’appartenance des échantillons aux lignées randomisée comme les OTUs. Si j’observais des probabilités inférieures à 0.05 pour les deux lignées alors l’hypothèse nulle de départ selon laquelle les microbiotes entre lignées sont identiques pouvait être rejetée et donc que les deux lignées peuvent être séparées selon certains OTUs représentés dans les composantes principales. Pour connaitre le nombre de composantes principales à partir desquelles les deux lignées peuvent être séparées j’ai fait des boucles de vérifications allant du nombre minimal de composantes principales au nombre maximum de composantes principales pour savoir à partir de combien de composantes principales les deux groupes pouvaient être ou pas séparés. Le nombre maximal de composantes principales possibles a été de 69 pour toutes les DAPC que j’ai réalisées dans ce rapport correspondant au nombre minimum entre le nombre d’échantillons et le nombre de variables étudiées les OTUs.
Une autre partie de la DAPC était comment prendre en compte les probabilités générées par les 500 simulations, pour cela j’ai réalisé trois types de simulations: en prenant les simulations inférieures ou égales à zéro, les simulations strictement inférieures à zéro et toutes les simulations différentes de zéro. Les résultats ces trois DAPC ont montré que c’est avec la simulation où l’on choisit les nombres inférieurs à zéro qui permet d’observer une très bonne séparation des groupes avec des probabilités inférieures au seuil de 5% en tenant bien sur compte du nombre de composantes principales. Ce type de simulation a été gardé pour toutes les analyses discriminantes réalisées dans ce rapport.
Une partie du script fait en R sera mis en annexe 2 pour montrer quelques boucles de vérifications du nombre de composantes principales.
4.5Analyse en corrélations canoniques régularisées(RCCA)
L’analyse des corrélations canoniques (CCA) est une approche multivariée exploratoire pour mettre en relief la corrélation entre deux jeux de données d’une même expérience. De la même manière que la PCA, la CCA recherche des combinaisons linéaires de variables appelées variables canoniques pour réduire la dimension des deux jeux de données tout en maximisant la corrélation entre les variables, appelée les corrélations canoniques. Une propriété importante des corrélations canoniques est qu’elles ne changent pas selon la transformation des variables, c’est la plus grande différence avec une analyse des corrélations ordinaires dépendant fortement de la manière dont les variables sont décrites (Gonzalez, Dejean, Martin, & Baccini, 2008).13
La CCA ne peut pas être effectuée lorsque le nombre d’expériences est inférieur au nombre maximum de variables dans les deux ensembles de données ce qui est le cas pour nos données zootechniques et la table d’abondance esprittree ou unique. Quand le nombre de variables augmente, la CCA tend à donner plusieurs corrélations canoniques proches de 1 indiquant que l’on ne peut découvrir aucune observation significative. En plus lorsque les variables sont fortement corrélées avec la CCA elles ont tendance à engendrer des erreurs conduisant au fait que l’on ne peut pas relier les deux data sets étudiés.
Un moyen pour éviter ce problème de la CCA avec les métadonnées a été d’introduire des paramètres de régularisation dans le calcul de la CCA, c’est la RCCA. Pour les deux jeux de données on aura un paramètre de régularisation appelé λ. Ces deux paramètres sont calculés par cross-validation en cherchant directement le maximum du paramètre dans les deux dimensions pour être sûr d’obtenir la valeur de λ optimale, cette méthode nécessite un espace de calcul important. Une autre méthode est de construire des grilles de taille raisonnable de valeurs de λ pour chaque table d’abondance pour évaluer par cross-validation pour chaque point de la grille et de choisir la meilleure combinaison de λ qui donne le CV-score (score de corrélation) le plus élevé. Cela se fait à l’aide de la fonction tune.rcc. Le choix spécifique des paramètres de régularisation reste ainsi un problème ouvert, comme il l'est dans d'autres méthodes des statistiques régularisées. (Gonzalez et al., 2008).
La RCCA est implémenté sous R dans le package ‘mixOmics’, il sert à l’étude des données provenant des technologies de séquençage à haut débit, tels que les données ‘omiques’ (transcriptomique, protéomique, métabolomiques). Cependant ‘mixOmics ’peut être appliqué à d’autres ensembles de données où le nombre de variables est plus grand que le nombre d’individus dans l’expérience. Pour cela de nombreuses sorties sous forme de graphique sont fournis par le package afin de mieux visualiser et interpréter les résultats.
Au vu du nombre d’OTU pour les deux tables d’abondance la RCCA m’a aidé pour mettre en relation les données zootechniques et les différentes tables d’abondance à partir de différents scripts que j’ai réalisés pour voir s’il existe des covariations entre les espèces bactériennes présentes dans le microbiote et les performances zootechniques. Pour les différentes corrélations que donne chaque analyse à l’aide d’une validation croisée j’ai mis en place en plus un script permettant de ressortir les corrélations étant significatives La première étape consistait à tout d’abord randomiser les performances zootechniques 1000 fois que j’ai récupérées dans une liste. Ensuite pour chaque randomisation des données zootechniques j’ai réalisé une RCCA où chaque résultat est intégré à une liste. L’étape suivante est de créer une liste dans laquelle
14
on répertorie toutes les matrices de corrélations que l’analyse des corrélations canoniques régularisée produit. La prochaine étape que j’ai mise en place était de créer une liste de n vecteurs où n correspond à la dimension de la matrice de corrélation étudiée. Dans chaque vecteur on retrouve 1000 valeurs de corrélations issues des randomisations pour chaque valeur de corrélation « vraie » estimée. Dans cette liste chaque vecteur a été trié de la plus faible corrélation vers la corrélation la plus élevée, ensuite à l’aide d’un code R que j’ai créé j’effectue un test à 5% pour voir à quelle position se situe la valeur de corrélation observée dans le vecteur des valeurs théoriques issues des 1000 randomisations. Je prenais en compte une valeur de corrélation comme significative si et seulement si je la trouvais comprise entre les positions 1 et 25 du vecteur ordonné ou entre les positions 975 et 1000 pour correspondre à un test à 5%. Cette estimation de seuil empirique a été faite à chaque fois qu’une RCCA a été mise en place.
