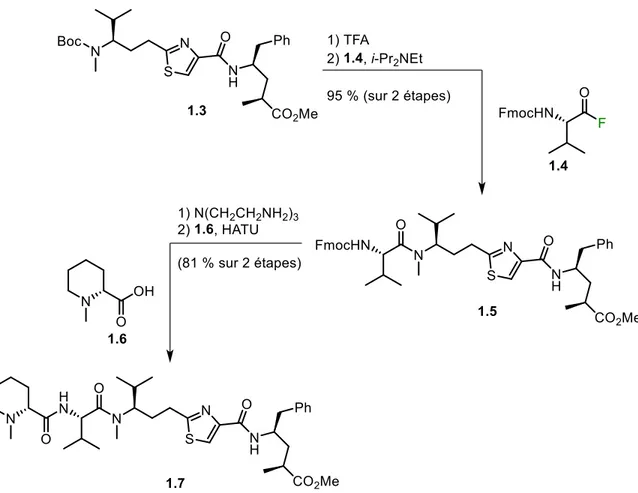
Utilisation du XtalFluor pour la déoxofluoration de
composés carbonylés
Mémoire
Marie Gonay
Maîtrise en chimie - avec mémoire
Maître ès sciences (M. Sc.)
Utilisation du XtalFluor
®pour la déoxofluoration
de composés carbonylés
Mémoire
Marie Gonay
Sous la direction de :
Résumé
OmegaChem, une compagnie québécoise, a développé en 2009 une nouvelle classe
d’agents de déoxofluoration : le XtalFluor®. Ce réactif, un sel cristallin de type aminodifluorosulfinium, représente une solution de rechange plus sécuritaire aux réactifs classiques tels que le DAST et le Deoxo-Fluor® parce qu’il est plus facile à manipuler et plus stable thermiquement. De plus, lorsqu’il est utilisé avec une source externe de fluorure, le XtalFluor® montre une excellente réactivité envers une grande variété d’alcools et de dérivés carbonylés. Cependant, les conditions initialement développées pour ce type de transformation sont loin d’être idéales du point de vue de la chimie verte parce qu’elles impliquent l’utilisation du dichlorométhane, un solvant potentiellement cancérigène, et de Et3N·3HF, une source de fluorure corrosive. Ce mémoire porte ainsi sur l’optimisation des conditions de déoxofluoration utilisant le XtalFluor®.
L’intérêt des molécules organiques fluorées sera abordé en guise de préambule à ce mémoire. Une présentation générale de la réaction de déoxofluoration incluant les caractéristiques du XtalFluor® de même qu’une courte introduction aux principes de la chimie verte seront ensuite dressées afin de bien comprendre le contexte dans lequel les projets ont évolué. Les travaux portant sur l’optimisation des conditions réactionnelles seront ensuite présentés pour la synthèse de deux motifs fluorés d’intérêts. Dans un premier temps, les caractéristiques des fluorures d’acyle seront abordées puisqu’ils représentent potentiellement d’excellents intermédiaires en chimie organique. La méthodologie ayant mené au développement d’une méthode de synthèse utilisant le XtalFluor® sera présentée (Chapitre 1). La déoxofluoration d’aldéhydes sera ensuite abordée en suivant la même méthodologie dans le but de rendre le groupement -CF2H plus facilement accessible au domaine pharmaceutique, qui l’utilise principalement pour faire varier la lipophilie d’un médicament (Chapitre 2).
Abstract
Developed by OmegaChem in 2009, XtalFluor® is an aminodifluorosulfinium fluorinating reagent which is safer than those typically employed for the deoxofluorination reaction (DAST, Deoxo-Fluor® and their counterparts). Indeed, its crystalline structure and its thermal stability make it easier to handle in comparison to the other reagents. In addition, when used with an external source of fluoride, XtalFluor® shows excellent reactivity toward a wide range of alcohols and carbonyl derivatives. However, the conditions generally employed for this type of transformation are far from being ideal as they imply the use of dichloromethane, a potentially carcinogenic solvent, and Et3N·3HF, a corrosive fluoride source. Therefore, this project aims at developing greener conditions for the deoxofluorination reaction using XtalFluor®.
To introduce this thesis, the motivations behind the growing interest for fluorinated organic molecules will be depicted. A general presentation of the deoxofluorination reaction, including the characteristics of XtalFluor®, and an introduction to the principles of green chemistry will serve to illustrate the general context in which the project has grown. The optimization of the reaction conditions will then be presented for the synthesis of two fluorinated motifs of interest. First, acyl fluorides characteristics will be portrayed because this motif potentially represents excellent intermediates for organic chemistry. The methodology that has led to the development of a synthetic way using XtalFluor® will be detailed in Chapter 1. The deoxofluorination of aldehydes will then be illustrated following the same methodology in order to allow access to difluorinated molecules more easily (Chapter 2). This motif is of interest especially for the pharmaceutical branch which uses it to moderate a drug’s lipophilicity.
Table des matières
Résumé ... ii
Abstract ... iii
Table des matières ... iv
Liste des abréviations ... ix
Remerciements ... xii Introduction ... 1 L’atome de fluor ... 1 Composés organofluorés ... 3 Lien C‒F ... 3 Propriétés ... 3 Applications ... 5 Synthèse ... 7 Déoxofluoration ... 8 Agents de déoxofluoration ... 9 XtalFluor® ... 10 Chimie verte ... 14 Objectifs généraux ... 15
Chapitre 1. Synthèse de fluorures d’acyle ... 16
1.1 Caractéristiques des fluorures d’acyle ... 17
1.2 État de l’art ... 19
1.3 Objectifs spécifiques ... 22
1.4 Optimisation ... 23
1.4.1 Effet du solvant ... 23
1.4.2 Choix de la source de fluorure ... 24
1.4.3 Optimisation de la température ... 25
1.4.4 Optimisation du temps, du XtalFluor-E® et de la concentration ... 26
1.4.5 Optimisation d’un système catalytique ... 27
1.5 Hypothèses mécanistiques ... 29 1.6 Étendue de réaction ... 30 1.6.1 Fluorures d’acyles ... 30 1.6.2 Amidation ... 34 1.7 Conclusion ... 36 1.8 Partie expérimentale ... 37
Chapitre 2. Déoxofluoration d’aldéhydes ... 49 2.1 Caractéristiques du groupement -CF2H ... 49 2.2 État de l’art ... 50 2.3 Objectifs spécifiques ... 53 2.4 Optimisation ... 54 2.4.1 Effet du solvant ... 54 2.4.2 Optimisation de la température ... 55
2.4.3 Choix de la source de fluorure ... 56
2.4.4 Optimisation XtalFluor-E® et de la concentration ... 57
2.4.5 Optimisation du temps ... 58 2.5 Hypothèses mécanistiques ... 59 2.6 Étendue de réaction ... 60 2.7 Conclusion ... 62 2.8 Partie expérimentale ... 63 Conclusion et perspectives ... 67 Bibliographie ... 70
Liste des figures
Figure I. 1.[18F]Fluorodeoxyglycose ... 2
Figure I. 2. Viroptic® (Trifluridine) ... 6
Figure I. 3. Groupement fluorés présents en chimie médicinale et en agrochimie ... 6
Figure I. 4. Différents composés fluorés utilisés en sciences des matériaux ... 7
Figure I. 5. Dérivés de trifluorure de dialkylamino soufre ... 9
Figure I. 6. Dérivés α-fluoroaminés ... 10
Figure I. 7. XtalFluor® ... 10
Figure I. 8. Thermogramme DSC du DAST, Deoxo-Fluor® et XtalFluor® ... 12
Figure 1. 1. Structure générale d’un fluorure d'acyle ... 17
Liste des tableaux
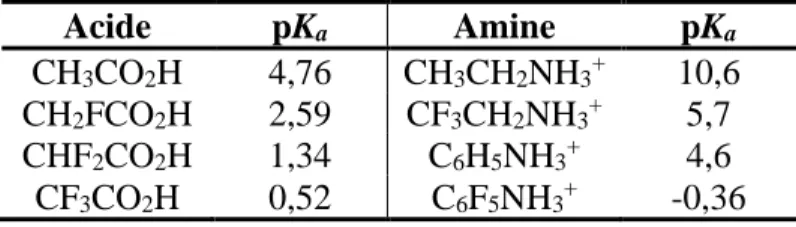
Tableau I. 1. Influence de l'atome de fluor sur le pKa ... 4
Tableau I. 2. Influence de l'atome de fluor sur la lipophilie d’un alcane (Log P) ... 4
Tableau I. 3. Les 12 principes de la chimie verte ... 14
Tableau 1. 1. Choix du solvant ... 24
Tableau 1. 2. Choix de la source de fluorure ... 25
Tableau 1. 3. Influence de la température ... 26
Tableau 1. 4. Optimisation du temps réactionnel, du XtalFluor-E® et de la concentration . 27 Tableau 1. 5. Optimisation du système catalytique ... 28
Tableau 1. 6. Tests contrôles ... 28
Tableau 1. 7. Système catalytique vs stœchiométrique ... 31
Tableau 2. 1. Choix du solvant ... 55
Tableau 2. 2. Influence de la température ... 56
Tableau 2. 3. Choix de la source de fluorure ... 57
Tableau 2. 4. Optimisation de la quantité de XtalFluor-E® ... 57
Tableau 2. 5. Optimisation de la concentration ... 58
Liste des schémas
Schéma I. 1. Fluoration de molécules organiques ... 8
Schéma I. 2. Réaction de déoxofluoration ... 8
Schéma I. 3. Voie de synthèse du XtalFluor® ... 11
Schéma I. 4.Utilisation du XtalFluor-E® comme agent de déoxofluoration- ... 12
Schéma I. 5. Utilisations du XtalFluor® ... 13
Schéma 1. 1. Synthèse de la rifamycine S ... 16
Schéma 1. 2. Synthèse d’un analogue de la pretubulysine D ... 17
Schéma 1. 3. Synthèse d'un amide stériquement encombré via un fluorure d'acyle ... 19
Schéma 1. 4. Stratégies de synthèse des fluorures d'acyle ... 20
Schéma 1. 5. Synthèse des fluorures d'acyle par le groupe de Schoenebeck ... 20
Schéma 1. 6. Voie de synthèse développée par Prakash... 21
Schéma 1. 7. Fluoration radicalaire d'un aldéhyde par le groupe de Martin et Britton ... 21
