© Noémie Jean-Leblanc, 2019
La voie canonique Wnt est nécessaire pour le maintien
de l'intégrité de la barrière hémato-encéphalique après
un accident vasculaire cérébral: impacts sur la thérapie
thrombolytique
Mémoire
Noémie Jean-Leblanc
Maîtrise en neurobiologie - avec mémoire
Maître ès sciences (M. Sc.)
iii
Résumé
L'accident vasculaire cérébral (AVC) déclenche une perturbation de la barrière hémato-encéphalique (BHE) et entrave la récupération des tissus en altérant le microenvironnement cérébral local. L'administration de l'activateur tissulaire du plasminogène (rtPA) dans une fenêtre thérapeutique étroite de 4,5 heures après l'AVC demeure le seul traitement existant. Au-delà de cette fenêtre, le tPA aggrave la perturbation de la BHE et provoque des transformations hémorragiques. La voie canonique Wnt est connue comme induisant la formation et la maturation de la BHE pendant l'ontogenèse. Nous émettons l'hypothèse que la voie est nécessaire pour maintenir l'intégrité de la BHE après un AVC et que son activation pourrait constituer une approche prometteuse pour améliorer le traitement par rtPA. Ainsi, nous avons d'abord évalué l'activité de la voie dans le cerveau de souris soumises à un modèle d’AVC. Ensuite, nous avons évalué l’effet de la désactivation de la voie sur l’intégrité de la BHE ainsi que son activation dans un contexte. d'administration retardée de rtPA. Nos résultats montrent que l'activité de la voie est induite spécifiquement dans les cellules endothéliales cérébrales après un AVC ischémique. La désactivation de la voie par un inhibiteur aggrave la dégradation de la BHE et augmente l'incidence des transformations hémorragiques spontanées sans affecter l’infarct. En revanche, l'activation de la voie par un activateur spécifique, la 6-bromoindirubine-3'-oxime (6-BIO), atténue la dégradation de la BHE et réduit l'incidence des transformations hémorragiques associées à l'administration retardée du rtPA en induisant l’expression d’une protéine des jonctions serrées (claudine 3) et atténue la perméabilité basale endothéliale en réprimant l'expression de PLVAP, sans affecter l'infarctus, la vascularisation ou l'inflammation du cerveau. Notre étude démontre que l'activation de la voie canonique Wnt constitue une stratégie cliniquement pertinente pour étendre la fenêtre thérapeutique du rtPA en atténuant la dégradation de la BHE via la régulation des mécanismes spécifiques à la BHE.
iv
Abstract
Stroke triggers blood-brain barrier (BBB) disruption and hampers tissue recovery by impairing the local brain microenvironment. Administration of recombinant tissue plasminogen activator (rtPA) within a therapeutic window of 4.5 hours after onset constitutes the only existing treatment. Beyond this window, tPA worsens BBB disruption and causes haemorrhagic transformation. Canonical Wnt pathway induces BBB formation during ontogeny. We hypothesize here that pathway activity is required to maintain BBB integrity after stroke and that its activation might constitute a promising approach to improve rtPA therapy via protection of the BBB. Therefore, we have first assessed pathway activity in the brain of mice subjected to transient middle cerebral artery occlusion (MCAo). Next, we have evaluated the effect of pathway deactivation early after stroke on BBB integrity. Finally, we have assessed the potential of pathway activation on BBB breakdown associated to the delayed administration of rtPA. Our results show that pathway activity is induced specifically in brain endothelial cells early after ischemic stroke. Early deactivation of the pathway using a potent inhibitor, XAV939, aggravates BBB breakdown, and increases the incidence of spontaneous haemorrhagic transformation, without affecting brain infarct. On the other hand, pathway activation using a potent specific activator, 6-Bromoindirubin-3’-oxime (6-BIO), attenuates BBB breakdown, and reduces the incidence of haemorrhagic transformation associated to delayed rtPA administration by inducing expression of the tight junction claudin-3, and attenuates endothelial basal permeability by repressing the expression of PLVAP, without affecting brain infarct, vascularization and inflammation. Our study demonstrates that activation of the canonical Wnt pathway constitutes a clinically relevant strategy to extend the therapeutic window of rtPA by attenuating BBB breakdown via regulation of BBB-specific mechanisms.