5. Résultats et Discussion
5.1 Statistiques descriptives des données
La première étape de l’analyse statistique que j’ai réalisée a été d’étudier les tables d’abondance dans leur globalité pour avoir une bonne visualisation du jeu de données.
5.1.1 Table d’abondance esprittree
Elle est composée de 26824 clusters avec 296 niveaux de taxonomie différents. J’ai remarqué que plus d’un quart des bactéries présentes dans les microbiotes étudiés correspondait à des Bacteroidetes/Prevotella appartenant à un des 5 phylums bactériens qui représente 95% du microbiote. J’ai aussi retrouvé un autre phylum bactérien majoritaire dans la composition du microbiote les Firmicutes qui correspondant à plus d’un quart des espèces bactériennes retrouvées dans le microbiote des lignées efficace et non-efficace. Ensuite j’ai observé qu’une très grande partie de ces bactéries correspondait à des espèces bactériennes des phylums Actinobacteria et Proteobacteria majoritaires dans la composition du microbiote.
5.1.2 Table d’abondance unique
C’est une table d’abondance composée de 104812 groupes correspondant chacune à des séquences que l’on retrouve dans les espèces bactériennes après que les différents traitements
aient été réalisé comme la normalisation par le bas et la suppression des chimères. J’ai observé comme principale différence entre les deux tables d’abondance à part la clusterisation des taux
15
d’abondances qui sont inférieurs en moyenne par échantillon pour la table d’abondance unique.
5.1.3 Performances zootechniques
Après les générations des tables d’abondance on a mis à ma disposition les données zootechniques pour observer s’il existe des liens de covariations entre les espèces bactériennes du microbiote chez les deux lignées et les différents caractères zootechniques. J’ai commencé par me poser une question qui me paraissait essentielle sur le fait de savoir si les différences entre lignées efficace et non-efficace étaient visibles pour les caractères zootechniques. Pour cela j’ai fait des comparaisons pour chaque caractère étudié entre les deux lignées pour voir si j’observais des différences significatives. Pour faire ces comparaisons il fallait savoir si les valeurs mesurées pour chaque caractère suivaient une loi normale. Ensuite j’ai réalisé des tests de student pour comparer les moyennes de chaque caractère zootechnique des deux lignées de porcs pour savoir si elles étaient significativement différentes. Les résultats obtenus sont
16
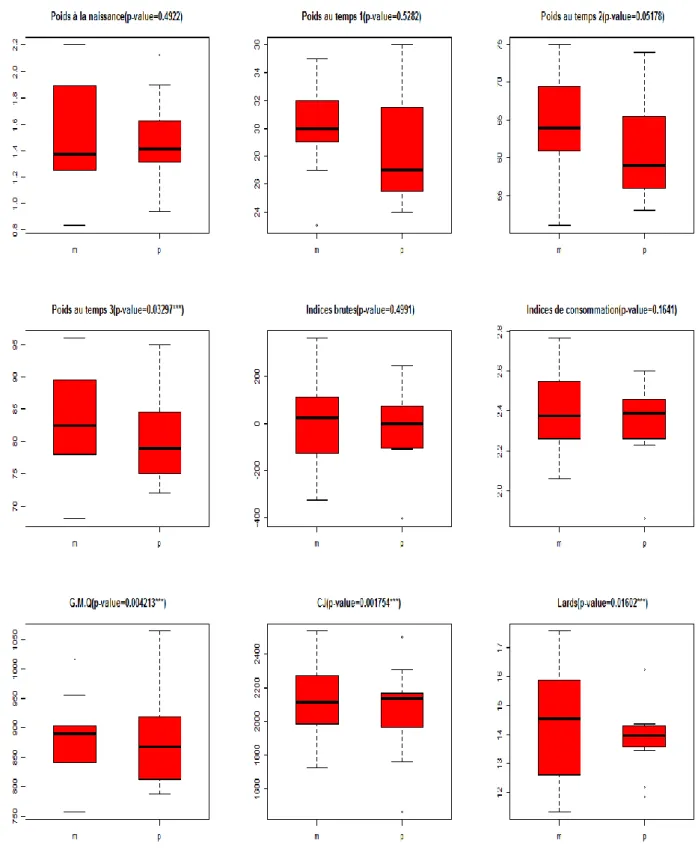
Figure 1 : Figure représentant les boxplot de 9 caractères zootechniques pour chaque lignée où m représente la lignée efficace et p la lignée non-efficace. Chaque boxplot a un titre au dessus identifiant le caractère étudié et entre parenthèse la p-value des tests de student.(CJ =Consommation journalière, GMQ = gain moyen quotidien)
17
A partir de ce test de student, j’ai observé que les moyennes entre lignée pour le poids au temps 3, le gain moyen quotidien, la consommation journalière, l’épaisseur de lard sont significativement différentes vu que pour ces caractères j’ai trouvé des p-values inférieures à 0.05. Pour les autres caractères les p-values sont supérieures à 0.05 signifiant que les moynnes pour ces caractères entre lignée ne sont pas significativement différentes. J’ai pu donc remarquer qu’au temps 3 la différence de poids entre lignée efficace et non-efficace séléctionné selon la CMJR est significative au temps 3 à 20 semaines alors qu’au temps 1 et 2 les porcs de lignée différentes doivent avoir des poids assez similaires.