Schéma 1. 8. Synthèse des fluorures d'acyle avec le XtalFluor® ... 22
Schéma 1. 9. Intermédiaire connu de la déoxofluoration d'alcools avec le XtalFluor-E® ... 29
Schéma 1. 10. Mécanismes supposés pour la déoxofluoration d'acides carboxyliques avec le XtalFluor-E® ... 30
Schéma 1. 11. Étendue de réaction des fluorures d’acyles ... 33
Schéma 1. 12. Étendue de réaction des amides ... 35
Schéma 2. 1. Stratégies d'incorporation d'un groupement difluorométhyle ... 51
Schéma 2. 2. Usage du Fluolead™ pour synthétiser des molécules difluorométhylées ... 52
Schéma 2. 3. Synthèse d'aryles difluorométhylés par le groupe de Sanford ... 52
Schéma 2. 4. Déoxofluoration d'un aldéhyde par le XtalFluor® ... 53
Schéma 2. 5. Mécanismes suggérés pour effectuer la déoxofluoration d'aldéhydes avec le XtalFluor-E® ... 60
Schéma 2. 6. Étendue de réaction de la déoxofluoration d'aldéhydes ... 61
Schéma C. 1. Conditions développées pour la synthèse de fluorures d'acyles ... 67
Liste des abréviations
Δ chauffage ° C degré Celsius Å angström Ac acétyle aq. aqueux Boc tert-butoxycarbonyle Bn benzyle BTFFH fluoro-N,N,N′,N′-bis(tétraméthylène)formamidinium hexafluorophosphate cat. catalytique Cbz chloroformiate de benzyleDAST trifluorure de N,N-diéthylaminosulfure DCC N,N'-Dicyclohexylcarbodiimide
DIPEA N,N-Diisopropyléthylamine
DMF N,N-diméthylformamide
DSC differential Scanning Calorimetry;
calorimétrie différentielle à balayage
éq./eq. équivalent
Et éthyle
Fmoc fluorénylméthoxycarbonyle
h heure
HATU hexafluorophosphate de
(diméthylamino)-N,N-diméthyl(3H-[1,2,3]triazolo[4,5-b]pyridin-3-yloxy)méthaniminium
HRMS high resolution mass spectometry;
spectromètre de masse à haute résolution
iPr isopropyle
IR infrarouge
LiHMDS bis(triméthylsilyl)amidure de lithium
Me méthyle mg milligramme mL millilitre mp melting point; température de fusion 2-MeTHF 2-méthyltétrahydrofurane
MTBE méthyl tert-butyl éther
NBS N-Bromosuccinimide
NFSI N-fluorobenzenesulfonimide
ppm partie par million
Ph phényle
pKa logarithme de la constante d’acidité
rdt rendement
RMN résonance magnétique nucléaire
t.a. température ambiante
TBAF fluorure de tétrabutylammonium
tBu tert-butyle
temp. température
THF tétrahydrofurane
« On ne fait jamais attention à ce qui a été fait ; on ne voit que ce qui reste à faire. »
Remerciements
Les études graduées ont été pour moi un chemin pavé d’émotions. C’est avec ambition et appréhension que je me suis lancée dans cette étape de ma vie qui, jusqu’à maintenant, représente le summum de mes études. Il s’agit de deux années qui m’ont permis, oui, d’explorer la chimie un peu plus en profondeur, mais surtout d’apprendre à me connaitre. J’y ai rencontré une personne déterminée et travaillante, mais qui se met également beaucoup de pression : lorsqu’on côtoie au quotidien des personnes qui sont faites pour accomplir de grandes choses, il devient facile de se sentir petit. Néanmoins, ce passage de ma vie m’a permis aussi de découvrir que j’avais une certaine aisance à parler en public et que cette force m’aidera pour ma prochaine étape : enseigner. C’est donc pour cette raison que, quand je regarde derrière moi et que je visualise tout ce que j’ai accompli et traversé, je suis fière.
Je suis aussi reconnaissante pour tous ceux qui m’ont permis de me rendre jusqu’ici, à commencer par mon directeur de recherche : Jean-François Paquin. Il m’a proposé une maitrise au sein de son laboratoire alors que le fait de penser à ce qui viendrait après le baccalauréat me faisait encore peur. Du moment où j’ai accepté, son sens de l’organisation hors pair m’a tout de suite étonnée et rassurée. J’ai toujours pu compter sur lui pour me donner ses impressions sur mon travail.
Les études graduées c’est aussi apprendre à connaitre de nouvelles personnes qui nous touchent toutes à leurs façons : Myriam, Jean-Denys, Mélissa, Marius, Majdouline, Audrey, Xavier, Raphaël et Camille. Camille fait partie de ces personnes à qui parler fait du bien. Au bout d’un temps, j’avais besoin de mon moment « caucus » avec elle pour pouvoir me plaindre quand c’était difficile, ou me venter quand j’étais fière de moi. Camille a été un pilier à ma maitrise et a contribué à l’alléger considérablement. Elle a mis de la joie et des rires dans mes journées et on a traversé ensemble les différentes étapes de la maitrise. Merci, Camille, d’avoir été là pendant ces deux années.
Travailler sur un projet de maitrise, c’est apprendre à travailler en équipe. J’ai eu la chance d’avoir une stagiaire postdoctorale, Chloé, pour m’aider sur mon projet. Chloé est tellement altruiste et respectueuse. Elle s’est mise à travailler avec moi en s’assurant toujours
de respecter ma vision tout en y mettant son grain de sel. Je n’aurais pas pu mieux espérer comme coéquipière. Chloé, merci d’avoir été attentive à mes idées, à mes besoins et à mes frustrations.
La maitrise m’a également permis de faire passer mon savoir à une stagiaire de premier cycle, Marie-Rose. C’est une expérience très enrichissante qui fait ressortir en nous nos forces et nos faiblesses. Marie-Rose a été une stagiaire formidable : curieuse et très intelligente. Elle est d’une aisance innée avec les gens qui ne cesse de m’épater et sa « bourgeoisie » était aussi drôle qu’inspirante. Merci, Marie-Rose, d’avoir partagé ta fraicheur et ta culture impressionnante.
Finalement, il est bien difficile d’entreprendre des études graduées sans le support de sa famille. Mes parents ont toujours cru en moi et ont toujours été fiers de moi. Tout au long de mon processus, ils se sont assuré que j’étais heureuse dans mes choix et que je n’abandonnais pas aux moindres difficultés. Merci aussi à mon grand frère qui m’a toujours laissée comprendre à quel point mon ambition et ma persévérance l’épataient. À vous trois : merci, je vous aime profondément. Merci aussi à Danny, qui est devenu bien plus que mon correcteur. Il était là au moment où j’en avais le plus besoin pour me pousser à me dépasser, pour me montrer que j’étais capable de faire bien plus que je ne l’aurais cru. Il a été là pour croire en moi quand j’oubliais de le faire. Il m’a aussi appris à me battre pour mes idées et mes principes et à ne pas me laisser de côté. Danny, merci d’être dans ma vie, simplement.
Introduction
L’atome de fluor
Le fluor élémentaire (F2) a été découvert en 1886 par le chimiste français Henri Moissan après avoir effectué l’électrolyse du bifluorure de potassium (KHF2). L’isolation de ce gaz jaune-vert toxique et corrosif, en plus de la conception du four à arc électrique, lui ont d’ailleurs valu le prix Nobel de chimie en 1906.1 Dans la nature, le fluor est présent presque exclusivement sous forme de fluorure (F-) et provient majoritairement de la fluorine (CaF2), un minéral. Il est également le 13e élément le plus abondant dans la croute terrestre et dans les océans, mais est absent de la biosphère.2
Le fluor est un élément qui suscite l’intérêt des chimistes parce qu’il se différencie des autres à plusieurs niveaux. Il est souvent comparé à l’atome d’hydrogène ou aux autres halogènes pour illustrer ses propriétés uniques. D’abord, le fluor est l’élément le plus électronégatif du tableau périodique. Cette électronégativité d’une valeur de 3,98 sur l’échelle de Pauling est, d’ailleurs, en partie responsable de sa petite taille puisque ses électrons sont fortement attirés par son noyau.2 Avec un rayon de van der Waals de 147 Å, l’atome de fluor se compare davantage en termes de taille à l’atome d’hydrogène (120 Å) qu’aux autres halogènes (175 Å, 185 Å et 198 Å pour le Cl, Br et I respectivement). Également, le nuage électronique autour de l’atome de fluor n’est pas facilement déformé en raison des deux propriétés tout juste énoncées, ce qui se traduit par une faible polarisabilité, soit 0,557 Å3. Il s’agit d’une différence assez marquée avec l’atome de chlore (2,18 Å3), qui est l’halogène suivant dans le tableau périodique. Encore une fois, les caractéristiques de l’atome de fluor sont plus près de celles de l’atome d’hydrogène (0,667 Å3).