v
Table des matières
Résumé ... iii
Table des matières ... v
Liste des figures ... vii
Liste des abréviations ... viii
Remerciements ... x
Avant-propos ... xi
Chapitre 1 ... 1
1. Introduction ... 1
1.1 Les accidents vasculaires cérébraux ... 1
1.2 L'ischémie cérébrale ... 1
1.2.1 L'unité neurovasculaire en condition physiologique ... 2
1.2.2 L'unité neurovasculaire en condition ischémique ... 7
1.3 Le système de l'activateur tissulaire du plasminogène ... 18
1.3.1 Fonction du système de l’activateur tissulaire du plasminogène ... 19
1.3.2 L'action de l'activateur tissulaire du plasminogène sur la barrière hémato-encéphalique ... 21
1.3.3 L'activateur tissulaire du plasminogène dans les accidents vasculaires cérébraux et ses limitations ... 23
1.4 La voie de signalisation Wnt/ β-caténine ... 23
1.4.1 Fonctions de la voie Wnt ... 25
1.4.2 Fonctions de la voie Wnt dans la barrière hémato-encéphalique ... 26
1.4.3 La voie Wnt et l'accident vasculaire cérébral ... 27
1.5 Hypothèse de travail et objectifs spécifiques ... 27
Chapitre 2 ... 30
2. Canonical Wnt pathway maintains blood-brain barrier integrity upon ischemic stroke and its activation ameliorates tissue-plasminogen activator therapy ... 30
2.1 Résumé ... 31
2.2 Abstract ... 32
2.3 Introduction ... 33
2.4 Material and Methods ... 35
2.4.1 Animal experiments ... 35
2.4.2 Tissue processing ... 36
2.4.3 Transmission Electron Microscopy ... 37
2.4.4 Isolation of brain capillaries ... 38
2.4.5 Analysis of brain injury, IgG extravasation and haemorrhagic transformation 38 2.4.6 Fluoro-jade B staining ... 39
2.4.7 ELISA assay ... 40
vi
2.4.9 Immunofluorescence analysis ... 41
2.4.10 Cell-based assays ... 42
2.4.11 Statistical analysis ... 44
2.5 Results ... 45
2.5.1 Pathway activity is detectible predominately in the brain endothelial cells and re-emerges upon ischemic stroke ... 45
2.5.2 Early pathway deactivation aggravates BBB breakdown and increases incidence of spontaneous haemorrhagic transformation ... 47
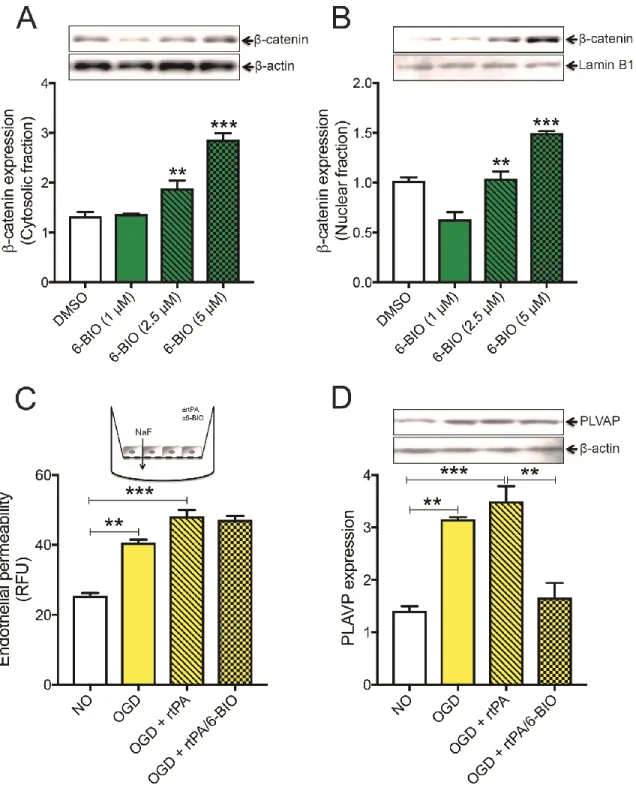
2.5.3 6-BIO dose dependently activates the pathway in brain endothelial cells and reverses OGD-induced endothelial basal permeability ... 47
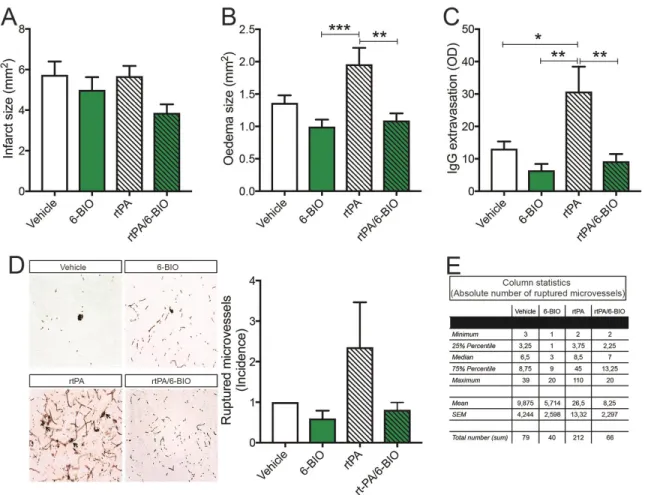
2.5.4 Pathway activation increases BBB integrity and reduces incidence of rtPA-induced perivascular bleeding ... 49
2.5.5 6-BIO administration does not influence ischemic stroke-associated neuronal injury, angiogenesis and inflammation ... 51
2.5.6 6-BIO systemic administration induces and stabilizes pathway activity specifically in brain microvasculature ... 53
2.5.7 Pathway activation attenuates tight junction disruption and basal endothelial permeability associated to delayed rtPA administration ... 54
2.6 Discussion ... 57 2.7 Acknowledgements ... 62 2.8 Disclosure statement ... 62 Chapitre 3 ... 63 3. Discussion ... 63 Chapitre 4 ... 70 4. Conclusion et perspectives ... 70 Références ... 73
vii
Liste des figures
Figure 1.1 La pénombre ischémique. ... 2 Figure 1.2. L'unité neurovasculaire ... 3 Figure 1.3. Schéma des jonctions serrées inter-endothéliales et des protéines intégrales impliquées dans la régulation paracellulaire ... 6 Figure 1.4. La cascade ischémique ... 8 Figure 1.5. Schéma des événements phasiques de la barrière hémato-encéphalique associés à l'ischémie cérébrale et à la reperfusion au cours du temps ... 15 Figure 1.6. Organisation schématique de la signalisation de la β-caténine dans les cellules endothéliales du système nerveux central ... 24 Figure 2.1. Canonical Wnt pathway activity is detectable predominantly in brain endothelial cells and is induced upon ischemic stroke.. ... 