Avec l’indice brut correspondant à la CMJR et l’indice de consommation je n’ai pas pu observer de différence significative entre les lignées alors qu’elles ont été sélectionnées selon la CMJR et que l’indice de consommation est un des paramètres de caractérisation de l’efficacité alimentaire, corrélé positivement avec la CMJR. Selon moi cela peut-être dû au nombre de porcs étudiés pour cette expérience qui n’est pas assez suffisant pour différencier significativement les paramètres de l’efficacité alimentaire. Cela nécessitera une augmentation du nombre de porcs pour une meilleure analyse des caractères zootechniques.
5.2 Transformation des tables d’abondance
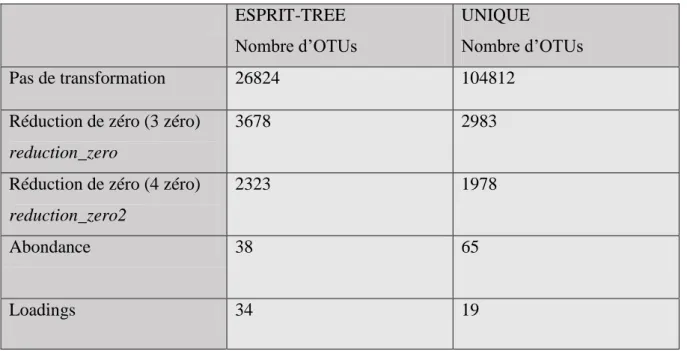
Pour les deux tables d’abondance esprittree et unique j’ai réalisé diverses transformations afin de réduire le jeu de données pour l’étudier dans son ensemble. En transformant les jeux de données la question de départ était toujours la même selon l’analyse. Pour la DAPC je voulais savoir si les OTUs générées avec la clusterisation et avec chaque transformation ou avec chaque temps me permettait de séparer de façon significative la lignée efficace de la lignée non-efficace. Pour la RCCA la question était de savoir suivant les temps étudiés et les transformations donc les OTUs si des covariations entre espèces bactériennes et caractères zootechniques existaient. Le nombre d’OTUs obtenu après chaque transformation est dans le Tableau2.
La première transformation que j’ai réalisée était de réduire le nombre de zéro des deux tables d’abondance avec deux seuils différents. A l’aide d’un script R j’ai compté le nombre de zéro pour chaque cluster c’est-à-dire l’absence d’une séquence d’un échantillon dans l’OTU. Ensuite j’ai créé une table d’abondance appelée reduction_zero en gardant seulement les OTUs où l’on trouvait au moins les séquences de trois échantillons différents et j’ai produit une autre table d’abondance appelée reduction_zero2 en gardant les OTUs où l’on observait au moins des séquences de quatre échantillons différents.
18
Une autre transformation a été de faire selon l’abondance des OTUs. Pour cela j’ai stocké l’abondance de chaque OTU dans une nouvelle colonne des tables d’abondance, j’ai ensuite ordonné les tables selon l’abondance des clusters du plus abondant au moins abondant. Puis j’ai tracé une figure représentant l’abondance en fonction des différents clusters, lorsque j’ai observé un palier c’est-à-dire une forte diminution de l’abondance j’ai réalisé une nouvelle table d’abondance appelée abondance en prenant tous les OTUs de ma table ordonnée en s’arrêtant
à l’OTU où le palier a été observé. Le nombre d’OTUs sélectionné est dans le Tableau2. Une autre transformation a été de faire selon les loadings. Ces derniers correspondent aux
clusters qui contribuent le plus à la séparation des lignées, pour cela j’ai utilisé une fonction de la DAPC appelée loadingplot qui donne les clusters contribuant le plus à la différenciation des lignées si et seulement si la séparation est effective. Le nombre d’OTUs sélectionné est dans le Tableau 2.
J’ai réalisé une autre transformation, la transformation logarithmique des tables d’abondance afin de réduire l’impact des OTUs les plus rares et d’augmenter l’impact des OTUs contribuant le plus sur la séparation des lignées. A noter que plus précisément c’est une transformation log +1 que j’ai réalisé pour éviter les problèmes liés aux zéro que les tables d’abondance contiennent. . Ensuite j’ai aussi voulu observer l’effet du temps sur le microbiote des lignées efficace et non-efficace : pour cela à partir des tables d’abondance initiales j’ai créé 3 nouvelles tables d’abondance en sélectionnant seulement les échantillons au temps 1, au temps 2 et au temps 3. Pour ces 3 nouvelles tables d’abondance je suis passé de 69 à 23 échantillons.
ESPRIT-TREE Nombre d’OTUs
UNIQUE
Nombre d’OTUs
Pas de transformation 26824 104812
Réduction de zéro (3 zéro) reduction_zero
3678 2983
Réduction de zéro (4 zéro) reduction_zero2
2323 1978
Abondance 38 65
Loadings 34 19
Tableau 2 : Tableau représentant le nombre d’OTUs suite aux transformations des tables d’abondance initiales
19
5.3. Analyse discriminantes en composantes principales
A partir de la DAPC j’ai pu générer divers résultats permettant de savoir si la composition en espèces bactériennes des microbiotes entre lignée efficace et non-efficace est significativement différente. D’autres résultats m’ont permis d’être plus précis et de comparer les lignées aux trois temps différents pour savoir si elles pouvaient être séparées et aussi d’étudier la sensibilité de la DAPC au nombre d’échantillons.
