HAL Id: jpa-00210326
https://hal.archives-ouvertes.fr/jpa-00210326
Submitted on 1 Jan 1986
HAL is a multi-disciplinary open access
archive for the deposit and dissemination of
sci-entific research documents, whether they are
pub-lished or not. The documents may come from
teaching and research institutions in France or
abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est
destinée au dépôt et à la diffusion de documents
scientifiques de niveau recherche, publiés ou non,
émanant des établissements d’enseignement et de
recherche français ou étrangers, des laboratoires
publics ou privés.
Singly and doubly charged clusters of (S2) metals. Ab
initio and model calculations on Mg+n and Mgn++n(n
≤ 5)
Geoffroy Durand, J.-P. Daudey, Jean-Paul Malrieu
To cite this version:
Geoffroy Durand, J.-P. Daudey, Jean-Paul Malrieu. Singly and doubly charged clusters of (S2) metals.
Ab initio and model calculations on Mg+n and Mgn++n(n
≤ 5). Journal de Physique, 1986, 47 (8),
pp.1335-1346. �10.1051/jphys:019860047080133500�. �jpa-00210326�
Singly
and
doubly
charged
clusters
of
(S2)
metals.
Ab initio
and model
calculations
onMg+n
and
Mgn++n
(n ~
5)
G.
Durand,
J.-P.Daudey
and J.-P. MalrieuLaboratoire de Physique
Quantique,
Université Paul-Sabatier, Unité Associée au C.N.R.S. n° 505,118, route de Narbonne, 31062 Toulouse Cedex, France
(Reçu le 7 janvier 1986, accepté le 7 avril 1986)
Résumé. 2014 Des calculs ab initio
précis
incluant la corrélation électronique ont été effectués pourMg+2
etMg++2.
Le deuxième
système
présente un minimum local. A l’aide d’un procédé de diabatisation, on a pu définir un hamil-tonien modèle simple porté par lesproduits
des configurationsatomiques
s2,
s1 et s0. L’hamiltonien modèle inclut les effets depolarisation
instantanée (ainsi que les effets à troiscorps)
et larépulsion
coulombienne descharges
positives pour les agrégats doublement chargés. Il peut être considéré comme une
représentation
de un ou deuxtrous actifs dans une bande s
polarisable.
Le modèleprédit
que la structure la plus stable deMg+3
etMg++3
est linéaire, que les structures carrées et triangulaires centrées sont pratiquementéquivalentes
pourMg+4
etMg++4.
Pour
Mg+5
etMg++5
existent également deux minima quasimentdégénérés :
le tétraèdre centré et le carré centré. L’existence de telles structures a été confirmée par des calculs Hartree-Fock, par minimisation directe de l’énergieet calcul de la matrice des constantes de force pour la conformation optimale, mais on démontre que dans l’approche
du modèle à
particules indépendantes
les longueurs de liaisons sont trop longues et les structures centrées sonttrop stables.
L’énergie
du minimum deMg++5
est située à 1,71 eV au-dessus de la somme desénergies
dessystèmes
Mg+3
etMg+2.
Les potentiels d’ionisation verticaux ont été calculés et on démontre queMg+4, Mg+5
etMg++5
peuvent évaporer un atome neutre.
Abstract 2014 Accurate ab initio CI calculations on
Mg+2
andMg++2
have beenperformed.
The second appears tohave a local minimum. Through a diabatization
procedure,
it waspossible
to define asimple
model Hamiltonianspanned by products of atomic
s2,
s1 and s0configurations.
The model Hamiltonian includes instantaneouspola-rization energies (and
three-body
effects) and Coulombic repulsion of the holes indoubly charged
clusters, and may be considered as treating one or two active holes in the polarizable s band. The modelpredicts
linearMg+3
and
Mg++3
structures and nearly degenerate square and centredequilateral
triangular structures forMg+4
andMg++4; Mg+5
andMg++5
both present nearly degenerate minima of centred tetrahedron and centred squarestructures. The existence of such structures has been confirmed by UHF ab initio (gradient + force) calculations;
it is shown however that
single-determinantal
approaches overestimate the bond lengths and unduly favour the centred structures; theMg++5
minimum is 1.71 eV above the(Mg++3
+Mg+2)
dissociation limit and 1.41 eV under the (3 Mg + 2Mg+)
dissociation limit. The vertical Ionisation Potentials(IP)
have been calculated and it is shown thatMg+4,
Mg+5
andMg++5
may evaporate a neutral atom.Classification
Physics Abstracts 36.40 -
31.20R - 31.20E
1. Introduction.
The atoms of column II of the
periodic
table haveground
stateS2(lS)
configurations.
The atoms may be considered tokeep
essentially
thisground
state structure in smallclusters,
despite
some weakhybri-dization with the
sp(3, 1 P)
configuration.