45 Figure 2.2. Figure 2. Deactivation of the canonical Wnt pathway in brain endothelial early after ischemic stroke aggravates brain oedema and causes haemorrhagic transformation ... 46 Figure 2.3. 6-BIO efficaciously activates the canonical Wnt pathway in brain endothelial cells and decreases the OGD-induced basal permeability ... 48 Figure 2.4. 6-BIO attenuates BBB breakdown and reduces the incidence haemorrhagic transformation associated to delayed rtPA administration after stroke ... 50 Figure 2.5. 6-BIO does not influence neuronal death, angiogenesis and inflammation after ischemic stroke ... 52 Figure 2.6. Delayed rtPA administration impairs canonical Wnt pathway activity in brain endothelial cells, which is rescued by 6-BIO systemic treatment ... 53 Figure 2.7. 6-BIO systemic administration attenuates tight junction disruption associated to delayed rtPA administration. ... 55 Figure 2.8. 6-BIO systemic administration recovers basal endothelial permeability after delayed rtPA administration ... 56 Figure 4.1 Niveaux de la protéine Dkk-1 chez la souris suite à un AVC ischémique ... 72
viii
Liste des abréviations
AVC Accident vasculaire cérébral 6-BIO 6-Bromoindirubine-3'-oxime
AMPA α-amino-3-hydroxy-5-méthylisoazol-4-propionate APC Protéine adenomatous polyposis coli
ATP Adénosine triphosphate
BDNF Facteur neurotrophique issu du cerveau
BEMC Cellules endothéliales microvasculaires primaires du cerveau bEnd3 Cellules endothéliales immortalisées dérivées de cerveaux murins BHE Barrière hémato-encéphalique
CASK Calcium calmoduline serine thréonine kinase CK1α Kinase caséine 1α
DISC Complexe d'induction de mort Dkk-1 Dickkopf-1
Dvl Protéine Dischevelled
FADD Protéine associée au domaine de mort Fas FDA Food and Drug Administration
Fzd Récepteur Frizzled
GCSF Facteur stimulant les colonies de granulocytes GDNF Facteur neurotrophe dérivé de la glie
GLUT1 Transporteur du glucose 1
GSK3β Glycogène syntéthase kinase 3β ICAM-1 Molécule d'adhésion intercellulaire 1 IL-10 Interleukine 10
IL-1β Interleukine-1β
JAM-A/-B/-C Molécule d'adhésion jonctionnelle A, B, C
LRP1/5/6 Protéine apparentée aux récepteurs des lipoprotéines à faible densité 1, 5, 6
LTD Dépression à long terme LTP Potentialisation à long terme MAC Pores de transition mitochondriale
ix
MAGI-1 Membrane-associée guanylate kinase MCAO Occlusion de l'artère cérébrale moyenne MCP-1 Protéine chimiotactique des monocytes 1 MMP-2/-3/-9 Métalloprotéinases matricielles 2, 3, 9 NADH Nicotinamide adénine dinucléotide NF- Кβ Facteur de transcription Кβ
NIHSS National Institute of Health Stroke Score
NMDA N-méthyl-D-aspartate
NO Oxyde nitrique
OGD Déprivation d'oxygène et de glucose ONOO- Peroxynitrite
PAF Facteur d'activation plaquettaire
PAI-1/-2 linhibiteur des activateurs du plasminogène 1, 2 Par-3/-6 Protéines de polarité 3, 6
PLVAP Protéine associée aux vésicules de plasmalemma PN-1 Protéase nexine 1
ROS Dérivés réactifs de l'oxygène
rtPA Activateur tissulaire du plasminogène recombiné SOD Superoxyde dismutase recombinante
STAIR Stroke Therapy Academic Industry Roundtable
TGF-β Facteur de croissance transformant β
TNFR1/2 Récepteur du facteur de nécrose tumorale 1, 2 TNF-α Facteur de nécrose tumorale α
tPA Activateur tissulaire du plasminogène
rtPA Activateur tissulaire du plasminogène, forme recombinante UNV Unité neuro-vasculaire
VCAM-1 Protéine d'adhésion vasculaire cellulaire 1
VEGF Facteur de croissance de l'endothélium vasculaire
VEGFR-2 Récepteur du facteur de croissance de l'endothélium vasculaire 2 ZO-1 Zonula occludens 1
x
Remerciements
Mon projet de maîtrise et le mémoire qui s'y rattache ne seraient pas les mêmes sans le support et l'aide incroyable que j'ai pu recevoir durant ces deux dernières années.