5.3.1 La séparation des lignées sur la base du microbiote
A partir des résultats de clusterisation et de transformation des table d‘abondance la question que je me suis posé était de savoir si les différents OTUs permettaient de séparer la lignée efficace de la lignée non-efficace.
5.3.1.1 Sans transformation avec les tables d’abondances initiales
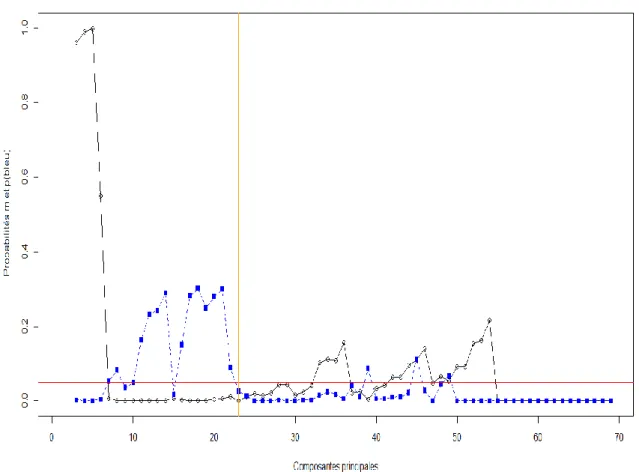
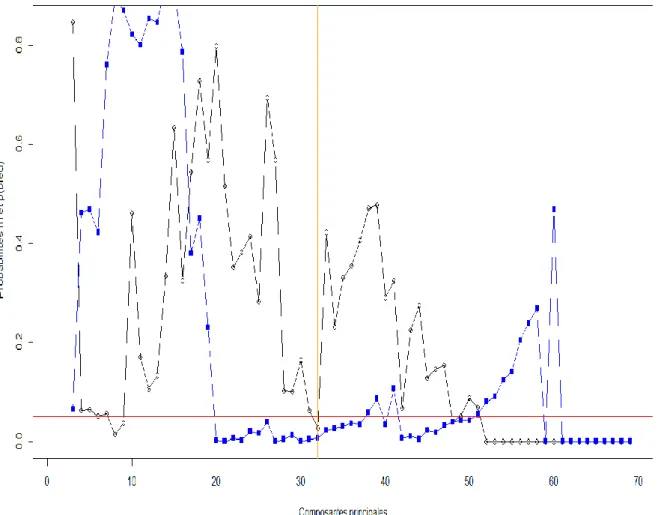
J’ai commencé à analyser par l’analyse discriminantes en composantes principales les tables d’abondance esprittree et unique brutes pour savoir si les clusters permettaient la distinction des deux lignées. Pour cela j’ai réalisé des boucles de vérification pour le nombre de composantes principales pour les deux tables. La Figure 2 représente les probabilités de m et de p d‘être du au hasard en fonction du nombre de composantes principales pour la table d’abondance esprittree brute. Il peut être observé qu’entre 23 et 32 composantes principales (en orange sur la Figure 2) il est possible de séparer les deux lignées efficace et non-efficace et donc d’accepter l’hypothèse alternative : ce qui est observé est dû à une réalité biologique et non au hasard.
20
Figure 2 Probabilités d’avoir une meilleure discrimination que le hasard de la lignée efficace ’m’ (noir) et de la lignée non-efficace ‘p’(en bleu) selon le nombre de composantes
principales de la DAPC pour la table esprittree brute.
Légende La séparation par DAPC des microbiotes entre porcs efficaces et non-efficaces est significative si l'ACP comprend entre 23 et 32 axes, qui capturent 98 et 99 % de la variabilité de la table d'abondance.
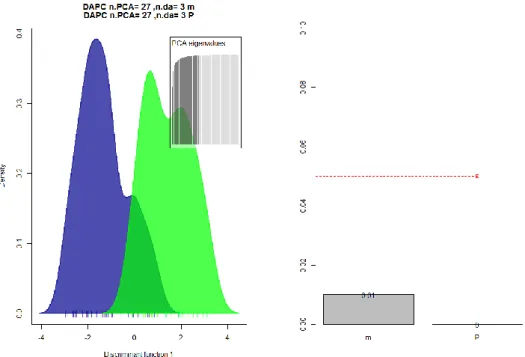
Suite à cela j’ai réalisé une autre DAPC en prenant comme paramètre pour le nombre de composantes principales 27. J’ai choisi de prendre 27 composantes principales car j’ai pu y observer les probabilités de la lignée efficace et de la lignée non-efficace très inférieures 0,05. La Figure 3 vient de l’analyse discrimante de la table esprittree brute résumée avec 27 composantes principales capturant 98% de la variabilité. En bleu c’est la courbe de densité qui correspond à la lignée efficace et en vert à la ligne m moins efficace. Sur cette courbe de densité fait avec 500 permutations il a été montré que les groupes biologiques sortis par Esprit-tree permettent une très bonne séparation des groupes suggérant que l’information de la table d’abondance esprittree basée sur les séquences 16S permet la distinction des deux groupes même avec un nombre d’individus de 23 assez faible .
21
Figure 3 A gauche la courbe de densité de la DAPC avec 27 composantes principales et capturant 98% de la variabilité pour la table esprittree. En bleu courbe de densité de la lignée efficace et en vert celle de la lignée non-efficace. A gauche un barplot représentant les probabilités d’avoir une meilleure discrimination que le hasard de la lignée efficace ‘m’ et non-efficace ‘p’ avec 27 composantes principales pour la table esprittree.