Le 19F est particulièrement utile en résonance magnétique nucléaire (RMN) en raison de ses propriétés spectroscopiques. Son spin nucléaire (I) de ½ lui permet d’être actif en RMN tandis que son abondance naturelle de 100 % de même que son rapport gyromagnétique
1Tressaud, A. Angew. Chem. Int. Ed. 2006, 45, 6792.
2 Kirsch, P. Introduction. Dans Modern Fluoroorganic Chemistry, 2e éd.; Wiley-VCH: Weinheim, Allemagne,
particulièrement élevé font de lui un atome hautement sensible en RMN avec une sensibilité relative équivalant 83 % de celle de l’hydrogène.3 Son étendue spectrale est également très large, ce qui permet d’identifier facilement et précisément les faibles variations dans son environnement à l’aide des déplacements chimiques.
Bien que le 19F soit le seul isotope naturel stable, il est tout de même possible de synthétiser le 18F. Cet isotope a un temps de demi-vie d’environ 110 min, ce qui le rend particulièrement utile dans le domaine de l’imagerie médicale, notamment en tomographie par émission de positrons (TEP). En effet, ce temps de demi-vie est assez long pour permettre d’effectuer les différentes analyses, mais assez court pour être complètement éliminé du corps en quelques heures.Il existe plusieurs radiotraceurs fluorés qui permettent d’effectuer de l’imagerie médicale. Entre autres, le [18F]fluorodeoxyglycose I.1 est un des radiotraceurs les plus connus et est fortement utilisé en oncologie, cardiologie et neurologie (Figure I. 1). Ces branches de la médecine agissent toutes sur des cellules qui se divisent plus rapidement que la moyenne (cellules cancéreuses, cérébrales et rénales), nécessitant conséquemment plus d’énergie et donc, plus de glucose. Le [18F]fluorodeoxyglycose étant facilement absorbé par ce type de cellule, il permet ainsi de localiser les différents systèmes d’intérêts (les tumeurs, par exemple) via la radiation émise et captée par un détecteur. Le 18F se désintègre en 18O qui est non toxique et le radiotraceur est alors éliminé de la même façon que le glucose.4,5
Figure I. 1.[18F]Fluorodeoxyglycose
3 Dolbier, W. R., Jr. General Introduction. Dans Guide to Fluorine NMR for Organic Chemists, 2eéd.; Wiley:
Hoboken, NJ, 2016, p. 1.
4 Ametamey, S. M.; Honer, M.; Schubiger, P. A. Chem. Rev. 2008, 108, 1501.
Composés organofluorés
Lien C‒F
La liaison chimique qui unit l’atome de carbone et l’atome de fluor présente plusieurs singularités.6 Elle est notamment la liaison simple la plus forte que le carbone peut faire et son énergie de dissociation de 439 kJ/mol est considérablement supérieure à celles de la liaison C‒H (414 kJ/mol) et C‒Cl (330 kJ/mol). La petite taille de l’atome de fluor et sa grande électronégativité influencent également la longueur du lien C‒F et font de lui un lien court (1,35 Å), dont la longueur se situe entre celles du lien C‒H (1,09 Å) et du lien C‒O (1,43 Å). La grande différence d’électronégativité entre l’atome de fluor (3,98) et l’atome de carbone (2,55) explique également que le lien C‒F soit fortement polarisé et donc peu polarisable.2 C’est cette faible polarisabilité qui rend également les composés organofluorés plus volatils puisqu’elle est à l’origine des faibles forces de cohésion entre les molécules fluorées.7 Toutes ces caractéristiques font de la liaison C‒F un lien particulièrement stable et relativement inerte. De même, plus il y a d’atomes de fluor sur un même atome de carbone, plus la force de la liaison C‒F sera importante : CF4 (485 kJ/mol) > CHF3 (479 kJ/mol) > CH2F2 (458 kJ/mol) > CH3F (448 kJ/mol). En effet, la présence de plusieurs atomes de fluor chargés négativement (δ-) appauvrit en électrons l’atome de carbone (δ+) sur lequel ils se rattachent, ce qui a pour effet d’augmenter les charges électrostatiques entre les deux atomes et ainsi renforcer la liaison.6
Propriétés
Les propriétés exceptionnelles de l’atome de fluor se font ressentir dans l’environnement dans lequel il se trouve. Conséquemment, l’introduction d’un tel atome au sein de sa structure modifie considérablement les différentes propriétés physicochimiques d’un composé organique.5 Tout d’abord, la forte tendance du fluor à attirer les électrons influence grandement le pKa du composé dans lequel il se trouve, ce qui s’exprime par une
modification de son acidité ou de sa basicité. Par exemple, l’acide trifluoroacétique est nettement plus acide que son analogue non fluoré. De même, l’introduction d’atomes de fluor
6 O'Hagan, D. Chem. Soc. Rev. 2008, 37, 308. 7 Lemal, D. M. J. Org. Chem. 2004, 69, 1.
au sein d’amines aliphatiques diminue de façon notable leur pKa, les rendant, par conséquent,
beaucoup moins basiques (Tableau I. 1).
Tableau I. 1. Influence de l'atome de fluor sur le pKa
Acide pKa Amine pKa
CH3CO2H 4,76 CH3CH2NH3+ 10,6 CH2FCO2H 2,59 CF3CH2NH3+ 5,7 CHF2CO2H 1,34 C6H5NH3+ 4,6 CF3CO2H 0,52 C6F5NH3+ -0,36
La lipophilie des composés organofluorés est également influencée par l’ajout d’un ou de plusieurs atomes de fluor. La lipophilie d’une molécule est souvent exprimée comme étant le coefficient de partition entre l’octanol et l’eau (Log P). Un composé lipophile se retrouvera majoritairement dans la phase de l’octanol tandis qu’un composé hydrophile aura tendance à être préférentiellement dans la phase aqueuse. Ainsi, plus la valeur du Log P est élevée, plus le composé est lipophile. La mono-, la di- et la trifluorométhylation terminale d’une molécule aliphatique tendent à diminuer sa lipophilie en raison de la création d’un dipôle plus important que pour le lien C‒H (Tableau I. 2) La fluoration aromatique, la polyfluoration et la fluoration adjacente aux insaturations, elles, ont plutôt l’effet inverse en raison de l’excellent recouvrement orbitalaire entre les orbitales du fluor et celles du carbone adjacent. Le lien C‒F est donc très peu polarisable et moins hydrophile.
Tableau I. 2. Influence de l'atome de fluor sur la lipophilie d’un alcane (Log P) Log P (octanol-eau)
CH3CH3 1,81
CH3CHF2 0,75
CH3(CH2)3CH3 3,11 CH3(CH2)3CH2F 2,33
Bien que la substitution d’un atome d’hydrogène par un atome de fluor amène des changements stériques mineurs en raison de leur rayon de van der Waals similaire, ce n’est pas vrai pour tous les groupements fluorés. Par exemple, l’introduction d’un groupement -CF3 peut apporter des changements drastiques étant donné que son volume de van der Waals (39,8 Å) ressemble plus à celui du groupement éthyle (38,9 Å) qu’à celui du
groupement méthyle (21,6 Å).8 En termes de taille, c’est plutôt le groupement -CF2H qui se rapproche le plus du groupement -CH3.9 Les effets électroniques sont également grandement influencés par la présence d’atomes de fluor. Le lien C‒F étant fortement polarisé, il a la capacité d’induire des dipôles, ce qui induit un changement conformationnel au sein du composé. Une des illustrations classiques des effets stéréoélectroniques sur la conformation d’un composé est le difluoroéthane, qui préfère adopter une conformation gauche plutôt qu’anti. Finalement, il a été montré que l’atome de fluor peut accepter faiblement des liaisons hydrogène et ainsi influencer le comportement chimique des composés organofluorés.10
Applications
À la lumière de tous ces phénomènes, il n’est pas étonnant de retrouver les composés organofluorés dans plusieurs sphères de la chimie malgré qu’ils soient absents des produits naturels. Par exemple, entre 20 et 25 % des médicaments sur le marché contiennent un atome de fluor tandis que ce pourcentage grimpe à 30-40 % en agrochimie.8,11 Les composés organofluorés trouvent même des applications dans les sciences des matériaux.12
L’effet du fluor sur les propriétés physicochimiques est tel que tester l’incorporation de ce petit atome sur les molécules ciblées est devenu une routine en chimie médicinale. Par exemple, l’influence de l’atome de fluor sur le pKa d’un composé organofluoré peut se faire
sentir sur la biodisponibilité d’un médicament. En effet, l’acidité de la molécule joue parfois un rôle sur le processus d’absorption, un phénomène qui permet de contrôler la dose du médicament qui circule dans le système circulatoire. De même, la lipophilie est une propriété importante lors du design de médicaments parce qu’elle permet de contrôler le passage des molécules à travers les membranes lipidiques tout en évitant qu’elles demeurent trappées dans la cellule. L’influence de l’atome de fluor dépasse cependant les aspects physicochimiques et se fait également sentir sur la stabilité métabolique d’un médicament. Principalement, sa présence rend difficiles les mécanismes d’oxydation et d’hydrolyse, ce
8 Meanwell, N. A. J. Med. Chem. 2011, 54, 2529.
9 Levi, N.; Amir, D.; Gershonov, E.; Zafrani, Y. Synthesis 2019, 51, 4549.
10 (a) Champagne, P. A.; Desroches, J.; Paquin, J.-F. Synthesis 2015, 47, 306. (b) Schneider, H.-J. Chem. Sci.
2012, 3, 1381.