The elec-tronicpopulation
of a neutral cluster of such elementsmay thus be seen as an
(s)
full band. Thepositive
ionmay
consequently
be treated as asingle
holeproblem,
delocalized on the different sites of the cluster
through
asingle-hole
operator. Adoubly
charged
cluster may be considered as a two-holesystem
in the sband,
and treated like a two-electron
problem,
including
correlation. The present paper proposes such
sim-plified
models forsingly
anddoubly
charged
clusters of(s’)
atoms. ForMg
clusters thediagonal
effectiveenergies
and interactions are extracted from accurateMO-CI calculations on
Mng2+
(n = 1, 2)
and from the’experimental Mg2
potential
curve.The
problem
received an increased interest fromthe recent observation of an
Hg5 +
doubly charged
cluster
[1],
from electronimpact
on a beam of mercuryclusters. The appearance at 22 eV of such a
peak
in the massspectrometer
analysis
wasquite surprising
since the Coulombicrepulsion
between the two holes shouldcompel
such a small cluster toexplode
intotwo
singly charged
fragments. Brechignac et
al.[1]
suggested
that thepositive charge
in this clustermight
be concentrated on the central atom of a compactcluster,
theHg+ +
atom(the
ionizationpotential
of which is 29eV) inducing
stabilizing polarization
forceson the
surrounding
neutral atoms.The
present
work does not concernHg
clusterssince the correct treatment of such a
heavy
atomrequires
the inclusion of relativisticeffects;
theirtreatment is in progress in our
laboratory through
relativistic
pseudopotentials
[2, 3],
which will beapplied
further to thisprecise problem through
calculations ofHg2,
Hg2 ,
H g2 I
potential
curves.The
present
paper is limited to the isoelectronicproblem
ofMg clusters,
in order to see wether anMg25 +
cluster may exist at least as a stable localminimum. The
question
is treatedthrough
both model and ab initio MOcalculations,
which comparefairly
well.2. A
simplified
model Hamiltonian forMgn
andMgn +
clusters.2.1 NEUTRAL INTERACTIONS. - Let
us assume that in the neutral
ground
state, each atomessentially
keeps
an(s2)
configuration.
This means that the clusteris
essentially
heldby dispersion
forces. This alsomeans that the cluster should have a compact
form,
maximising
the number of nearestneighbours through
a
packing
of tetrahedra. The dissociation energy ofMgn
into nMg
atoms should bedirectly
related tothe
number p
of nearestneighbours. Assuming
anadditivity
ofrepulsion energies
anddispersion
forces(i.e.
cancellation betweenthree-body repulsions
andthree-body dispersive energies)
the dissociation energyper atom
De(n)
of the cluster should bewhere
De
is the dissociation energy of the dimer. Thisquantity
is rather weak forMg2
(De
= 0.05eV)
andequation (1) certainly
underestimates the dis-sociation energy of the cluster since the cohesiveenergy of the metal is 1.1
eV,
muchlarger
than(12/2)
De/2,
which would be theasymptotic
limit ofequation (1).
This may be due to the involvement of the pband,
to attractive interaction with secondneighbours and/or
an attractive balance betweenmany-body’§orces.
It is clear however that the disso-ciation energyof Mgn
is much smaller than that of thecorresponding positive
clusterMg,,,
which involves delocalization andpolarization
energies.
Theprecise
research of thegeometry
and energy of the weakMgn
neutral cluster is not in the scope of our
model,
and we
simply
assume that its wave function isdomi-nated
by products
of(s’)
atomicconfigurations
where A is an
antisymmetrizer,
l/Ji is eitheran si si
product
of 3s atomicorbitals,
or anangularly
cor-related function
The interaction energy between neutral atoms
4 i
at distance rij issupposed
to beequal
to the inter-action energy of the dimerR(r;) = Rij
and the totalcohesive energy would be
2.2 MODEL HAMILTONIAN FOR THE SINGLY POSITIVE CLUSTER. - For the
positive
clusterMg’
one mayconsider, according
to the Valence Bond(VB)
des-cription,
a series of n localizedfunctions,
where thehole concerns the various atoms. If the hole is on the
ith atom
where
ai is
an annihilationoperator
of a fl spin
electronon the site i. Then the wave function of the cluster
is a linear combination of the
4Jt’s
The
matrix elements of the n x n model HamiltonianH are
easily
obtained as follows. For thediagonal
elements,
one may writeRjk
represents
the instantaneous interaction betweenneutral centres,
supposed
to beequal
to that in thecorresponding
neutralsystems.
Rit
is the interactionbetween the
positive
atom i and the neutral atomj.
Its determination will be
given
below,
andIP,
is the lowest ionizationpotential
of the atom.The
off-diagonal
elementrepresents
thehopping integral
between atoms iwhere F is the Fock operator of the
ground
state.In fact
(see below)
we have used effectiveFit
integrals
which introduce some
reorganization
effects.For
Mg’
theproblem
reduces to a 2 x 2 matrixwhich generates two
roots 2 Eu+,
2 Eg+
and thus the functions
R1+2
andF 2
areimmediately
extracted from the
knowledge
of the exactpotential
curves of the diatomic cation
Two
things
should be noticed :i)
the method isidentical,
at this step, to a Diatomin Molecules
(DIM) procedure [4];
ii)
it includespolarization
forces sinceR 2
involves both apurely repulsive
part and anal/2
r’,
polariza-tion energy of the
adjacent
atomby
thepositive charge.
Since thesingly
ionic clusteronly
bears onepositive
charge
thepolarization
energykeeps
an additive form.2.3 MODEL HAMILTONIAN FOR THE DOUBLY CHARGED CLUSTERS. - For
a two-hole
cluster,
one mayima-gine
twotypes
ofelementary
VB situations.- The two holes
may be on different atoms,
generating
afunction l/Ji; +.
For Sz
= 0 one must introduce two determinants :Their number is thus
n(n -
1).