Premièrement, j'aimerais remercier mon directeur de recherche, mentor, collègue et ami, Ayman ElAli. Il a su m'offrir un support et une écoute incroyable dans un milieu qui n'était pas toujours facile. Merci de m'avoir fait confiance et de m'avoir accepté dans ton laboratoire en tant que première étudiante. Les connaissances et l'expertise que j'ai acquises sont inestimables et me serviront pour le reste de ma vie, tant professionnelle que personnelle. Je suis fière du laboratoire que tu as su créer et les prochains étudiants seront chanceux d'étudier sous ta direction.
Merci à tous les membres de l'équipe du Dr Serge Rivest pour leur support incroyable particulièrement Marie-Michelle Plante, Paul Préfontaine et Nataly Laflamme qui ont pu m'aider avec toutes mes petites questions techniques fatiguantes.
Merci aux membres de mon équipe, Revathy et plus récemment Romain et Maxime. Parce qu'une équipe de 4 c'est pas mal mieux qu'une équipe de 1!
Merci à ma famille et mes amis, de leur support et de leur curiosité scientifique à l'égard de mon projet. Pouvoir répondre à vos questions scientifiques et voir la compréhension illuminer vos faces est plus gratifiant que la meilleure des notes dans un bulletin.
Finalement, merci au comité d'évaluation pour leur temps et leur contribution pour la version finale de ce mémoire.
xi
Avant-propos
L’article intitulé Canonical Wnt pathway maintains blood-brain barrier integrity upon ischemic stroke and its activation ameliorates tissue-plasminogen activator therapy est en révision dans le journal Brain Pathology. Cet article représente le second chapitre du mémoire. Lors de ce projet, j’ai effectué l’intégralité des expériences mis à part les chirurgies de MCAO qui ont été réalisées par Revathy Guruswamy et les expériences en microscopie électronique qui ont été réalisées par Katherine Picard et Geneviève Parent. J’ai également accompli l’analyse des résultats, puis interprété ceux-ci en discutant avec Ayman ElAli. Suite à la discussion scientifique, j’ai rédigé l’article scientifique avec l'aide de Ayman ElAli, article qui fut ensuite corrigé par l'ensemble des co-auteurs. Le projet lui-même a été conçu et encadré par Ayman ElAli.
1
Chapitre 1
1. Introduction
1.1 Les accidents vasculaires cérébraux
L'accident vasculaire cérébral (AVC) est un trouble cérébral courant qui se classe parmi les trois principales causes de mortalité au monde et constitue la cause la plus importante d'incapacité à long terme chez les adultes (1). L'hypertension, l'hypercholestérolémie, l'obésité, le diabète, l'inactivité physique et l'âge constituent les facteurs de risques les plus communs (2). Chaque année, 50 000 nouveaux cas d'AVC sont recensés au Canada dont 15 à 30% souffriront d'incapacité à long terme (2). Le vieillissement grandissant de la population laisse présager une augmentation de l'incidence des AVC. Le pronostic suivant un AVC varie en fonction du type et de la gravité de l'atteinte, de l'emplacement de l'occlusion ainsi que de l'efficacité et de la disponibilité du traitement anti-thrombolytique. 15 à 30% des individus ayant subi un AVC doivent composer avec des incapacités permanentes et 20% seront placés dans des institutions pour des soins à long terme (2). Les AVC engendrent des coûts astronomiques sur le système de santé. Par exemple, il est estimé que les coûts directs et indirects sont montés à plus de 40.1 milliards de dollars américains entre 2013 et 2014 aux États-Unis (2). Compte tenu de la prévalence des AVC, des effets à long terme sur la population, de sa nature multifactorielle et du fardeau économique qu'il engendre, il est impératif pour les chercheurs de développer des thérapies plus efficaces pour non seulement prévenir les AVC, mais aussi pour améliorer la récupération subséquente.