11 Fujiwara, T.; O’Hagan, D. J. Fluorine Chem. 2014, 167, 16.
12 (a) Vincent, J.-M. Chem. Commun. 2012, 48, 11382. (b) Berger, R.; Resnati, G.; Metrangolo, P.; Weber, E.;
qui ralentit l’élimination de la molécule fluorée. Cette stratégie permet également d’éviter la racémisation in vivo d’un composé bioactif, donc un meilleur contrôle de son activité biologique. Finalement, puisque les composés organofluorés ont des propriétés assez différentes, leur utilisation en tant qu’inhibiteur d’enzymes est également reconnue. La trifluridine I.2, commercialisée sous le nom Viroptic®, est d’ailleurs utilisée comme tel dans la lutte contre les infections oculaires provenant de l’herpès viral (Figure I. 2).
Figure I. 2. Viroptic® (Trifluridine)
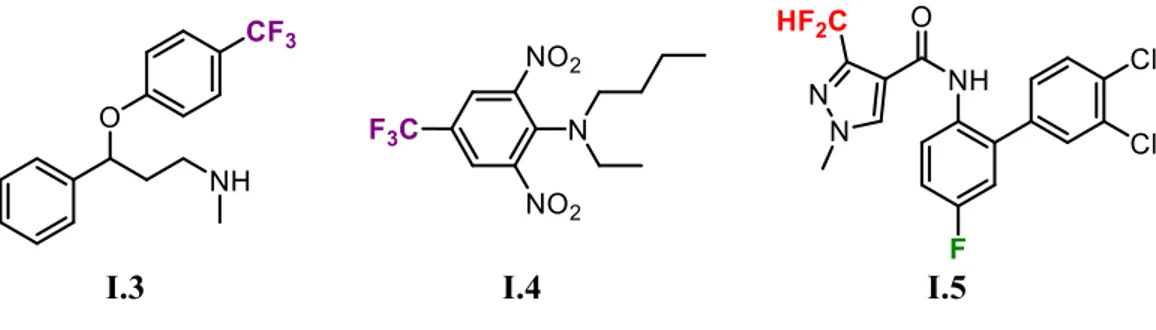
Différents groupements fluorés sont utilisés pour moduler l’activité des composés destinés à une application médicinale ou agrochimique. Notamment, la fluoxétine I.3 et la benfluraline I.4 contiennent tous deux un groupement -CF3, mais le premier est utilisé comme antidépresseur alors que le deuxième est un herbicide. Le bixafen I.4, lui, comporte à la fois un atome de fluor et un groupement -CF2H et est utilisé comme fongicide (Figure I. 3).
Les sciences des matériaux utilisent également des composés organofluorés. Alors que les deux premiers domaines se basent principalement sur les propriétés physicochimiques et la stabilité métabolique qu’induit un atome de fluor, les matériaux tirent surtout avantage de la grande stabilité chimique et thermique du lien C‒F.13 Depuis l’invention du polytétrafluoroéthylène (PTFE) dans les années 30, les molécules organofluorées sont fortement présentes dans les recherches sur les matériaux. En plus des polymères, ce type de composés est notamment utilisé dans les cristaux liquides, les réfrigérants et les cellules photovoltaïques (Figure I. 4).13
Figure I. 4. Différents composés fluorés utilisés en sciences des matériaux
Synthèse
Avec l’intérêt grandissant pour les composés organofluorés, plusieurs nouvelles méthodes de synthèse ont été développées au cours des dernières années.14 Bien que la création d’un lien C‒F comporte plusieurs défis, particulièrement parce qu’il est quasiment absent de la nature, plusieurs stratégies ont été développées pour fluorer des molécules organiques.5 Les trois principales sont la fluoration nucléophile, électrophile et radicalaire (Schéma I. 1). L’appellation « nucléophile » vient du fait que la fluoration se fait via l’attaque d’un ion fluorure sur un substrat agissant comme électrophile. Il s’agit de la forme de fluoration la plus fréquente. À l’inverse, la fluoration électrophile représente un substrat nucléophile qui attaquera un ion « F+ ». Il est cependant important de noter qu’un tel ion n’existe pas réellement : le transfert de l’atome de fluor se fait plutôt via une réaction SN2 ou
13 Harsanyi, A.; Sandford, G. Green Chem. 2015, 17, 2081.
14 Exemples de revues de la littérature sur la synthèse de composés organofluorés : (a) Furuya, T.; Kamlet, A. S.; Ritter, T. Nature 2011, 473, 410; (b) Liang, T.; Neumann, C. N.; Ritter, T. Angew. Chem. Int. Ed. 2013, 52, 8214; (c) Ma, J.-A.; Li, S. Org. Chem. Front. 2014, 1, 712; (d) Champagne, P. A.; Desroches, J.; Hamel, J.-D.; Vandamme, M.; Paquin, J.-F. Chem. Rev. 2015, 115, 9073; (e) Chatalova-Sazepin, C.; Hemelaere, R.; Paquin, J.-F.; Sammis, G. M. Synthesis 2015, 47, 2554; (f) Campbell, M. G.; Ritter, T. Chem. Rev. 2015, 115, 612.
via un transfert d’électron non apparié. Finalement, le dernier type de fluoration nécessite la formation in situ d’un carbone radicalaire qui réagira avec une source de fluor.
Schéma I. 1. Fluoration de molécules organiques
Déoxofluoration
La réaction de déoxofluoration est une forme de fluoration nucléophile qui permet de remplacer un atome d’oxygène par un atome de fluor. Ce type de réaction convertit donc les alcools en fluorures d’alkyle, les carbonyles en dérivés gem difluorés et les acides carboxyliques en fluorures d’acyle (Schéma I. 2).
Agents de déoxofluoration
Le SF4 était le réactif communément employé dans les années 60 pour effectuer de la déoxofluoration.15 Les dangers associés à ce gaz (toxicité et corrosion extrêmes) ont cependant incité à développer de nouveaux agents de déoxofluoration afin de faciliter les manipulations. Ainsi, en 1975, le groupe de Middleton a développé le trifluorure de diéthylaminosulfure (DAST), un réactif qui montre une grande réactivité envers différents substrats (Figure I. 5).16 Cependant, il a vite été établi que le DAST est un réactif instable thermiquement, voire explosif.17 Quelques années plus tard, le groupe de Lal a développé le Deoxo-Fluor®, un réactif qui se dégrade considérablement moins violemment que le DAST (Figure I. 5).18 Les deux analogues comportent cependant des dangers similaires, à savoir qu’ils réagissent violemment avec l’eau en plus de relâcher du HF libre, ce qui rend difficile l’utilisation de matériel fait de borosilicates.
Figure I. 5. Dérivés de trifluorure de dialkylaminosulfure
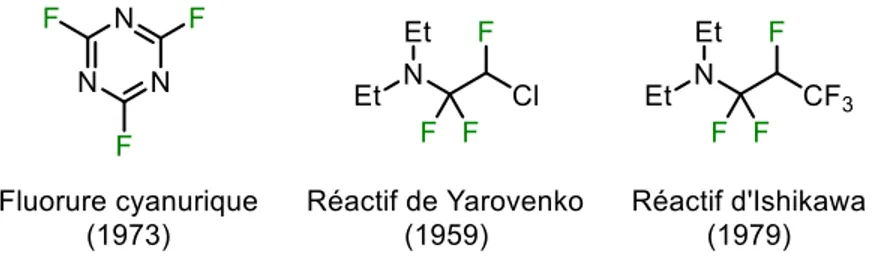
Les composés de types α-fluoroaminés ont été développés en parallèle, dont le fluorure cyanurique, le réactif de Yarovenko (2-chloro-N,N-diéthyl-1,1,2-trifluoroéthanamine) et le réactif d’Ishikawa (N,N-diéthyl-1,1,2,3,3,3-hexanfluoropropylamine), tous illustrés à la Figure I. 6.19,20,21 Bien que thermiquement plus stables, leur réactivité demeure cependant moins étendue que celle des réactifs de type trifluorure de dialkylaminosulfure présentés ci-haut.
15 Hasek, W. R.; Smith, W. C.; Engelhardt, V. A. J. Am. Chem. Soc. 1960, 82, 543. 16Middleton, W. J. J. Org. Chem. 1975, 40, 574.
17 (a) Middleton, W. J. Chem. Eng. News 1979, 57, 43; (b) Cochran, J. Chem. Eng. News 1979, 57, 74. 18 Lal, G. S.; Pez, G. P.; Pesaresi, R. J.; Prozonic, F. M.; Cheng, H. J. Org. Chem. 1999, 64, 7048. 19 Olah, G. A.; Nojima, M.; Kerekes, I. Synthesis 1973, 487.
20 Yarovenko, N. N.; Raksha, M. A. Zh. Obshch. Khim. 1959, 29, 2159. 21 Takaoka, A.; Iwakiri, H.; Ishikawa, N. Bull. Chem. Soc. Jpn. 1979, 52, 3377.
Figure I. 6. Dérivés α-fluoroaminés
XtalFluor®
Le XtalFluor® est un sel de type aminodifluorosulfinium qui a été développé en 2009 par la compagnie OmegaChem comme alternative plus sécuritaire aux réactifs de déoxofluoration connus jusqu’alors.22,23,24 Il en existe deux types : XtalFluor-E® et XtalFluor-M®, qui ont tous deux une structure cristalline (Figure I. 7).