Their effective energyinvolves,
besides the energy2IP1
needed to ionizetwo atoms, the interactions between the
instanta-neously
neutral atoms, the interactions between thepositive
and the neutral atoms, and the Coulombicrepulsion
between theholes,
Rit
+ whichweakly
deviates fromriJ 1.
The last term6 pol
is a correctionto the
polarization
energy which is notadditive,
since the field created
by
the two holes on the neutralpolarizable
atom k must be combinedvectorially.
Let us call
Eki = rkilrli
the field createdby
the hole i on the neutral atom k.Then the
polarization
energy isSince
Rkt
involved a term isgiven by
It
gives
thethree-body
correction to thepolarization
energy.
- The two holes
may be on the same atom, which is now
s°,
while the others are neutral(s’).
Thecor-responding
VB function isThe
diagonal
energy associated with this function isIP2
is the double-ionizationpotential
R’ ’
is an effective interaction betweenMg
andMg+
+,
and its
practical
extraction will begiven
below. One may conceive two main types ofoff-diagonal
interactions :The
hopping integral corresponding
to thejump
ofa hole from the
site j
to the site k may be takenequal
to its value for the
singly positive
cluster.Actually
where
Ji
is the Coulombic operator relative to the(3s)
atomic orbital of the atom i; the three-centreintegral
should be smallcompared
to the monoelectronic(essentially kinetic) integral.
One may
neglect
the bielectronicintegrals
(cor-responding
to 2 e-jump)
such as(even
when i = q and p =k) ;
thisapproximation
is usual inchemistry,
in the various Zero DifferentialOverlap (ZDO)
systematics [5, 6],
and in Solid StatePhysics
in the Hubbard Hamiltonian[7]
which is the crudest version of thisapproximation;
the ZDOapproximation
consists inneglecting
the bielectronic interactioninvolving overlap
distributions.from a
doubly charged
atom to an(adjacent)
neutralatom, this
jump
creating
apair
ofpositively
charged
atoms. In a
simple
model one may writeIn that
precise
case theintegral i
I Ji Ii>
cannot beneglected,
since itonly
involves two centres, andF å + =1=
F i; .
.The two-hole model
requires
theknowledge
of threesupplementary
functions,
Ri++,
R;) +
andF;) + .
This information may be obtained from thepotential
curves of the
doubly charged
diatomMg++.
In thatcase one has two electrons in two
orbitals,
and theproblem
isisomorphic
to theH2
problem. There
arefour
determinants
1 ab I, 1 ba I,
1 a-a
I
and
I bb
where a
and b are the two 3s AO’s of the two atoms A and B.The matrix takes the form
The bicentric
exchange integrals Kab
may beneglected.
The
triplet
state(I ab I -
ba
1)/.,/2,
of3 Iu+
sym-metry has an energy
which determines
Rat +
(or
the deviation of therepul-sion from its coulombic
part).
Thesinglet
’Z,,’
state(I
a-a I -
bb
1)/.,,/2-
has an energywhich determines the function
R 2 + .
The
(If 9 +)
ground
state energy isgiven
by
a seconddegree equation
and the
knowledge
of the1 E ’
ground
state energyE(’,E 9 ’)
will determine the value ofF ab
as a functionof the interatomic distance
2.4 DIATOMIC POTENTIAL CURVES OF
Mg2,
Mgi
ANDMg2 + AND
r DEPENDENCE OF THE 6 PARAMETERS OF THE MODEL HAMILTONIAN. - The model involves6
para-meters R,
R +, F +,
R + +,
R2+,
F 2 +.
The first is identical to theground
statepotential
energy of the neutral dimer and rather thancalculating
it,
afunction fitted to the
experimental spectroscopic
parameters [8-9]
has been usedwhere A is a Morse function
involving
threepara-meters a1 a2 a3
(see
TableI).
The second(R +)
and third(F + )
functions were determined from accurateMO-CI calculations on
Mg2+ ,
using
a[3s
2p
ld]
basisset. The
2Eg
and2Eu
potential
curves were inagreement with a
previous
calculationby
Stevens andKrauss
[10].
Theresulting
functions have been fittedas follows :
where A’
is a Morsefunction,
al thepolarizability
of neutral atoms
and f1 (r)
a short rangescreening
of the electric field. The three last
parameters
comefrom calculations on
Mg",
which were achievedin the same basis set
(and
complete
CI).
The mainproblem
concerned the definitionof R2+,
the energy of the(I aa
I -
bb
0/J2
state,dissociating
intoMg2+
+Mg(3s2)
(IP2
= 22.67eV),
due to a curvecrossing
with a statedissociating
intoMg+(3p)
+Mg+(3s),
at 19.72 eV aboveMg(3s2)
+Mg(3s2),
i.e. 2.9 eV below thepreceding
dissociation limit(IP2).
An ab initio diabatizationprocedure [11]
has beenapplied
to thatproblem
in order to definenearly
diabatic12:+
and1 Eg+
excited stateskeeping
theMg(3s2)
+Mg + + (3s0)
character from infinitedis-tances to the relevant short range
region [12].
Theresulting
parameters
have been fitted as followswhere
R + +
involves a Coulombicr-1
repulsion,
aMorse
potential
and a(screened) polarizability
part,a2
being
thepolarizability
ofMg +
atoms.R 2 +
has been fittedby
asimple
Morse function.Table Ia. - R
parameters
of
the model. M are Morsefunctions :
a2[(’ -
exp(- a3(r -
ai)))2 - 1]
and fare
screening
functions :
1 -a4
exp[-
a5(r -
a6 )2].