1.2 L'ischémie cérébrale
Un AVC est caractérisé par une perte soudaine de la fonction cérébrale suite à l'interruption du débit sanguin cérébral causé par un blocage des vaisseaux sanguins (AVC ischémique) ou par une rupture (AVC hémorragique). Les AVC ischémique représentent la majorité des cas (87%) et surviennent suite à un rétrécissement des artères engendré par l'accumulation de plaques
2
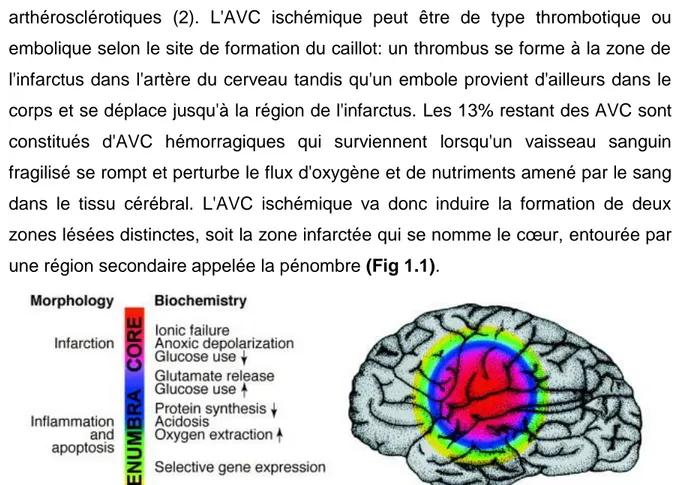
arthérosclérotiques (2). L'AVC ischémique peut être de type thrombotique ou embolique selon le site de formation du caillot: un thrombus se forme à la zone de l'infarctus dans l'artère du cerveau tandis qu'un embole provient d'ailleurs dans le corps et se déplace jusqu'à la région de l'infarctus. Les 13% restant des AVC sont constitués d'AVC hémorragiques qui surviennent lorsqu'un vaisseau sanguin fragilisé se rompt et perturbe le flux d'oxygène et de nutriments amené par le sang dans le tissu cérébral. L'AVC ischémique va donc induire la formation de deux zones lésées distinctes, soit la zone infarctée qui se nomme le cœur, entourée par une région secondaire appelée la pénombre (Fig 1.1).
Figure 1.1. La pénombre ischémique. Une région cérébrale de faible perfusion dans laquelle les cellules ont perdu leur
potentiel membranaire («cœur») est entourée d'une zone où la perfusion intermédiaire prédomine («pénombre») et les cellules se dépolarisent par intermittence («dépolarisation péri-infarctus»). Dès le début du déficit de perfusion focale, le cœur et la pénombre sont dynamiques dans l'espace et dans le temps. Il existe des seuils de perfusion au-dessous desquels certaines fonctions biochimiques sont bloquées (échelle de couleur). (3) [tiré intégralement]
Le cœur est la zone la plus faiblement irriguée, caractérisée par un débit sanguin inférieur à 10ml/min/100g, et sera donc là où la survie cellulaire sera compromise puisqu'elle correspond à une lésion structurale. La pénombre est caractérisée par un débit sanguin légèrement plus élevé (10 à 18 ml/min/100g) et est aussi à risque de devenir nécrosée mais la survie cellulaire y est assurée dans un premier temps (3). Les régions du cœur et de la pénombre sont dynamiques et leurs dimensions évoluent dans le temps (4).
1.2.1 L'unité neurovasculaire en condition physiologique
Le cerveau consomme jusqu'à 20% des nutriments (principalement du glucose) et de l'oxygène présent dans le sang, rendant les neurones complètement
3
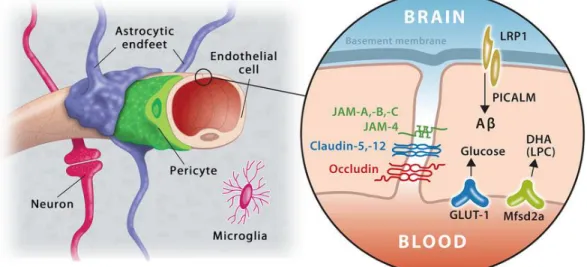
dépendantes du système vasculaire pour assurer leur survie (5). Ainsi, la microvasculature cérébrale doit interagir fonctionnellement et structurellement avec le parenchyme cérébral pour assurer le fonctionnement adéquat du cerveau. Cet échange est gouverné par l'unité neurovasculaire (UNV). Le concept d'UNV a été pour la première fois défini par le Stroke Progress Review Group qui l'a décrit comme étant une triade composée des neurones, des cellules gliales (astrocytes et microglies) et de la vasculature adjacente (artérioles et capillaires) (6). Les cellules endothéliales, en étant scellées entre elles, forment la barrière hémato-encéphalique (BHE) (Section 1.2.1.1 La barrière hémato-hémato-encéphalique) , et interagissent physiquement et chimiquement avec les protéines de la matrice extracellulaire, les péricytes, les pieds astrocytaires, les microglies et les neurones afin de créer une barrière fonctionnelle qui régule le mouvement des molécules qui rentrent et qui sortent du cerveau (Fig 1.2) (7). L'UNV est donc un site dynamique d'échanges biochimiques et cellulaires qui a pour but d'assurer l'homéostasie et le fonctionnement optimal du cerveau en influençant le microenvironnement cérébral. Le contrôle du couplage neurovasculaire (8), l'ajustement local du flot sanguin cérébral (9) et le réglage des paramètres et des fonctions de la BHE (10) sont donc assurés par les composantes de l'UNV afin de répondre au besoin du cerveau en tout temps (5).