Figure I. 7. XtalFluor®
Il est possible de synthétiser ce type de composés à partir du DAST ou de son analogue morpholino I.6 en utilisant l’éthérate de trifluorure de bore (BF3·Et2O) comme accepteur de fluorure (Schéma I. 3a). Une autre stratégie a cependant été développée pour éviter de distiller préalablement le DAST, une opération risquée. Dans cette seconde stratégie, le DAST est généré in situ à partir de diéthylamine triméthylsilylée I.7 et de SF4
22 Beaulieu, F.; Beauregard, L.-P.; Courchesne, G.; Couturier, M.; LaFlamme, F.; L’Heureux, A. Org.
Lett. 2009, 11, 5050.
23 L’Heureux, A.; Beaulieu, F.; Bennett, C.; Bill, D. B.; Clayton, S.; LaFlamme, F.; Mirmehrabi, M.; Tadayon,
S.; Tovell, D.; Couturier, M. J. Org. Chem. 2010, 75, 3401.
24 Mahé, O.; L’Heureux, A.; Couturier, M.; Bennett, C.; Clayton, S.; Tovell, D.; Beaulieu, F.; Paquin, J.-F. J.
(Schéma I. 3b). Dans les deux cas, les sels peuvent ensuite être récupérés par précipitation. La seconde voie de synthèse n’a cependant été appliquée qu’au XtalFluor-E®.
Schéma I. 3. Voie de synthèse du XtalFluor®
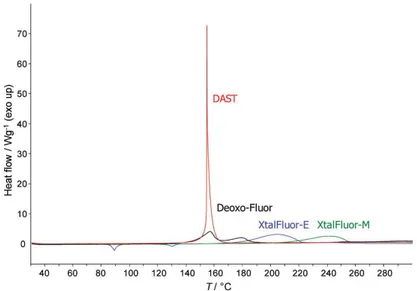
Contrairement à ses analogues, le XtalFluor® est nettement plus intéressant à manipuler puisqu’il est stable thermiquement. Selon les études de calorimétrie différentielle à balayage (en anglais, Differential Scanning Calorimetry ou DSC), le XtalFluor® se dégrade à des températures supérieures à celles du DAST et du Deoxo-fluor® et c’est d’autant plus vrai pour le XtalFluor-M® (Figure I. 8).23 De plus, les deux sels relarguent considérablement moins d’énergie lors de leur décomposition que le DAST. Leur forme cristalline les rend également moins contraignants à manipuler que l’aspect liquide et fumant des deux autres réactifs. Le XtalFluor-E® et le XtalFluor-M® sont également disponibles commercialement pour un prix raisonnable de 0,89 $/mmol et 1,08 $/mmol respectivement, ce qui les rend plus accessibles que le DAST et le Deoxo-Fluor® qui sont de moins en moins disponibles commercialement.25
Figure I. 8. Thermogramme DSC du DAST, Deoxo-Fluor® et XtalFluor® 23
En dépit du fait que leur structure soit très semblable, le DAST et le XtalFluor® ont des réactivités qui diffèrent. Alors que le premier est couramment utilisé seul, le second nécessite l’emploi d’un additif, généralement Et3N·3HF, parce qu’il ne libère pas d’ions fluorures après avoir été attaqué par le substrat. Le XtalFluor® s’avère également efficace lors de la fluoration de plusieurs types de composés (alcools primaires et secondaires variés, aldéhydes, cétones cycliques, acides carboxyliques) et est généralement plus sélectif que le DAST, qui tend à faire plus d’élimination (Schéma I. 4).
Bien qu’il ait été développé comme un agent de déoxofluoration, le XtalFluor-E® est également utilisé dans une vaste gamme de réactions organiques.26 Il peut aussi bien être utile dans des réactions de déshydratation (Schéma I. 5a),27 de formylation (Schéma I. 5b)28 ou d’expansion de cycle (Schéma I. 5c)29 que comme agent activant pour les alcools (Schéma
I. 5d)30 et acides, par exemple. Ce sont cependant les réactions d’halogénation qui seront concernées dans cet ouvrage.
Schéma I. 5. Utilisations du XtalFluor®
26 Pour une revue de la littérature sur l’usage du XtalFluor®, voir Mohammadkhani, L.; Heravi, M. M. J.
Fluorine Chem. 2019, 225, 11.
27 Keita, M.; Vandamme, M.; Mahé, O.; Paquin, J.-F. Tetrahedron Lett. 2015, 56, 461. 28 Roudias, M.; Vallée, F.; Martel, J.; Paquin, J.-F. J. Org. Chem. 2018, 83, 8731. 29 Cochi, A.; Pardo, D. G.; Cossy, J. Eur. J. Org. Chem. 2012, 2023.
Chimie verte
Depuis plusieurs années maintenant, la communauté scientifique s’intéresse de plus en plus à introduire le concept de développement durable au laboratoire dans le but de redorer l’image de la chimie. En effet, l’industrie chimique a tendance à être associée à différents effets néfastes sur l’environnement et l’humain. Ainsi, dans le but de ne pas compromettre les besoins des générations futures tout en comblant ceux des présentes générations, le concept de chimie verte a été mis au point. Pour faciliter l’application d’un tel concept, un guide regroupant 12 principes a d’ailleurs été développé dans les années 90 (Tableau I. 3).31
Tableau I. 3. Les 12 principes de la chimie verte
1. Prévention
2. Économie d’atomes
3. Produits chimiques utilisés moins dangereux 4. Design de produits plus sécuritaires
5. Emploi de solvants et auxiliaires plus sécuritaires 6. Demande énergétique minime
7. Emploi de ressources renouvelables
8. Quantités minimes de dérivés (réduire le nombre d’étapes d’une synthèse) 9. Catalyse
10. Dégradation des produits
11. Analyse en temps réel pour prévenir la pollution 12. Risques d’accident réduits
Parmi ces principes, certains sont plus facilement accessibles que d’autres. Par exemple, utiliser le XtalFluor® plutôt que le DAST répond au troisième et au dernier principe puisqu’il est moins toxique et plus stable thermiquement. Également, le choix de solvant est un critère qui peut facilement faire une différence. D’ailleurs, plusieurs guides ont été créés pour faciliter le remplacement d’un solvant nocif en prenant en considération plusieurs facteurs comme la sécurité et la toxicité qui y sont associées.32 Finalement, considérant que
31 Anastas, P. T.; Warner, J. C. Green Chemistry: Theory and Practice, Oxford University Press: New York,
NY, 1998, p. 30.
32 (a) Prat, D.; Hayler, J.; Wells, A. Green Chem., 2014, 16, 4546; (b) Alfonsi, K.; Colberg, J.; Dunn, P. J.;
Fevig, T.; Jennings, S.; Johnson, T. A.; Kleine, H. P.; Knight, C.; Nagy, M. A.; Perry, D.A.; Stefaniak, M.
Green Chem., 2008, 10, 31; (c) Henderson, R. K.; Jimenez-Gonzalez, C.; Constable, D. J. C.; Alston, S. R.;
Inglis, G. G. A.; Fisher, G.; Sherwood, J.; Binks, S. P.; Curzons, A. D. Green Chem., 2011, 13, 854; (d) Alder, C. M.; Hayler, J. D.; Henderson, R. K.; Redman, A. M.; Shukla, L.; Shuster, L. E.; Sneddon, H. F. Green Chem.,
la demande énergétique prend en considération le temps réactionnel et la température de chauffage d’une réaction, il devient assez accessible de modifier ces paramètres pour qu’ils respectent le sixième principe de chimie verte. Ces principes peuvent d’ailleurs être appliqués à la réaction de déoxofluoration avec le XtaFluor®.
Objectifs généraux
Alors que l’efficacité du XtalFluor® en tant qu’agent de déoxofluoration a été largement démontrée par OmegaChem, il n’en demeure pas moins que les conditions réactionnelles présentées sont loin d’être optimales dans un contexte de chimie verte. Certes, l’emploi du XtalFluor® réduit les risques associés aux autres réactifs de déoxofluoration communs tels que le DAST et le Deoxo-Fluor®, mais il comporte tout de même quelques dangers. Premièrement, le solvant utilisé est le dichlorométhane (CH2Cl2), un solvant toxique et potentiellement cancérigène. Également, pour que la réaction fonctionne, il doit y avoir une source de fluorure dans le milieu. Dans ce cas, la source choisie par OmegaChem est Et3N·3HF. Bien qu’il soit moins dommageable pour le matériel borosilicaté que le HF libre, il s’agit tout de même d’un réactif corrosif et certains dangers y sont donc associés. Ainsi, ce projet de maitrise visait à développer ces conditions pour qu’elles soient plus sécuritaires. Cette optimisation sera présentée pour la synthèse de deux motifs fluorés : les fluorures d’acyle33 (Chapitre 1) et le groupement difluorométhyle (Chapitre 2).
Chapitre 1. Synthèse de fluorures d’acyle
Parmi les nombreux motifs fluorés, les fluorures d’acyle sont loin d’être les premiers à venir à l’esprit. Pourtant, ils gagnent à être connus parce qu’ils peuvent être particulièrement utiles comme intermédiaire en chimie organique. Par exemple, il est possible d’employer le fluorure de benzoyle dans la synthèse de rifamycines, des antibiotiques montrant une activité anti-inflammatoire et antitumorale et qui sont utilisés dans le traitement de la tuberculose (Schéma 1. 1).34
Schéma 1. 1. Synthèse de la rifamycine S
Les tubulysines, qui représentent des agents anticancéreux, peuvent également être synthétisées à partir de fluorures d’acides dérivés d’acides aminés. Le groupe de Nicolaou a d’ailleurs fait la synthèse d’une centaine d’analogues de tubulysines.35,36 La synthèse d’un analogue de la pretubulysine D est illustrée au Schéma 1. 2.36
34 Zakzeski, J.; Fan, I. S.; Bell, A. T. Appl. Catal., A 2009, 360, 33.
35 Nicolaou, K. C.; Yin, J.; Mandal, D.; Erande, R. D.; Klahn, P.; Jin, M.; Aujay, M.; Sandoval, J.; Gavrilyuk,
J.; Vourloumis, D. J. Am. Chem. Soc. 2016, 138, 1698.