The
explicite representation
of thepolarization
energy in
equation
(26)
is notcompulsory
forsingly
charged
clusters. The determination of ascreening
function for the electric field was
required
for a correctestimate of the
three-body
term inequation (15).
Thescreening
function has been determinedby comparing
the
energies
ofMgA
+MgB
(3s
frozen)
in minimal and extended basis sets forMgA.
Ananalogous
procedure
has been used forMgA
+[MgB(+)
(3s
frozen)].
Thepotential
curves used for theparame-trization have been
plotted
infigure 1 a), c).
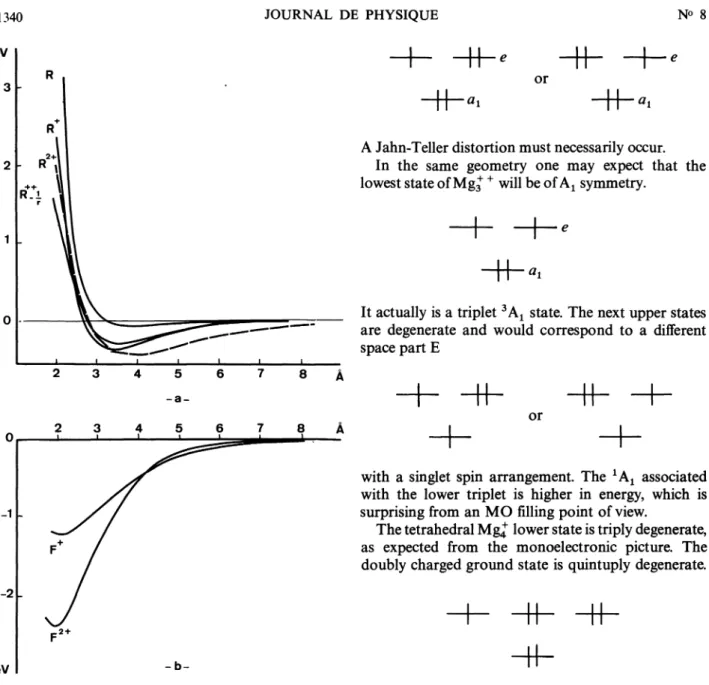
Theground
statepotential
curveof Mg+ +
isrepulsive
withrab 1 behaviour,
but it presents a local minimum for r = 2.93A
which is due tothe
mixing
between the«
pseudo-neutral »
(I
ab +
I b-ajl
configuration
with the
doubly
ionic( aa
I +
bb
I)V2-
configuration
(as
inH2), through
thehopping
integral
F 2 +.
TheFig. 1. -
a)
Experimental
Mg2potential
curve. b) Ab initioMg2 (2 Lu+
and2 Lg+)
potential
curves. c) Ab initio adiabatic(-) and diabatic (---)
Mg+ ’ potential
curves.parameters
have beenplotted
as functions of theinteratomic distance in
figure
2.They
have theexpect-ed
r-dependence.
One should notice however that for shortr values,
theF 2 +
integral
has alarger amplitude
than
F + ;
this increase may be due either to the effect of the hole(cf.
Eq.
(20))
or to the indirect role of theMg(3p)
+Mg(3s)
state whichbelongs
to the samerange of energy. Standard deviation from
experimental
data for the R
parameter
ofMg2
and from ab initiocalculations for the
Rk+
andFk+ parameters
of Mg2
andMg+ +
is less than10- 3
hartrees.3. Results.
3.1 VERTICAL IONIZATION POTENTIALS. -
Assuming
compact structures for the neutral clusters
(i.e.
equi-lateral
triangle
forMg3,
tetrahedron forMg4
andregular
trigonal
bipyramid
forM95
with r =re(Mg2)
= 7.35 a.u. interatomicdistances),
the vertical ionizationpotentials
have been calculated andthey
appear in table II for bothsingle
and double ionizations. Notice therapid
decrease of the ionizationpotentials;
for aregular
linear chain orcycle
of natoms the ionization
potential
would decrease as n-1(as
shown from Huckeltype
analytic
derivations[13]).
If,
for agiven
value of n, the cluster becomes more compact, theincreasing
number of nearestneighbours,
and
consequently
ofhopping integrals,
results in alowering
of the ionizationpotential.
This increasedcompactness
explains
therapid
decrease of the calculated ionizationpotentials.
It may be
interesting
toanalyse
the space andspin
symmetries
of the lowest states of theion,
aspredicted
by
ourmodel,
andcompared
to the monoelectronicpicture,
which suggests toempty
thehighest occupied
MO(or
MO’s if adegeneracy
occurs).
The lowest state of the
triangular
Mg+
is of coursedegenerate, according
to the monoelectronicpicture
(to
which our model Hamiltonian is hereidentical).
Fig.
2. -r-dependence
of the various parameters of the model Hamiltonian. a) diagonal terms (for sake ofcompa-rison, the r-1 1
repulsion
has been subtracted from R ++).
b) off-diagonal
integrals.
Table II. - Vertical and adiabatic ionization
poten-tials
(in eV).