Figure 1.2. L'unité neurovasculaire comprend les cellules endothéliales, les péricytes et les astrocytes qui, ensemble,
confèrent des propriétés uniques à la barrière hémato-encéphalique. Les cellules endothéliales qui bordent le système vasculaire cérébral ont de nombreux transporteurs transcellulaires en plus de jonctions serrées qui limitent la diffusion paracellulaire passive de tous les solutés et des ions, sauf ceux de très petite taille. (11) [tiré intégralement]
4 1.2.1.1 La barrière hémato-encéphalique
Tel que mentionné plus haut, la BHE est formée des cellules endothéliales jointes ensemble en une barrière qui sépare le sang du cerveau. Les jonctions serrées lient les cellules endothéliales adjacentes entre elles et poussent les molécules à emprunter la route transcellulaire pour atteindre le parenchyme cérébral, créant donc une réelle barrière physique (12). Des gaz comme l'O2 et le CO2 peuvent
diffuser librement à travers la membrane ainsi que de petits agents lipophiles tels que l'éthanol, mais ce sont principalement la présence de systèmes de transporteurs spécifiques qui sont capables de réguler le trafic des molécules hydrophiles de petites tailles (13). Les jonctions serrées induisent aussi la polarité nécessaire de la BHE qui est caractérisée par la présence de deux côtés fonctionnellement distincts, un côté luminal faisant face à la circulation sanguine et un côté abluminal faisant face au parenchyme cérébral (7). Finalement, une barrière métabolique est créée par la présence d'enzymes intracellulaires et extracellulaires, tels que la monoamine oxydase et le cytochrome P450 qui sont capables d'inactiver plusieurs composants neuroactifs et toxiques (14). L'endothélium cérébral exhibe une activité d'endocytose et de transcytose quasi inexistante en comparaison au reste de l'endothélium périphérique (15).
La BHE rempli plusieurs rôles. Elle facilite l'entrée des nutriments nécessaires au cerveau et régule la sortie des composants potentiellement nocifs et des déchets métaboliques (13). Elle restreint les mouvements ioniques et fluidiques entre le sang et le cerveau afin de protéger des fluctuations dans la composition ionique qui pourraient perturber la signalisation synaptique (16). Elle permet aussi de séparer les neurotransmetteurs qui agissent de manière centrale ou périphérique afin d'éviter un effet redondant (17).
Les cellules endothéliales qui composent la BHE sont en contacts étroits avec les péricytes (18, 19) et les pieds astrocytaires (17). Cette proximité induit une communication bidirectionnelle. La communication entre les deux côtés est essentielle pour la préservation de l'homéostasie du cerveau. Du côté luminal, les cellules endothéliales sont exposées à des facteurs de stress présents dans la circulation sanguine tels que des médiateurs inflammatoires et des toxines
5
environnementales qui peuvent compromettre l'intégrité de la barrière (5). Le côté abluminal contient de nombreux récepteurs et protéines membranaires qui contribuent au relai du signal émis par les péricytes et les pieds astrocytaires dans l'espace périvasculaire vers les cellules endothéliales (17, 18). Ce sont ces signaux périvasculaires qui contrôlent la formation des jonctions cellulaires entre les cellules endothéliales et régulent l'abondance et l'activité des transporteurs membranaires (20).