36 Nicolaou, K. C.; Erande, R. D.; Yin, J.; Vourloumis, D.; Aujay, M.; Sandoval, J.; Munneke, S.; Gavrilyuk, J.
Schéma 1. 2. Synthèse d’un analogue de la pretubulysine D
1.1 Caractéristiques des fluorures d’acyle
Les fluorures d’acyle 1.8 sont des dérivés des acides carboxyliques et contiennent donc un carbonyle fluoré (Figure 1. 1).
Ces composés ont des propriétés particulières qui les avantagent par rapport aux autres intermédiaires de synthèse typiques, tels que les chlorures d’acide et les anhydrides.37 Notamment, parce que le lien C‒F est plus stable que le lien C‒Cl, les fluorures d’acide sont moins réactifs face aux atomes d’oxygène neutres, tels que l’eau et les alcools. Ils ont, cependant, la même réactivité que leurs analogues chlorés face aux amines nucléophiles et aux atomes d’oxygène chargés. D’ailleurs, l’emploi des fluorures d’acyle permet parfois d’éviter des réactions secondaires ou de la racémisation. Leur stabilité supérieure fait également d’eux des intermédiaires plus facilement isolables par colonne chromatographique. La petite taille de l’atome de fluor représente aussi un autre avantage des fluorures d’acyle parce qu’elle réduit l’encombrement stérique lors de l’approche d’un nucléophile. L’accès à différents composés fortement encombrés tels que des amides, des esters et des thioesters est donc rendu possible. Le groupe d’Ulven a d’ailleurs montré que l’utilisation de fluorures d’acyle pour le couplage de l’acide triphénylacétique 1.9 avec la propargylamine 1.10 a permis d’obtenir l’amide 1.11 avec un rendement nettement supérieur à ceux qui ont été obtenus avec d’autres méthodes de couplage (Schéma 1. 3).38 Entre autres, pour la même réaction, l’utilisation du N,N'-Dicyclohexylcarbodiimide (DCC) permettait d’obtenir un rendement de 50 %, ce qui représente une perte de presque 30 % de rendement par rapport à l’utilisation d’un fluorure d’acyle.39 C’est d’ailleurs pourquoi la synthèse peptidique est une des principales applications des fluorures d’acyle. Utiliser ce type de composé dans ce contexte représente une méthode efficace, que ce soit sur support solide ou en solution. Ce motif fluoré est également utilisé comme peptidomimétique ou encore dans d’autres contextes, comme la construction d’hétérocycles.40
37 (a) Barda, D. A. Cyanuric fluoride. Dans Encyclopedia of Reagents for Organic Synthesis, John Wiley &
Sons, Ltd.: Hoboken, NJ, 2002; p 1. (b) Carpino, L. A.; Beyermann, M.; Wenschuh, H.; Bienert, M. Acc. Chem.
Res. 1996, 29, 268. (c) Montalbetti, C. A. G. N.; Falque, V. Tetrahedron 2005, 61, 10827.
38 Due-Hansen, M. E.; Pandey, S. K.; Christiansen, E.; Andersen, R.; Hansen, S. V. F.; Ulven, T. Org. Biomol.
Chem. 2016, 14, 430.
39 Kizjakina, K.; Bryson, J.M.; Grandinetti, G.; Reineke, T. M. Biomaterials, 2012, 33, 1851.
40 Dovgan, I.; Ursuegui, S.; Erb, S.; Michel, C.; Kolodych, S.; Cianferani, S.; Wagner, A. Bioconjugate Chem.
Schéma 1. 3. Synthèse d'un amide stériquement encombré via un fluorure d'acyle
1.2 État de l’art
Les méthodes classiques de synthèse de fluorures d’acyle recourent à l’échange d’halogènes à partir de chlorures d’acyle (Schéma 1. 4a). Pour se faire, des réactifs fluorés tels que le fluorure de potassium (KF), le bifluorure de potassium (KHF2) et le fluorure d’hydrogène (HF) sont couramment utilisés.41 En raison de l’instabilité des chlorures d’acyle, la synthèse de fluorures d’acide se fait, cependant, de plus en plus à partir des acides carboxyliques.42 Les réactifs de déoxofluoration présentés en introduction se sont montrés efficaces pour effectuer ce type de transformation (Schéma 1. 4b). Par exemple, le fluorure de benzoyle a été obtenu avec de bons rendements que ce soit en utilisant le DAST (85 %), le Deoxo-Fluor® (96 %), le fluorure cyanurique (97 %) ou encore le réactif d’Ishikawa (86 %).18,19,21,43 Encore aujourd’hui, il s’agit d’un motif fluoré d’intérêt pour la communauté scientifique et de nouvelles voies synthétiques sont régulièrement mises de l’avant.
41 Chen, C.; Chien, C.-T.; Su, C.-H. J. Fluorine Chem. 2002, 115, 75.
42 Pour une revue sur les méthodes de synthèse des fluorures d’acyles, voir Gonay, M.; Batisse, C.; Paquin, J.-F.
Soumis le 03/09/2020.
43 Hudlicky, M. Fluorination with diethylaminosulfur trifluoride and related aminofluorosulfuranes. Dans
Schéma 1. 4. Stratégies de synthèse des fluorures d'acyle
Le groupe de Schoenebeck a d’ailleurs développé un nouveau réactif soufré, le Me4NSCF3, qui permet d’obtenir d’excellents rendements sur une grande variété de fluorures d’acyle 1.8 à partir d’acides carboxyliques 1.12.44 Selon leurs conditions réactionnelles, la réaction se fait dans le dichlorométhane (CH2Cl2), à température ambiante et sur une courte période (Schéma 1. 5). Ils présentent ainsi une vingtaine de fluorures d’acyle synthétisés via cette méthode avec des rendements variant entre 69 et 98 %. Ce réactif, comparé au DAST et au Deoxo-Fluor® est plus stable en plus d’être sous forme solide. Il est également tolérant aux différents groupements fonctionnels, ne nécessite pas d’additif et peut être purifié par filtration sur silice. Cependant, il s’agit d’un réactif dispendieux (126 $/mmol)45 et difficile à synthétiser.46
Schéma 1. 5. Synthèse des fluorures d'acyle par le groupe de Schoenebeck
44 Scattolin, T.; Deckers, K.; Schoenebeck, F. Org. Lett. 2017, 19, 5740. 45 Prix en vigueur pour 1 g le 31/08/2020 par AstaTech en CAD.
Le groupe de Prakash, lui, utilise plutôt des réactifs beaucoup plus communs, tels que la triphénylphosphine (PPh3) et le N-Bromosuccinimide (NBS), combinés à une source de fluorure.47 Les conditions réactionnelles sont très semblables à celles présentées par le groupe de Schoenebeck (Schéma 1. 6). Cette méthode est également efficace pour plusieurs types de fluorures d’acyle et permet d’obtenir de bons rendements en général.
Schéma 1. 6. Voie de synthèse développée par Prakash
Il est également possible de synthétiser des fluorures d’acyle par fluoration radicalaire. Un des réactifs spécifiques de ce type de fluoration est le N-fluorobenzenesulfonimide (NFSI). C’est d’ailleurs le réactif que Martin et Britton utilisent pour fluorer un aldéhyde.48 En effet, leurs travaux présentent l’utilisation du décatungstène de sodium (Na4W10O32), un photocatalyseur, pour effectuer l’abstraction de l’hydrogène d’un aldéhyde 1.13 et ainsi former un acétyle radicalaire 1.14 qui peut trapper un atome de fluor. (Schéma 1. 7). Cette méthode est efficace pour plusieurs types d’aldéhydes, à l’exception des aldéhydes hétéroaromatiques en raison de l’incompatibilité du NFSI avec les atomes d’azote nucléophiles.
Schéma 1. 7. Fluoration radicalaire d'un aldéhyde par le groupe de Martin et Britton
47 Munoz, S. B.; Dang, H.; Ispizua-Rodriguez, X.; Mathew, T.; Prakash, G. K. S. Org. Lett. 2019, 21, 1659. 48 Meanwell, M.; Lehmann, J.; Eichenberger, M.; Martin, R. E.; Britton, R. Chem. Commun. 2018, 54, 9985.
OmegaChem a montré que le XtalFluor® peut également être utilisé pour synthétiser des fluorures d’acyle aromatiques 1.16 et aliphatiques 1.18.22-24 Bien qu’il ne s’agisse que de deux exemples, les rendements obtenus sont toutefois excellents, que ce soit avec le XtalFluor-E® ou le XtalFluor-M® (Schéma 1. 8). Les groupes de Zhang et de Schoenebeck ainsi que le groupe de Riant en collaboration avec Gagosz ont également utilisé cette méthode afin de synthétiser des fluorures d’acyles aromatiques.49 Les problèmes associés à la méthode sont cependant les mêmes que présentés en introduction, ce qui la rend loin d’être idéale pour une application industrielle.