(The
numbers inparentheses
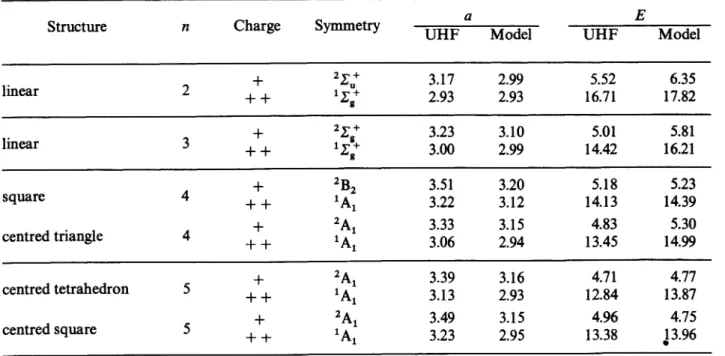
are secondary minima.)A Jahn-Teller distortion must
necessarily
occur.In the same
geometry
one mayexpect
that the lowest stateof Mg3 +
will beof A1
symmetry.
It
actually
is atriplet
3 Ai
state. The next upper statesare
degenerate
and wouldcorrespond
to a different spacepart
Ewith a
singlet spin
arrangement. ThelAi
associatedwith the lower
triplet
ishigher
in energy, which issurprising
from an MOfilling point
of view.The tetrahedral
Mg4+
lower state istriply degenerate,
as
expected
from the monoelectronicpicture.
Thedoubly charged ground
state isquintuply degenerate.
For the
bipyramid configuration
theMg’
lowerstate is
2A1
withlarger charges
on the comers of thepyramids.
Thedoubly charged
lowest state ist At
andcorresponds
to two holes in the samehighest
MO;
twodegenerate triplet
states areonly
0.016 eVabove.
The occurrence of low
lying
open shell states,sometimes
contradicting
the MOfilling
rule,
exam-plifies
theimportance
of therepulsion
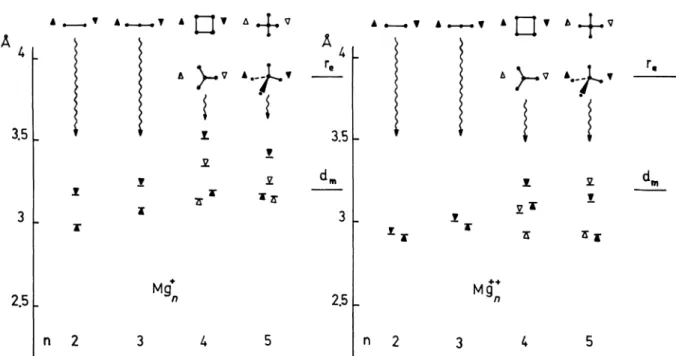
between the3.2 RELAXED GEOMETRIES AND ADIABATIC IONIZATION POTENTIALS. -
The results appear in table
III,
figu-res 3-4.
The
Mg’
andMg2 +
structures havealready
beengiven,
and one may see(Fig.1 c))
that the latter is a localminimum in the
repulsive ground
statepotential
curve; this local minimummight
accept
up to 6vibrational
levels,
the barrierbeing
0.35 eV.The
ground
stategeometries
ofMg’
andMg3 +
are both linear with
longer
distances(3.1
A)
inMg’
than inMg3 +
(3.0
A) ;
this is ageneral
trend in theseries. The states are of
2 E:
and1 E:
symmetry
in agreement with the orbital model(ionizations
from thehighest occupied
MO).
For both
Mg4+
andMg4+ +
twogeometries
are incompetition, namely
the square and the centredtriangle.
Both are real minima on thepotential
surfaces,
the formerbeing
0.6 eV below forMg:,
0.07 eV below forMg1 +.
The nearestneighbour
distances are shorter in the centred
triangle
than in thesquare,
especially
for thedoubly charged species
(Ar
= 0.2A).
The states are of2A1
and1 A1
symme-tries for the centredtriangle,
2B2g
and’A,,,
for thesquare, in
agreement
with the MOpicture.
For
Mgs
andMg5 +
twonearly degenerate
minimawere
found,
corresponding
to the centred tetrahedron(more
stableby
0.02 eV inMg5+ )
and centred square(preferred
inMg’ ’
by
0.09eV). They
all arefully
symmetric,
andcorrespond
to ionizations of thehighest
valence MO in the delocalized MOpicture.
Fig.
3. - Intrinsicenergies
of Mg.,Mg:
andMg" +
clusters(model Hamitonian), relative to n
separated Mg
atomsfor 0 neutral
ground
state conformation. A and A, relaxedconformations of the ions.
Table III. -
Energies
E (eV)
and nearestneighbour
distances a(A)
ofm gg+ and Me
+
clusters
(the
zeroof energy
Fig. 4. - Interatomic
distances between nearest neighbours in positive ions (relaxed conformations). V, V UHF ab initio
calculation. A, A model
calculation. re Mg2 experimental
re. dm metallic nearest neighbour distance.It seems
likely
that forlarger
andlarger
clusters the number ofcompeting
minima will increase and theirexhaustive
research will become a realproblem.
Inthese structures, whose
topology
issimilar,
the inter-atomic distances are almost identical.For centred structures, the
positive charge
tends tobe concentrated on the central atom; this
charge
isclose to one for
doubly charged species;
this may beunderstood
by
the decrease in electronicrepulsions
when the central atom has no valenceelectron,
by
thepolarization
of thesurrounding
neutral electrons andby
the role of cross-roadsplayed by
the centralatom in the hole delocalization.
The relaxation
energies (differences
between vertical and adiabatic ionizationpotentials)
arequite large :
There is no clear trend in this series.