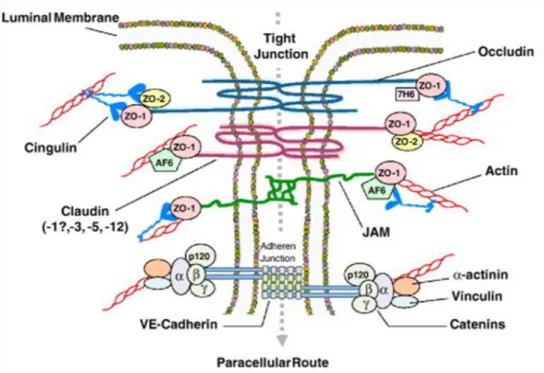
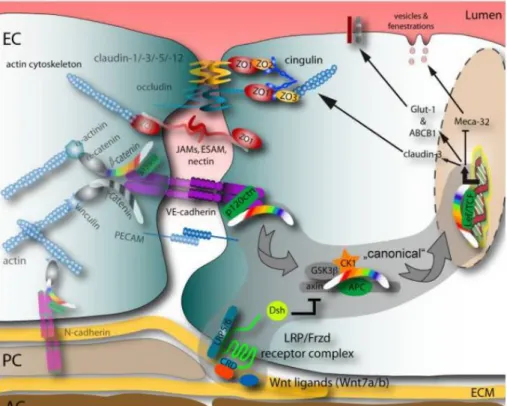
Il est maintenant reconnu que l'endothélium cérébral diffère de l'endothélium des tissus périphériques en raison de caractéristiques spécifiques qui contribuent à ses propriétés de barrières (physiques ou chimiques). Par exemple, les jonctions serrées sont nettement plus complexes dans l'endothélium cérébral et occluent la fente intercellulaire plus efficacement (Fig 1.3) (12). Parmi les composants identifiés ayant un rôle essentiel pour la structure des jonctions serrées, on retrouve les protéines transmembranaires occludines et claudines. Occludine est une protéine qui est capable de se lier avec la protéine de liaison au cytosquelette zonula occludens 1 (ZO-1). La fonction principale de la protéine occludine semble être la régulation des jonctions serrées (21). La délétion de la partie N terminale de la protéine induit un effet dramatique sur l'intégrité des jonctions serrées, caractérisé par une faible résistance électrique transcellulaire, une augmentation du flux paracellulaire de petits traceurs de masse moléculaire et la présence d'espaces dans les jonctions serrées en analyse en microscopie électronique (22). Les claudines, quant à elles, forment une famille de multigènes comprenant plus de 20 membres qui composent les jonctions serrées via une interaction homophile claudine-claudine (23). À la BHE, c'est principalement l'expression des claudines 3, 5 et 12 qui semblent contribuer le plus à la forte résistance électrique transendothéliale (12). Chaque claudine régule la diffusion d'un groupe de molécules d'un certain poids moléculaire (24). D'autres protéines de jonctions comme JAM-A, JAM-B et JAM-C sont aussi présentes dans les cellules endothéliales du cerveau et sont impliquées dans la formation et la maintenance des jonctions serrées (25). Les protéines transmembranaires sont connectées du côté cytoplasmique à un réseau complexe de protéines membranaires
6
périphériques qui forment de grands complexes protéiques, les plaques cytoplasmiques. À l'intérieur de ces plaques se retrouvent des protéines adaptatrices telles que ZO-1, la calcium calmoduline serine thréonine kinase (CASK), la membrane-associée guanylate kinase (MAGI-1) ou encore les protéines de polarité Par-3 et Par-6 (5). Les interactions cellules-cellules à la jonction serrée sont stabilisées par les jonctions adhérentes qui sont entremêlées avec les jonctions serrées.
Figure 1.3. Schéma des jonctions serrées inter-endothéliales et des protéines intégrales impliquées dans la régulation
paracellulaire, telles que définies dans le texte. (26) [tiré intégralement]
Les jonctions serrées restreignent fortement le transport paracellulaire et forcent donc les molécules hydrophiles à emprunter la voie transcellulaire. De manière générale, le transport transcellulaire à travers la BHE peut être classifié en cinq grandes catégories: le passage par transporteur, le passage d'ions, le transport actif d'efflux, le transport médié par un récepteur et le transport médié par les cavéoles (5). Les transporteurs qui assurent le passage des nutriments au cerveau incluent le transporteur de glucose GLUT1, plusieurs transporteurs d'acides aminés ainsi que des transporteurs de nucléosides, nucléobases et autres substances (13). De même, les cellules endothéliales expriment des transporteurs spécifiques qui permettent d'éliminer les déchets du système nerveux central vers
7
la circulation sanguine (27). Ces différents transporteurs peuvent être bidirectionnels, tel que GLUT1, et transporter les substances à l'encontre du gradient de concentration et se retrouver du côté luminal ou abluminal, ou les deux. Ainsi, la polarité apicale-basale des cellules endothéliales cérébrales se reflète donc dans leur fonction de transport polarisé et est maintenue en condition physiologique. Les cellules endothéliales du cerveau expriment aussi de manière très faible une activité de transcytose qui est cependant augmentée en condition de dysfonction de la BHE lors de conditions pathologiques. La transcytose à travers les cellules endothéliales est médiée par des vésicules de cavéoline. Cavéoline-1 est exprimée par toutes les cellules endothéliales du corps mais son expression est augmentée suite à un traumatisme crânien ou en conditions hypoxiques (28). De plus, l'expression de la protéine associée aux vésicules de plasmalemma (PLVAP) est normalement élevée dans les cellules endothéliales de la périphérie et relativement faible dans les cellules endothéliales du cerveau. Cette protéine est impliquée dans le trafic de vésicules ainsi que dans la formation de fenestration et son expression semble être augmentée dans l'endothélium cérébral dans une variété de conditions pathologiques où il y a atteinte à la BHE, tel que l'ischémie (29). Par conséquent, l'absence d'expression de PLVAP dans les cellules endothéliales du système nerveux sain semble être importante pour limiter la perméabilité de la BHE.