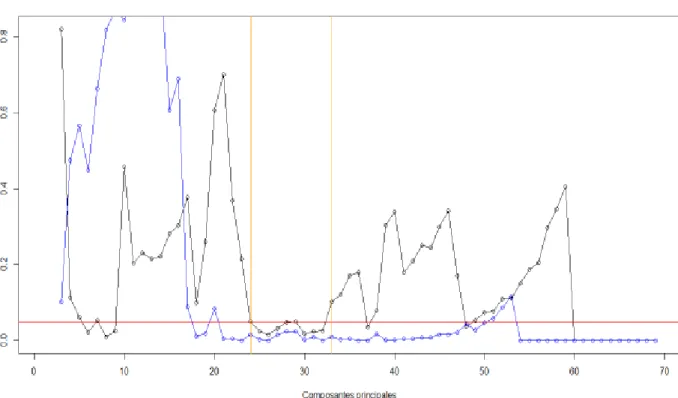
Pour la table d’abondance unique j’ai appliqué le même principe que pour la table d’abondance esprittree. Ce qui change est le paramètre seuil qui est pour la table unique égale au nombre de clusters générés par la clusterisation 104 812. La boucle de vérification permet de générer le graphe sur la Figure 4 où j’ai pu observer que si l’on prend 32 composantes principales en capturant 85% de la variabilité de l’information, une bonne séparation des groupes efficaces et non efficaces a été observée. Cela suggére que l’information de la table d’abondance unique basée sur les séquences 16S permet la distinction des deux groupes même avec un nombre d’individus de 23 assez faible . En conclusion tout cela signfie qu’il existe des différences significatives dans le microbiote des porcs entre une lignée efficace et une lignée non-efficace.
Par rapport à la table esprittree où l’on a un intervalle assez élevé du nombre de composantes principales pour la séparation des groupes avec la table unique la question de la robustesse du résultat se pose vu que c’est que pour un nombre de 32 composantes principales qu’une séparation significative entre groupe peut-être observé. A partir de composantes principales élevées j’ai observé que les probabilités pour la lignée efficace et la lignée non-efficace sont
22
nulles car en augmentant fortement les composantes principales on augmente fortement le nombre d’OTUs et donc que l’on trouvera toujours un moyen de séparer les deux lignées.
Figure 4 Probabilités d’avoir une meilleure discrimination que le hasard de la lignée
efficace’m’ (noir) et de la lignée non-efficace ‘p’(en bleu) selon le nombre de composantes principales de la DAPC pour la table unique brute.
Légende La séparation par DAPC des microbiotes entre porcs efficaces et non-efficaces est significative si l'ACP comprend 32 axes, qui capturent 85 % de la variabilité de la table d'abondance.
5.3.1.2 En réduisant le nombre de zéro des tables d’abondance
Pour les deux tables d’abondance esprittree et unique j’ai réduit le nombre d’OTUs en réduisant le nombre de zéro des tables en gardant uniquement les OTUs possédant des séquences d’au moins trois échantillons différents dans un premier temps et dans un second temps les OTUs
23
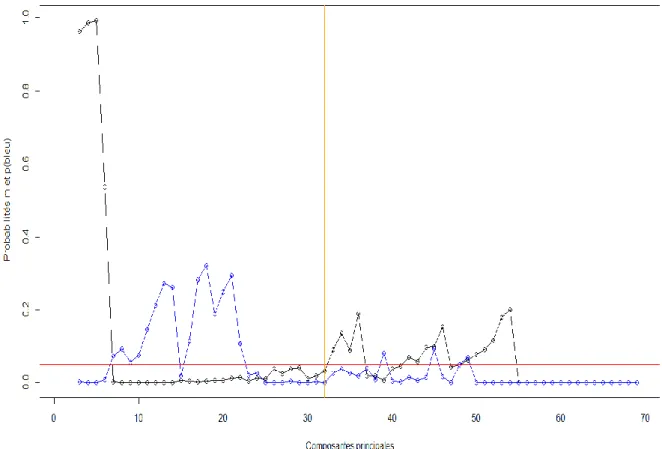
possédant des séquences d’au moins quatre échantillons différents afin d’éliminer les OTU très faiblement représentées. La Figure 5 représente les probabilités de la lignée efficace et non-efficace d’avoir une meilleure discrimination que le hasard pour une nouvelle analyse discriminante en composantes principales appliquée aux 3678 OTUs obtenues avec la réduction de trois zéro j’ai effectué une nouvelle analyse discriminante en composantes principales. J’ai remarqué qu’entre 21 et 29 composantes principales je pouvais séparer la lignée efficace et la lignée non-efficace signifiant que les 3678 sont suffisants à la distinction de les deux lignées et que l’information basée sur les séquences 16S dans ces OTUs permet une la séparation des lignées. Avec l’autre réduction de zéro en passant de 28264 à 2323 OTUs et entre 20 et 29 composantes principales j’ai pu constater que l’on pouvait distinguer la lignée efficace de la lignée non-efficace à l’aide de l’information basée sur les séquences 16S du microbiote.