The adiabatic ionization
potentials
appear in table IIThey
decreaseregularly
with n, and it is clear that oneis still
quite
far from the asymptote, which would be the metallic values(one
and two extractionpotentials
of themetal).
3.3
EVAPORATION,
AGGREGATION ANDFRAGMENTA-TION PROCESSES. -
Figure
3gives
the intrinsicenergies
of
Mg.,
Mg/
andMg."
taking
the energy of nMg
separated
atoms as the zero of energy. Thispicture
makes easy a direct discussion of theenthalpy
varia-tions in the processes
(neglecting
the zero vibrationcontributions).
Theaggregation
process(a)
occurs from the relaxedstructure of
M ± 1
(k
=1, 2),
toward a relaxed
geo-metry
of Mgn+
(k
=1, 2).
The decrease of the relaxedgeometry
energies
infigure
3 indicates that theaggregation
process isalways exoenergetic.
We havenot
analysed
the existence of barriers in thesepro-cesses, but it is
quite
clear -as summarized in
figure
5- that the
aggregation
of a neutral atom on the centralatom of the centred structure of
Mgn 1
andMgn i
shouldproceed
withoutdifficulty,
i.e. in the samesymmetry
and with small rearrangements. Thepro-cesses which
change
thesymmetry,
i.e. thoseinvolving
the square
geometries,
are moreproblematic
and the research of activation barriers should be per-formed.The
evaporation
processes areonly possible
from the vertical ionizations.They
areonly exoenergetic
for
4+-+3+
+ 1 and5+ -+ 4+
(square)
+ 1 and for5 + + -+ 4++
(square)
+ 1. The energy excess isvery low
(see Fig. 3)
and the existence of barriers ispossible
in view of thelarge
geometry
(and
symmetry)
changes.
The
possible fragmentation
ofMg+ ’
intoMg+
+Mg+
has beenanalysed.
Theexoenergeticity
is 1.72 eV whenstarting
from the lowest energy tetrahedralgeometry
toward the relaxedgeometry
Mg’
andMg+
ions. For afragmentation pathway keeping
C2,
symmetry,
the intersection between the relevantA, (Mg 5 +
+)
and iB2(Mg3
+Mgi)
potential
surfaces has its lowest energy 0.21 eV above the tetrahedronMg5 +
energy minimum. Thisquantity
may beFig. 5. - Net
charges,
symmetries
of the clusters, and schematic views ofpossible aggregation
andevaporation
processes. ---+ allowed processes. --->hypothetic
processes
(exoenergetic
but withpossible
barriers).Another
fragmentation
process,has also been
explored
Itsexoenergeticity
is 1.09eV,
, lower than the
preceding
one. It isquite
clear that thedoubly charged species
tend toexplode
intoequal
sizefragments,
due to the ndependence
of thestability
ofMgn
clusters.The barrier for the extraction process of the central
atom
of Mg5 +
isonly
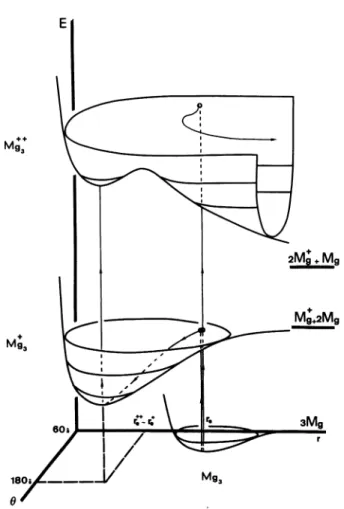
0.14 eV.3. 4 OBSERVABILITY OF DOUBLY CHARGED CLUSTERS.
-8 The existence of local minima in the
potential
energyhyper-surface
ofMgn+ +
is not sufficient to insure theobservability
of theseclusters,
since these minimaare
significantly higher
than the result of afragmen-tation into
Mgp
+Mgq ( p
+ q =n).
Another factorwhich may
prevent
the observation of thedoubly
charged
clusters is the great difference between thegeometry
of the neutral cluster and that of this local minimum. A vertical double ionization would notlead to the
doubly charged
cluster since itbrings
animportant
energy excess and the system couldhardly
achieve the travel from theoriginal
neutral cluster conformation to that of thedoubly charged
minimum and stay in thatregion
which isonly
surroundedby
weak barriers.This statement is
schematically
illustrated infigure
6,
representing
for instance theMg (k+)
(k
=0, 1,
2)
potential
curves as functions of two parametersonly
r =
rAB =
rBc and 0 =
ABC.
It isquite
clear that the vertical double ionizationgives
a smallprobability
to
stay
in the local minimum of the upper surface. As a veryimportant
fact,
one should notice that inall the cases studied
here,
the stable conformations ofMg+
andMgn +
arequite
similar. This means that anionization from
Mg.’
toMg,n+ +
would benearly
vertical,
and would lead close to the bottom of theMg/+
structure,giving
anopportunity
for thiscluster to have a
significant
lifetime. One must thusassume that the
ejections
of the two electrons under the electronimpact
are not simultaneous.During
the timeseparating
the first and second electronemission,
the cluster
Me
should rearrange toward the stableM g,7 configuration,
which is an absoluteminimum,
the second emission
proceeding
withoutimportant
geometry
changes
towards thesecondary
minimum of theMe +
potential hypersurface
which holds theMgn+ +
cluster(cf. Fig.