Schéma 1. 8. Synthèse des fluorures d'acyle avec le XtalFluor®
1.3 Objectifs spécifiques
Tel qu’énoncé dans les objectifs généraux, ce projet de maitrise visait l’amélioration des conditions présentées par OmegaChem pour rendre ce type de transformation plus accessible dans une optique de chimie verte. Concrètement, cela correspond à miser sur l’utilisation des sels comme source de fluorure et sur un solvant moins nocif pour synthétiser différents fluorures d’acyle. Idéalement, le projet visait également à éviter les températures extrêmes dans le but de minimiser la demande énergétique de la réaction. La même optique
49 Han, J.; Zhou, W.; Zhang, P.-C.; Wang, H.; Zhang, R.; wu, H.-H.; Zhang, J. ACS Catal. 2019, 9, 6890; (b)
Keaveney, S. T.; Schoenebeck, F. Angew. Chem., Int. Ed. 2018, 57, 4073; (c) Boreux, A.; Indukuri, K.; Gagosz, F.; Riant, O. ACS Catal. 2017, 7, 8200.
a été appliquée au temps réactionnel. Une étendue de réaction était également envisagée pour élargir la bibliothèque de fluorures d’acyle accessibles via l’utilisation du XtalFluor®. Ainsi, les conditions optimisées ont été appliquées à des acides carboxyliques aromatiques, hétéroaromatiques et aliphatiques.
1.4 Optimisation
Pour optimiser les conditions réactionnelles, il fallait s’assurer de contrôler tous les paramètres de la réaction et de n’en faire varier qu’un seul à la fois. Ainsi, les paramètres étudiés étaient le choix de solvant, la concentration, le nombre d’équivalents des réactifs, le temps de réaction et la température. Pour chacun des tests, l’acide 4-phénylbenzoïque 1.19 a été choisi comme substrat modèle parce que le dérivé fluoré 1.20 était connu de la littérature et qu’il s’agit d’un substrat simple, sans groupements fonctionnels sensibles et peu volatil. Sa réactivité peut donc se généraliser à plusieurs autres substrats plus facilement. Pour optimiser les différents paramètres, seuls les rendements RMN 19F ont été comparés pour simplifier les manipulations. Pour ce faire, il a fallu choisir un étalon interne qui ne réagirait pas dans les conditions réactionnelles en plus d’avoir un déplacement chimique dans une plage semblable à celle des fluorures d’acide. Le 2-fluoro-4-nitrotoluène était un bon choix dans ce contexte avec un déplacement se situant à -112,9 ppm en RMN 19F. Il suffisait d’ajouter une quantité connue précisément de cet étalon interne au milieu réactionnel pour ensuite calculer le ratio des intégrations des pics du produit et de la référence. Ce ratio, en fonction du nombre de moles de chacun des composés, permet ensuite d’obtenir un rendement de réaction. Par conséquent, pour le même nombre d’équivalents de la référence interne et du fluorure d’acyle, un ratio d’intégration de 1:1 signifierait un rendement RMN de 100 %.
1.4.1 Effet du solvant
Le premier paramètre qui a été optimisé est le choix de solvant. Pour ce faire, une gamme de solvant a été sélectionnée en fonction des dangers qui sont associés à chacun d’entre eux. L’acétate d’éthyle (AcOEt) a été choisi parce qu’il s’agit d’un solvant vert. Il est
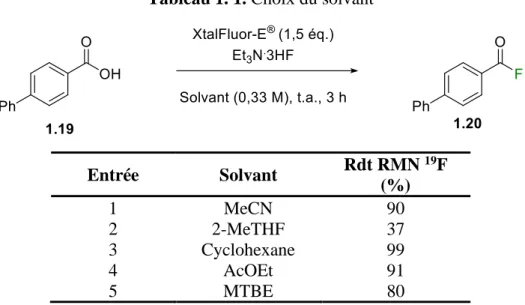
donc plus sécuritaire à manipuler et moins dangereux pour la santé et pour l’environnement. Les alcools, qui sont généralement des solvants verts, n’ont pas été étudiés en raison de leur réactivité envers le XtalFluor-E®. Les autres solvants sélectionnés ne sont pas considérés comme verts, mais ils sont tout de même moins dommageables que le dichlorométhane.32 Chaque solvant a ensuite été testé dans les conditions d’OmegaChem pour bien identifier celui qui donnait les résultats les plus prometteurs (Tableau 1. 1). À la lumière des résultats obtenus, le cyclohexane est le solvant qui permettait d’obtenir le meilleur rendement RMN (entrée 3). Cependant, étant donné que l’AcOEt est un solvant beaucoup plus sécuritaire et qu’il permet d’obtenir un excellent rendement RMN de 91 % (entrée 4), il semblait évident de continuer l’optimisation avec ce solvant.
Tableau 1. 1. Choix du solvant
Entrée Solvant Rdt RMN 19F (%) 1 MeCN 90 2 2-MeTHF 37 3 Cyclohexane 99 4 AcOEt 91 5 MTBE 80
1.4.2 Choix de la source de fluorure
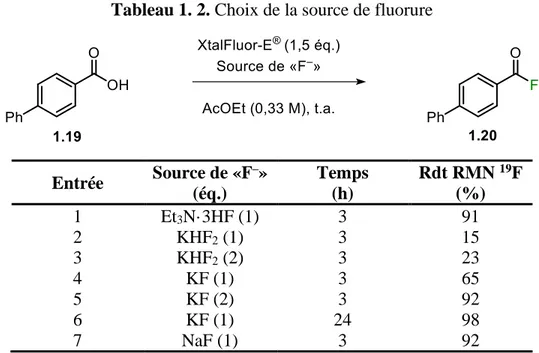
Une fois le solvant choisi, il a été possible de s’intéresser à la source de fluorure. Tel que mentionné dans les objectifs du projet, un sel de fluorure était préférable à l’emploi de Et3N·3HF. Afin de mieux comparer l’effet de la source de fluorure dans l’AcOEt, l’entrée 4 du tableau précédent été reportée au Tableau 1. 2 (entrée 1). KHF2 a été le premier à être testé, mais les rendements RMN obtenus étaient particulièrement faibles, et ce, même en doublant le nombre d’équivalents (entrée 2 et 3). Le fluorure de potassium (KF) permet
d’obtenir de meilleurs rendements RMN, particulièrement avec deux équivalents ou en augmentant le temps de réaction (entrées 4-6). Cependant, le résultat le plus intéressant a été obtenu avec le fluorure de sodium (NaF) avec un rendement RMN de 92 % pour un seul équivalent en seulement 3 h (entrée 7). Ce sel a donc été sélectionné pour la suite de l’optimisation.
Tableau 1. 2. Choix de la source de fluorure
Entrée Source de «F –» (éq.) Temps (h) Rdt RMN 19F (%) 1 Et3N·3HF (1) 3 91 2 KHF2 (1) 3 15 3 KHF2 (2) 3 23 4 KF (1) 3 65 5 KF (2) 3 92 6 KF (1) 24 98 7 NaF (1) 3 92 1.4.3 Optimisation de la température
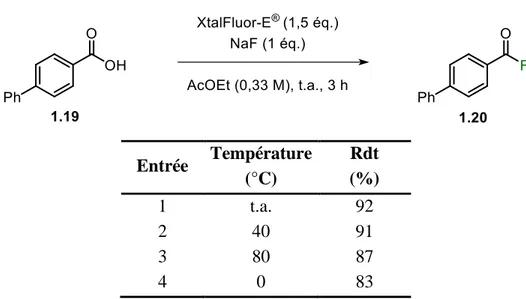
L’influence de la température sur la réaction a ensuite été étudiée. Chauffer la réaction ne semble pas affecter grandement les rendements (différence de maximum 5 % par rapport au rendement obtenu à température ambiante, Tableau 1. 3, entrées 2 et 3), tandis que son refroidissement accuse une baisse de rendement (entrée 4). Ainsi, les résultats obtenus précédemment à température ambiante semblent être les plus prometteurs.
Tableau 1. 3. Influence de la température Entrée Température (°C) Rdt (%) 1 t.a. 92 2 40 91 3 80 87 4 0 83
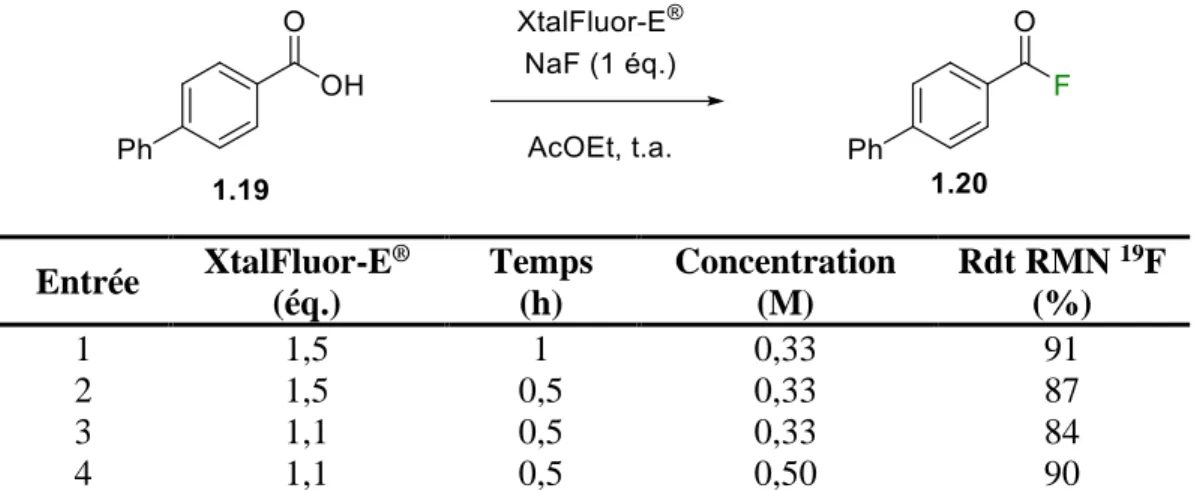
1.4.4 Optimisation du temps, du XtalFluor-E® et de la concentration
La prochaine étape a été d’évaluer l’impact d’un temps réactionnel plus court sur le rendement RMN (entrées 1 et 2). Il s’est avéré que sensiblement le même rendement a été obtenu en une heure et une légère baisse de rendement a été observée après 30 minutes, mais rien d’assez significatif pour justifier de continuer l’optimisation avec 1 h. La quantité de XtalFluor-E® a ensuite été diminuée à 1,1 équivalent, ce qui a amené une baisse minime de rendement (entrée 3).50 Il faut cependant garder en tête que ce projet vise, entre autres, une application industrielle dans un contexte de chimie verte. Ainsi, utiliser 0,4 éq. de moins pourrait économiser une quantité non négligeable de réactif (et d’argent) lors d’une synthèse sur grosse échelle. Ces conditions sont donc nettement plus intéressantes dans ce contexte. Finalement, la concentration du substrat a été étudiée (entrée 5). Il faut savoir qu’une concentration plus élevée signifie que moins de solvant est employé. Encore une fois, concentrer légèrement la réaction pourrait avoir un gros impact pour l’industrie. Il s’est avéré qu’une concentration de 0,50 M permet d’ailleurs d’augmenter le rendement RMN jusqu’à 90 %.