J’ai ensuite effectué une DAPC cette fois-ci sur les tables d’abondance transformées à partir de la table unique avec les deux réductions de zéro en sélectionnant les OTUs possédant des séquences d’au moins trois échantillons différents dans un premier temps et dans un second temps les OTUs possédant des séquences d’au moins quatre échantillons différents. Sur ces deux nouvelles table d’abondance contenant 2983 OTUs avec la première réduction de zéro et à 1978 OTUs avec la seconde réduction de zéro (Figure 6), les probabilités de la lignée efficace et non-efficace d’avoir une meilleure discrimination que le hasard selon le nombre de composantes principales pour la table unique transformée sont inférieures à 5% entre 25 et 33 composantes principales. Les deux lignées peuvent donc être séparées, indiquant que l’information présente dans le 2983 OTUs générés avec l’information des séquences 16S des microbiotes est suffisante à cette séparation. J’ai constaté des résultats similaires en étant plus strict avec la seconde réduction de zéro avec seulement 1978 OTUs.
J’ai pu donc conclure que ces deux transformations m’ont permis de réduire le jeu de données en diminuant fortement le nombre d’OTUs pris en compte dans les analyses et qu’ils sont suffisants pour séparer nos deux lignées suggérant donc des différences de microbiote entre les porcs efficaces et non-efficaces. J’ai aussi remarqué que le souci de robustesse que j’avais pour la DAPC avec la table d’abondance unique (Figure 4) disparaît en diminuant le nombre d’OTUs. Cette diminution a permis d’avoir un intervalle de composantes principales où la lignée efficace et la lignée non-efficace était distincte à partir de l’information des séquences 16S des microbiotes.
24
Figure 5 Probabilités de la lignée efficace’m’ (noir) et de la lignée non-efficace ‘p’(en bleu) d’avoir une meilleure discrimination que le hasard selon le nombre de composantes principales de la DAPC pour la table esprittree transformée en sélectionnant seulment les OTUs possédant au moins la présence de trois échantillons distincts( 3678 OTUs)
Légende. La séparation par DAPC des microbiotes entre porcs efficaces et non-efficaces est significative si l'ACP comprend entre 21 et 29 axes, qui capturent 98,6 et 99 % de la variabilité de la table d'abondance.
25
Figure 6 Probabilités de la lignée efficace’m’ (noir) et de la lignée non-efficace ‘p’(en bleu) d’avoir une meilleure discrimination que le hasard selon le nombre de composantes principales de la DAPC pour la table unique transformée en sélectionnant seulment les OTUs possédant au moins la présence de trois échantillons distincts( 2978 OTUs)
Légende La séparation par DAPC des microbiotes entre porcs efficaces et non-efficaces est significative si l'ACP comprend entre 24 et 34 axes, qui capturent 97 et 99 % de la variabilité de la table d'abondance.
5.3.1.3 Transformation selon l’abondance
A partir de la table d’abondance esprittree brute, j’ai récupéré uniquement les 38 OTUs les plus abondants dans une nouvelle table d’abondance appelée abondance. Ensuite j’ai fait une DAPC pour cette table réduite toujours sur le même principe de départ en réalisant une boucle de vérification pour avoir à partir de combien de composantes principales la séparation des lignées si elle existe peut être observée. Comme l’indique la Figure 7 représentant les probabilités de la lignée efficace et de la lignée non-efficace d’avoir une meilleure discrimination que le hasard j’ai pu conclure que les lignées pouvaient être séparées significativement entre 23 et 32
26
composantes principales. Cela indique que l’information présente dans ces 38 OTUs basée sur des séquences 16S permet de dire qu’il existe une différence de microbiotes entre lignée efficace et lignée non-efficace.
Sur la table d’abondance unique, j’ai sélectionné de la même façon que sur la table esprittree les 68 clusters les plus abondants. Sur cette dernière j’ai réalisé une DAPC où pour chaque composante principale j’ai pu observer si la lignée efficace et la lignée non-efficace pouvaient être séparées (Figure 8). De cette figure j’ai donc conclu qu’entre 27 et 30 composantes principales des probabilités pour la lignée efficace et non-efficace inférieures à 5% indiquant que les 68 OTUs les plus abondantes permettent la distinction des lignées selon l’efficacité alimentaire et impliquant des différences entre la composition du microbiote entre les lignées.
Figure 7 Probabilités de la lignée efficace’m’ (noir) et de la lignée non-efficace ‘p’(en bleu) d’avoir une meilleure discrimination que le hasard selon le nombre de composantes principales de la DAPC pour la table esprittree transformée en sélectionnant seulment les OTUs les plus abondants(38OTUs)
27
significative si l'ACP comprend entre 24 et 32 axes, qui capturent 98 et 99 % de la variabilité de la table d'abondance.
Figure 6 Graphe des probabilités de la lignée efficace’m’ (noir) et de la lignée non-efficace ‘p’(en bleu) d’avoir une meilleure discrimination que le hasard selon le nombre de
composantes principales de la DAPC pour la table unique transformée en sélectionnant seulment les OTUs les plus abondants( 68 OTUs)
Légende La séparation par DAPC des microbiotes entre porcs efficaces et non-efficaces est significative si l'ACP comprend entre 27 et 30 axes, qui capturent 89 et 95 % de la variabilité de la table d'abondance.
5.3.1.4 Transformation logarithmique
Pour les deux tables d’abondance esprittree et unique brute, j’ai réalisé une transformation logarithmique. Suite à ces transformations j’ai pu observer que les lignées pouvaient être
28
séparés selon les mêmes composantes principales qu’en prenant les tables brutes. Cela m’a permis de suggérer que l’information de la table d’abondance esprittree et de la table unique basée sur les séquences 16S permet la distinction des deux groupes même avec un nombre d’individus de 23.