6).
These remarks are crucial for small
clusters,
and forall cases where the
charges
arequite
concentrated,
inducing important
rearrangements
of thepositive
cluster withrespect
to the neutral one. For verylarge
clusters the rearrangementsmight
become small(vide infra)
and creations ofdoubly charged
clustersFig.
6. - Schematic view of the lowestpotential
surfaces ofMg(,")
(k = 0, 1, 2) as functions of r = rAB = ’Be and 0= ABC.
4.
Comparison
with ab initio SCFcalculations ;
metho-dological
discussion4.1 RESULTS OF SCF CALCULATIONS. - Valence
only
[14]
SCF calculations have beenperformed
on bothsingly
anddoubly charged
clusters,
using (3s, 2p)
AO basis sets(i.e.
deleting
the 3d AO involved in the MO-CI calculation onMg’
andMg2 +).
A UHF(Unrestricted
HartreeFock)
formalism was used withSz = 1/2
forMg’
andSz
= 0 forMg’
+,
but for the latters the solutions werealways
puresinglets.
These calculations have been
performed
tocheck;
i)
the occurrence of minima close to the structurespredicted by
ourmodel;
ii)
thepossible importance
of p
AO’s which do not appearexplicitly
in our scheme.The
energies
andgeometries
appear in tableIII;
they
concern real minima obtained from agradient
program and force constants calculations.
They
con-firm that all the
predicted
minima also appear in a monoelectronicapproach.
Thisagreement
is not toosurprising
since,
for the studiedminima,
the modelwave-functions were consistent with
single
and doubleionizations from the
highest occupied
MO.Significant
differences appear however :i)
the ionizationpotentials
arealways
muchlowqr
than in our model. This is
already
true for n =1,
since the correlation of the
S2
pair
inMg
is veryimportant, increasing
the ionizationpotential by
about 1eV;
this defect is stillacting
in the cluster UHFcalculation;
ii)
the relative energyordering
of the two minima forMg’, Mg4 +
andMg5+
is reversed with respect toour
model;
one may notice that forMg4+
andMg4++
the monoelectronic calculation favours the centred structure;iii)
the bond interatomic distances are muchlarger
(see
Fig.
4).
These two
points
ofdisagreement
will be discussed later. The electronicpopulations
calculatedaccord-ing
to the Mulliken criterion would indicate a moreeven net
charge
distribution;
they
also would indicatea
significant
ppopulation
on the central atom.However a careful examination of the MO’s shows
that p, AO’s do not appear with coefficients
larger
than0.15;
thehybridization
is therefore rather weak and theapparent
ppopulation
isessentially
dueto the
overlap
of the p, AO’s of the central atom with the s AO’s of thesurrounding
atoms, in atypical
artifact of Mulliken’s
population analysis.
This is confirmedby
the fact that the Pcdiagonal
element of thedensity
matrix isnegligible
(
0.03eV).
4.2 COMPARISON BETWEEN MODEL AND ab initio SCF
CALCULATIONS. - The
discrepancies
between ourmodel and SCF results may be
easily
understood It is clear that ab initio SCF calculations introducemany-body repulsive
effects which are not taken intoaccount in our model. However these
many-body
effects are not verylarge
andthey
cannotexplain
the differences in bond distances and in the relative
energy
orderings
of theMgi
andMg: ’
isomers.i)
Regarding
the interatomicdistances,
one shouldnotice that in a cluster the
probability
for twoadjacent
atoms to be both neutral is 1- Ifor
singly charged
n
clusters ai for
doubly charged
clus-ters. In both cases this
probability
goesrapidly
to one.One may therefore
expect
that when n increases themean interatomic distances tend toward that of the
neutral cluster
(i.e.
the metal forlarge
n).
Since the monoelectronicpicture
does not take into accountthe
dispersion
energy between neutral atoms, it will let the clusterexplode
and the interatomic distances will tend toinfinity.
This failure will also occur inpositive
clusters since the delocalization energy of the hole isbound
by
kF +
(k being
ascalar,
depending
on the compactness of thecluster)
as are the upper or lowerlimits of a band Since the
repulsive
interactions ERincrease as n, the delocalization of the
hole,
included in the monoelectronicscheme,
cannotfight efficiently
against
theincreasing repulsions,
and thepositive
cluster is more and moreloosely
bound withlarger
andThis
defect,
which does not appear in ourmodel,
is very difficult to overcome in ab initio
calculation,
since it would
require
the calculation ofdispersion
forces -a task which is
already
difficult forMg2
[15].
ii)
Regarding
the relativeenergies
of centred versussquare structures in
Mg+
andMg+ +,
one shouldnotice that the Hartree-Fock
approximation only
takes into account the static held Inhomogeneous
structures, as is the square
cluster,
thecharge
isequally
spread,
the static field is weakeverywhere;
on the contrary if thecharge
is well concentrated on oneatom - as occurs for the central atom of the centred
triangle
- the static field isimportant
on thesur-rounding
atoms, and the HFapproximation
is able totake this effect into account.
Our model deals with instantaneous
repolarizations
in all VB local structures, and therefore takes intoaccount an effect which would
only
appearthrough
double excitations in the MO scheme
(change
of the hole from thehighest occupied
MO to lower MOplus
one excitation to the outer
polarization space).
These comments - which will be
developed
in detail in aforthcoming
paper[16]
-explain
thepreference
of the monoelectronic calculation for centredstruc-tures, as due to an unbalanced
purely
static treatmentof
polarization
effects.5. Conclusion.