50 À noter que le XtalFluor-M® est généralement moins efficace que le XtalFluor-E®. C’est pour cette raison
Tableau 1. 4. Optimisation du temps réactionnel, du XtalFluor-E® et de la concentration Entrée XtalFluor-E ® (éq.) Temps (h) Concentration (M) Rdt RMN 19F (%) 1 1,5 1 0,33 91 2 1,5 0,5 0,33 87 3 1,1 0,5 0,33 84 4 1,1 0,5 0,50 90
1.4.5 Optimisation d’un système catalytique
À la lumière de ces excellents résultats, un système réactionnel dans lequel NaF serait présent en quantité catalytique a été étudié (Tableau 1. 5). L’intérêt d’un tel système réside dans le fait qu’il permet d’utiliser une quantité moindre de réactif. Le premier test a été réalisé en 3 h avec 0,25 éq. de NaF et a permis d’obtenir un rendement RMN assez prometteur pour chercher à pousser les conditions réactionnelles (entrée 1). Il s’est avéré qu’un temps réactionnel plus long permettait d’augmenter considérablement le rendement RMN (entrées
2-5). Bien que les résultats semblaient déjà excellents à partir de 8 h de réaction, un temps de
24 h a été préféré pour des raisons pratiques et également pour s’assurer d’avoir des conditions qui pourraient s’appliquer à plus de substrats, donc plus universelles. La charge catalytique a ensuite été diminuée à 0,10 et 0,05 éq. (entrées 6 et 7). Comme des rendements presque quantitatifs ont été obtenus dans les deux cas, ce sont plutôt des raisons pratiques en laboratoire qui ont mené au choix d’utiliser une charge catalytique de 0,10 éq. En effet, utiliser une charge de 0,05 éq. pour une échelle réactionnelle de 1 mmol revient à peser 2 mg de NaF, une masse infime qui rend les manipulations difficiles.
Tableau 1. 5. Optimisation du système catalytique Entrée NaF (éq.) Temps (h) Rdt RMN 19F (%) 1 0,25 3 47 2 0,25 5 86 3 0,25 8 94 4 0,25 16 94 5 0,25 24 98 6 0,10 24 99 7 0,05 24 99
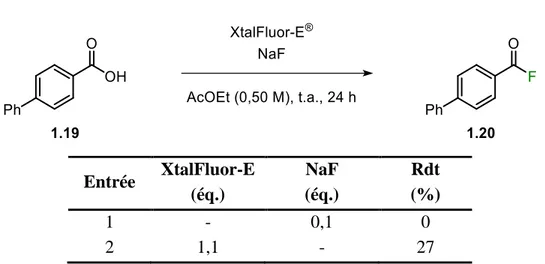
Des tests contrôles ont été effectués à cette étape afin de bien évaluer l’impact des deux réactifs (Tableau 1. 6). Selon les résultats obtenus, le XtalFluor-E® est bel et bien essentiel à la réaction puisque, même avec un léger excès de NaF, le rendement demeure nul sans lui. Également, NaF améliore grandement l’efficacité de la méthode : le faible rendement obtenu sans NaF laisse croire que le XtalFluor-E® relargue modérément des ions fluorures, mais pas assez pour compléter la réaction.
Tableau 1. 6. Tests contrôles
Entrée XtalFluor-E (éq.) NaF (éq.) Rdt (%) 1 - 0,1 0 2 1,1 - 27
1.5 Hypothèses mécanistiques
Après avoir constaté qu’un système catalytique fonctionne aussi bien qu’un système stœchiométrique, des hypothèses mécanistiques basées sur des intermédiaires connus ont été émises pour essayer de justifier cette observation.43,51 Premièrement, il faut savoir que le mécanisme de déoxofluoration d’alcools 1.21 débute par l’attaque du groupement hydroxyle sur l’atome de soufre du XtalFluor-E®, permettant dans ce cas le rabat des électrons de la double liaison sur l’atome d’azote, tel qu’illustré au Schéma 1. 9. Ainsi, il est tout à fait raisonnable d’envisager que le mécanisme de fluoration d’un acide carboxylique pourrait inclure un intermédiaire semblable.
Schéma 1. 9. Intermédiaire connu de la déoxofluoration d'alcools avec le XtalFluor-E®
Il a été montré avec les trifluorures de dialkylaminosulfures que le mécanisme de déoxofluoration d’acides carboxyliques 1.12 est analogue à celui de la déoxofluoration d’alcool.43 Dans le cas du XtalFluor®, il est ainsi possible d’envisager que la première étape soit la même que pour la déoxofluoration d’alcools, c’est-à-dire l’attaque du groupement hydroxyle sur l’atome de soufre du XtalFluor-E® pour former l’intermédiaire 1.23 (Schéma
1. 10, voie A). Cependant, il pourrait aussi s’agir de la formation du DAST in situ par
l’attaque du NaF sur le XtalFluor-E®.52 Le DAST réagirait ensuite avec l’acide carboxylique pour revenir au même intermédiaire (voie B). Pour vérifier cette hypothèse, OmegaChem a étudié la réaction d’un mélange 1 : 1 de XtalFluor-E® et de Et3N·3HF dans le dichlorométhane deutéré et n’a jamais observé le pic caractéristique du DAST par RMN.22 En conséquence, la voie B est peu probable, mais les études mécanistiques à ce sujet sont
51 Sutherland, A.; Vederas, J. C. Chem. Commun., 1999, 17, 1739.
52 (a) Mahé, O.; Desroches, J.; Paquin, J.-F. Eur. J. Org. Chem., 2013, 20, 4325; (b) Orliac, A.; Gomez Pardo,
D.; Bombrun, A.; Cossy, J. Org. Lett., 2013, 15, 902; (c) Pouliot, M.-F.; Angers, L.; Hamel, J.-D.; Paquin, J.-F.
Org. Biomol. Chem. 2012, 10, 988; (d) Vandamme, M.; Bouchard, L.; Gilbert, A.; Keita, M.; Paquin, J.-F. Org. Lett. 2016, 18, 6468.
encore trop peu nombreuses pour pouvoir éliminer totalement cette voie. Quoi qu’il en soit, dans les deux cas, un ion fluorure provenant du NaF (voie A) ou du DAST (voie B) pourrait ensuite attaquer le carbone du carbonyle pour former le fluorure d’acyle 1.8. Ce faisant, le XtalFluor-E® formerait le dérivé sulfoxide 1.24 en relarguant un ion fluorure. Ce dernier pourrait ensuite perpétuer le cycle catalytique, ce qui expliquerait les résultats présentés précédemment.
Tel que mentionné plus haut, les tests contrôles ont révélé que la présence du XtalFluor-E® est essentielle à la réaction. Ce résultat concorderait donc avec l’hypothèse émise selon laquelle le XtalFluor-E® activerait l’acide carboxylique, que ce soit sous sa forme initiale ou sous forme de DAST.
Schéma 1. 10. Mécanismes supposés pour la déoxofluoration d'acides carboxyliques
1.6 Étendue de réaction
1.6.1 Fluorures d’acyles
Après avoir formulé une hypothèse quant au fonctionnement du système, l’étape suivante était de vérifier l’applicabilité de la méthode à une variété d’acides carboxyliques. Tel que montré par l’optimisation, deux systèmes différents ont été développés : un stœchiométrique et un catalytique. Le premier présente l’avantage d’être particulièrement rapide (30 minutes) tandis que le deuxième permet d’utiliser une quantité moindre de NaF
(0,1 éq.), deux caractéristiques qui peuvent être précieuses pour une application à grosse échelle. Ainsi, les deux systèmes ont d’abord été testés en parallèle pour chaque acide carboxylique étudié. Après avoir analysé quelques substrats, il a vite été constaté que le système catalytique permettait d’obtenir des rendements généralement meilleurs (Tableau
1. 7). Il a donc été décidé de poursuivre l’étendue de réaction avec une quantité catalytique
de fluorure de sodium.
Tableau 1. 7. Système catalytique vs stœchiométrique
Entrée Substrats Produits Rendements
a Stœchiométrique Catalytique 1 1.19 80 94 2 1.25 65 91 3 1.26 67 83 4 1.27 73 96 5 1.28 89 87 6 1.29 17 76
a Conditions stœchiométriques : 1 éq. de NaF pendant 0,5 h; conditions