5.3.1.5 Loadings
Ils correspondent aux clusters contribuant le plus à la séparation des deux lignées, pour la table d’abondance esprittree ils sont au nombre de 34 et 19 pour la table unique. J’ai observé que sur chaque transformation que j’ai réalisée ces clusters étaient toujours retrouvés, pouvant expliquer le fait de pouvoir séparer la lignée efficace de la lignée non-efficace à partir des OTUs générés à l’aide des séquences 16S du microbiote. Cela m’a permis de conclure sur le fait qu’il existe des différences significatives entre le microbiote d’un porc efficace et d’un porc non-efficace.
Mon hypothèse est que les différences entre microbiote ne sont pas des différences liées à la présence d’espèces bactériennes particulières mais à des différences plus subtiles comme l’abondance des espèces bactériennes à l’intérieur du microbiote.
Une autre remarque a été de s’apercevoir que dans toutes les transformations que j’ai réalisées, dans les OTUs permettant d’expliquer la différence entre lignée je retrouvais toujours les OTUs correspondant aux loadings. Dans ces loadings j’ai observé que la majorité d’entre eux correspondaient à des OTUs avec une forte abondance et à des OTUs composés de séquence de tous les échantillons.
5.3.2 Comparaison des temps
J’ai produit un script sous R qui a permis d’aller récupérer pour chaque temps les OTUs présents au moins une fois à partir de la table d’abondance esprittree. Je me suis donc retrouvé avec trois tables pour chaque temps composé de 23 échantillons à partir desquelles j’ai réalisé des DAPC sur le même principe que les précédentes avec des boucles de vérification. La Figure 9 montre le résultat de DAPC pour le temps 1 où j’ai observé que l’on ne pouvait pas séparer les deux lignées à partir de l’information basée des séquences 16S suggérant qu’au temps 1 aucune différence significative au niveau du microbiote n’existe. Sur la Figure 10 montrant le résultat de la DAPC pour le temps 2 j’ai remarqué qu’entre 6 et 9 composantes principales la lignée efficace peut être distinguée de la lignée non-efficace à partir de l’information des séquences 16S du microbiote. Pour le temps 3, les résultats de la DAPC ont montré que pour 6
29
composantes principales la lignée efficace et la lignée non-efficace peuvent être seulement distinguées à l’aide de l’information contenue dans les séquences 16S du microbiote. Au temps 1 l’hypothèse est de dire que le microbiote est encore pas assez développé et encore jeune pour permettre l’observation de différences avec ce nombre réduit d’échantillons ou de dire que le changement d’environnement d’élevage une semaine avant le temps 1 a généré des perturbations du microbiote qui ne sont pas encore stabilisées
Figure 9 Graphe des probabilités de la lignée efficace’m’ (noir) et de la lignée non-efficace ‘p’(en bleu) d’avoir une meilleure discrimination que le hasard selon le nombre de composantes principales de la DAPC pour la table esprittree au temps 1
Légende La séparation par DAPC des microbiotes entre porcs efficaces et non-efficaces n’est pas significative
30
Figure 10 Graphe des probabilités de la lignée efficace’m’ (noir) et de la lignée non-efficace ‘p’(en bleu) d’avoir une meilleure discrimination que le hasard selon le nombre de
composantes principales de la DAPC pour la table esprittree au temps 2.
Légende La séparation par DAPC des microbiotes entre porcs efficaces et non-efficaces est significative si l'ACP comprend entre 6 et 9 axes, qui capturent 99 % de la variabilité de la table d'abondance.
A noter que à chaque fois que j’ai conclu sur la séparation des groupes je capturais au minimun avec les DAPC 85% de la variabilité. En conclusion des DAPC réalisées j’ai pu constater que l’information récoltée basée simplement sur les séquences 16S permet de suggérer qu’il existe des différences significatives entre le microbiote d’une lignée efficace et le microbiote d’une lignée non-efficace.
31
5.4 RCCA ; Analyse des corrélations entre table d’abondance
et performances zootechniques
Après le fait de savoir qu’il existait des différences significatives entre le microbiote des deux lignées, l’étape suivante que j’ai réalisée consistait à savoir s’il existait des covariations entre les espèces bactériennes du microbiote et les performances des caractères zootechniques mesurées. Pour cela j’ai tout d’abord comparé aux différents temps si des covariations étaient observées à partir des tables d’abondances transformées avec une réduction du nombre de zéro pour les OTUs.
5.4.1 Comparaison des trois temps
Avant de commencer tout en analyse de RCCA il faut que le nombre et le nom des échantillons pour les deux tables soit exactement identique afin d’obtenir des résultats que je pourrais commenter. J’ai donc fait ces manipulations pour les tables d’abondance en gardant seulement les échantillons au temps que je voulais étudier et les trier de la même façon que la table des performances zootechniques pour faire correspondre les lignes. Après chaque RCCA j’ai appliqué un script R me permettant de sortir des matrices de corrélation les valeurs significatives comme expliqué dans le Matériels et Méthodes.
Au temps 1 j’ai réalisé la RCCA avec la table transformée en gardant seulement les OTUs qui ont une forte contribution (loadings) et les performances zootechniques. En sortie de cette analyse j’ai obtenu une heatmap que l’on peut observer sur la Figure 11. A partir de ces corrélations les dendrogrammes sont créés pour les OTUs et les caractères en fonction de la ressemblance des profils. L’intervalle des corrélations observées est assez faible, allant de -0.66 à -0.66, ensuite avec mon script de vérification j’ai trouvé seulement 8 corrélations qui sont significatives que l’on retrouve dans le Tableau 3.