A careful extraction of information from the
potential
curves of the diatoms
Mg2,
Mg2
andMg2
+,
corres-ponding
to the stateskeeping
an sP +fl(p
+ q =4, 3,
2, p, q
2) character,
permits
to define asimple
modelHamiltonian of low size for the
study
ofsingly
anddoubly charged
clusters. The Hamiltonian involvesonly
6parameters
and its size isn2
fordoubly charged
clusters. The
parameters
being analytic
functions of theinteratomic distances
gradient techniques
may beeasily applied
Since the extraction of the parameters has been done from calculations
involving
basis sets andCI,
they
shouldprovide
some information forlarge
clusterstudies,
which are unattainable from direct MO-CIcalculations since
they rapidly require
reductions of the basis setand/or
severe CI truncations. Thesuperio-rity
of our model over ab initio SCF calculationsappeared clearly
in thepreceeding
discussion on both the internuclear distances(unreliable
in SCFcalcu-lations)
and the energyordering
of various minima(unduly preferred
as centred structures in SCFcalcu-lations).
The
present
work has shown stable structures forMe
andMg,.+ +
for n
5. It will beinteresting
toanalyse
thegeneral topology
of thepotential
energyhypersurface(s), taking
benefit of some recent theo-retical advances[17J.
This wouldgive
better insuranceregarding
the number ofminima,
aproblem
whichwill be of
increasing importance
forlarger
clusters. It will also be worthwile toanalyse
the existence andheights
of barriers foraggregation, evaporation
andfragmentation
processes.Extension to
larger
clusters ofMg
atoms is inprogress. An isoelectronic series may be
treated,
namely
theHg’
orHg/ +
clusters studied inrefe-rence
[1].
The extraction of the basic parameters for thisheavy
atom is in progress.Finally
one should mention that a direct extension to rare gas clusters iseasy to
perform,
the number of AObeing
then increasedto 3 n
(and
the size of the matrix to(3
n)2
fordoubly
positive
clusters).
In that last case theproblem
ofhybridization,
which may lead to difficulties forlarge
values of n inS2
atoms, would be avoidedReferences
[1] BRECHIGNAC, C., BROYER, M., CAHUZAC, Ph., DELA-CRETAZ, G., LABASTIE, P. and WÖSTE, L., Chem.
Phys. Lett. 118 (1985) 174.
[2] ERMLER, W. C., LEE, Y. S., CHRISTIANSEN, P. A. and
PITZER, K. S., Chem.
Phys.
Lett. 81 (1981) 70.CHRISTIANSEN, P. A., BALASUBRAMANIAN, K. and
PITZER, K. S., J. Chem. Phys. 76 (1982) 5087.
[3] TEICHTEIL, Ch., PÉLISSIER, M. and SPIEGELMANN, F.,
Chem. Phys. 81 (1983) 273, 283.
[4] KUNTZ, P. J., Chem. Phys. Lett. 16 (1972) 581 ; TULLY,
J. C., Modern Theoretical Chemistry, Vol. 7,
G. A.
Segal ed.
(New York, Plenum), 1977,p.173.
[5] PARISER, R. and PARR, R. G., J. Chem. Phys. 21 (1953)466, 767 ;
POPLE, J. A., Trans. Far. Soc. 49 (1953) 1375.
[6] POPLE, J. A., SANTRY, D. P. and SEGAL, G. A., J. Chem.
Phys. 43 (1965) 129;
BAIRD, N. C. and DEWAR, M. J. S., J. Chem. Phys. 50
(1969) 1262.
[7] HUBBARD, J., Proc. R. Soc. London A 276 (1963) 238.
[8] HUBER, K. P. and HERZBERG, G., Constants
of Diatomic
Molecules (Van Nostrand, N. Y.) 1979.[9] SCHEINGRABER, H. and VIDAL, C. R., J. Chem. Phys. 66
(1977) 3694.
[10] STEVENS, W. J. and KRAUSS, M., J. Chem. Phys. 67
(1977) 1977.
[11] SPIEGELMANN, F. and MALRIEU, J. P., J. Phys. B 17
(1984) 1259 ;
CIMIRAGLIA, R., MALRIEU, J. P., PERSICO, M. and
SPIEGELMANN, F., J. Phys. B 18 (1985) 3073.
[12] DURAND, G., BERNIER, A. and SPIEGELMANN, F., to be
published.
The effectivecoupling
between the[Mg++
+Mg(3s2)]
and[Mg+(3s)
+Mg+(3p)]
configurations
is a 3s|F |3pz > hopping
the diabatic
potential
curves to touch the adiabaticones in this
region.
[13] See for
example :
SALEM, L., The Molecular Theory ofConjugated
Systems(Benjamin,
N. Y.) 1965.[14] DURAND, Ph. and BARTHELAT, J. C., Chem. Phys. Lett.
27 (1974) 191.
BARTHELAT, J. C. and DURAND, Ph., Gazzetta Chim. Ital. 108 (1978) 225.
[15] PURVIS, G. D. and BARTLETT, R. J., J. Chem. Phys. 71
(1979) 548.
[16] MALRIEU, J. P., DURAND, G., DAUDEY, J. P., to be
published.
[17] LIOTARD, D., Symmetries and Properties of
non-rigid
molecules : AComprehensive
Survey.Proceedings
of an International