Tableau 3 : Tableau représentant les valeurs de corrélation significatives de la RCCA avec la table d’abondance esprittree contenant les OTUs contribuant le plus à la séparation des lignées au temps 1 et les performances zootechniques de 9 caractères
32
Figure 11 Heatmap représentant le résultat de corrélation d’une RCCA entre la table d’abondance esprittree au temps 1 transformée en gardant seulement les OTUs contribuant à la séparation des lignées (loadings) possédant au moins la présence de trois échantillons distincts avec les performances zootechniques de 23 porcs
Les résultats de la RCCA avec les valeurs significatives de chaque matrice de corrélation pour les temps 2 et 3 à partir des loadings que j’ai réalisé pour la table esprittree sont en annexe 3. Ce que j’ai pu tiré de ces différents résultats c’est qu’il existe des covariations entre certaines espèces bactériennes et certains caractères zootechniques. Ensuite j’ai remarqué que la plupart des valeurs de corrélations qui sont significatives correspondent à des Prevotella et les caractères zootechniques sont ceux caractéristiques de l’efficacité alimentaire. Au temps 3 les
33
valeurs significatives identifiées sont au nombre de 13, plus nombreuses que celles identifiées au temps 1 et temps 2. Ceci peut-être causé par le fait qu’au temps 3 la différenciation du microbiote entre la lignée efficace et non-efficace est plus prononcée au temps 1 et au temps 2.
J’ai réalisé d’autres RCCA en utlisant toujours la même table des performances zootechniques avec trois tables d’abondance différentes correspondant aux trois temps issue de la table d’abondance esprittree en réduisant le nombre de zéro pour chaque OTU. Ensuite les valeurs significatives ont été identifiées et la plupart correspondent aux résultats obtenus avec les loadings, OTUs contribuant le plus à la séparation des lignées.
Sur ces résultats j’ai pu conclure sur le fait qu’il existe des covariations entre les espèces bactériennes présentes dans le microbiote dans les lignées efficaces et non-efficace avec les caractères zootechniques. Les matrices de corrélation sont obtenues à l’aide de validation croisée, j’ai décidé de vérifier les corrélations avec un test de 5% avec 1000 randomisations des performances zootechniques. Une autre technique pour voir si ma méthode de vérification est satisfaisante aurait été d’introduire un caractère zootechnique que j’aurais échantillonné avec une forte corrélation avec un OTU que j’aurais choisi et de voir si mon script l’identifie comme valeur significative.
J’ai observé qu’entre le temps 1, le temps 2 et le temps 3 les OTUs corrélés significativement correspondent souvent à des Prevotella que ce soit avec les tables d’abondance contenant seulement les loadings ou les tables d’abondance où j’ai réduit le nombre de zéro. Elle correspond à une des espèces bactériennes les plus représentées dans la flore microbienne d’après la littérature scientifique. J’ai retrouvé d’autres OTUs qui sont fortement corrélés aux caractères zootechniques de l’efficacité alimentaire de la famille des Firmicutes, une famille très abondante dans le microbiote. Un des axes de recherches a développé est une étude plus fine de ces corrélations essayant de caractériser plus distinctement les différences de composition de microbiote existantes entre une lignée efficace et une lignée non-efficace
34
6. CONCLUSION
L’objectif de mon stage était d’évaluer le rôle du microbiote intestinal dans l’efficacité alimentaire du porc à partir de données de séquençage de l’ARNr 16S et de mesures sur certains caractères zootechniques. Pour cette analyse j’ai fait deux clusterisations différentes avec Esprit-tree et une autre en récupérant les uniques, j’ai observé un nombre d’OTUs cinq fois plus élevé avec Unique qu’avec Esprit-tree issue d’une différence de méthodologie dans le seuil de clusterisation. A partir de ces tables et à l’aide de la DAPC j’ai pu montrer que l’information basée sur les séquences 16S étaient suffisante dans un premier temps pour séparer mes deux lignées d’un point de vue statistique suggérant des différences de compositions de flore intestinale entre la lignée efficace et la lignée non-efficace. Sur les différentes analyses discriminantes en composantes principales réalisées à partir des tables d’abondance esprittree et unique brutes j’ai pu découvrir pour chaque table d’abondance les clusters contribuant à la distinction des lignées. Cela permettant de suggérer que l’information basée sur les séquences 16S avec seulement 23 échantillons permet d’identifier des différences de flore intestinale entre une lignée dite efficace et une lignée non-efficace. Ces différents OTUs sont retrouvés dans les tables d’abondances transformées car ce sont les plus abondants, ce sont ceux qui possèdent presque au moins une séquence de tous les échantillons et ce sont ceux qui expliquent la variabilité de la flore intestinale entre. J’ai exécuté un test de student pour comparer la moyenne des abondances par échantillon sur ces OTUs et aucune différence significative n’a été identifiée. Ensuite à l’aide d’analyses en corrélations canoniques régularisées , j’ai observé qu’il existait des covariations entre les espèces bactériennes présentes dans les microbiotes des différents porcs et les caractères zootechniques de l’efficacité alimentaire permettant de faire un lien entre les différences de composition supposées et les caractères phénotypiques de l’efficacité alimentaire.
Pour connaître à quel niveau de composition de microbiote les différences observées existent, il serait bien dans un premier temps d’accroitre le nombre d’individus étudiés afin d’augmenter la puissance des traitements statistiques réalisés. Ensuite une étude transcriptomique sur les OTUs contribuant le plus à la distinction de la lignée efficace et de la lignée non-efficace permettrait de faire un lien plausible entre le niveau d’expression des gènes et les caractères zootechniques de l’efficacité alimentaire. Une autre étude intéressante selon moi serait de travailler de façon plus approfondie sur les fonctions des bactéries et leurs liens connus avec les performances zootechniques.