1.2.2 L'unité neurovasculaire en condition ischémique
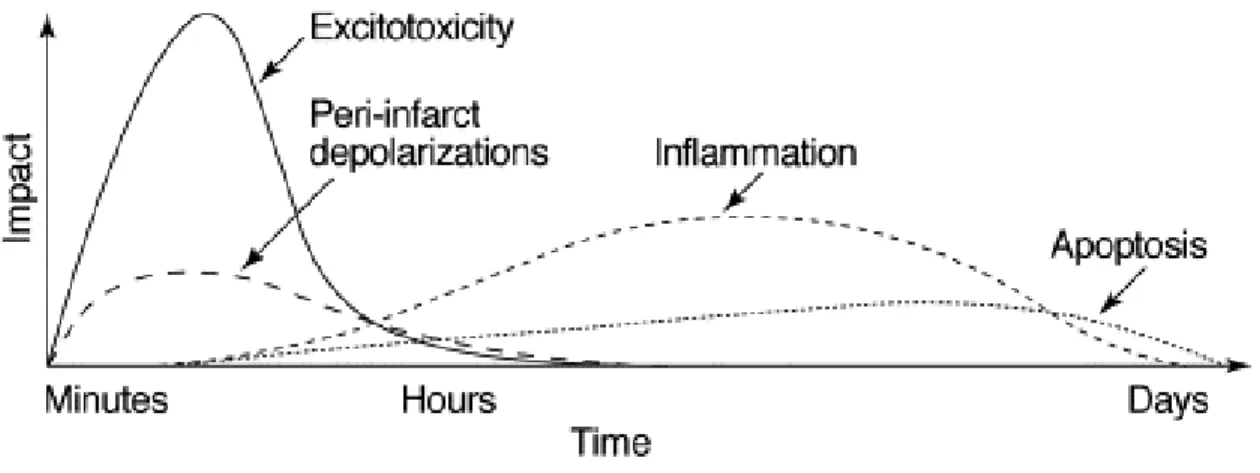
Ainsi, l'homéostasie créée par la synergie des composants de l'unité neurovasculaire ainsi que par le contrôle fin de la perméabilité par la BHE est nécessaire au bon fonctionnement du cerveau. Cependant, en conditions pathologiques, l'unité devient dysfonctionnelle et peut détériorer les mécanismes de réparation du cerveau (30, 31). Par exemple, l'ischémie cérébrale va initier une cascade complexe d'évènements pathophysiologiques qui évolueront dans le temps et l'espace et qui incluent plusieurs mécanismes qui aggraveront le dommage initial: excitoxicité, dépolarisations péri-infarctus, inflammation et mort cellulaire programmée (Fig 1.4) (3).
8
Figure 1.4. La cascade ischémique. Cascade putative d'événements dommageables dans l'ischémie cérébrale focale.
Très tôt après le début du déficit de perfusion focale, les mécanismes excitotoxiques peuvent endommager létalement les neurones et les autres cellules. En outre, l'excitotoxicité déclenche un certain nombre d'événements qui peuvent contribuer davantage au dommage du tissu. De tels événements comprennent les dépolarisations péri-infarctus et les mécanismes plus retardés de l'inflammation et de la mort cellulaire programmée. L'axe des abscisses reflète l'évolution de la cascade dans le temps, tandis que l'axe des ordonnées vise à illustrer l'impact de chaque élément de la cascade sur le résultat final. (3) [tiré intégralement]
Perte d'énergie et excitoxicité
L'arrêt brusque de l'irrigation sanguine causée par un caillot sanguin lors d'un AVC va diminuer de manière drastique les niveaux d'oxygène et de glucose ainsi que la disponibilité des substrats pour la phosphorylation oxydative tels que la nicotinamide adénine dinucléotide (NADH) (32). Le cerveau est incapable de survivre en respiration cellulaire anaérobie et dépend donc uniquement de la phosphorylation oxydative pour subvenir à ses besoins élevés en adénosine triphosphate (ATP) pour son fonctionnement normal (3). Ainsi, 30 minutes après un AVC, la concentration en ATP dans les cellules est déplétée et la plupart des processus cellulaires ATP-dépendant sont compromis. Particulièrement, les pompes ioniques Na+/K+ et Ca2+/H nécessaires au maintient d'un gradient d'ions et
au potentiel de membrane des cellules arrêtent de fonctionner (33). L'accumulation de sodium, de calcium et de bicarbonate à l'intérieur des cellules induit la diffusion passive d'eau dans le cytoplasme et cause un œdème cytotoxique (34). L'œdème est un facteur crucial dans la mortalité de patients puisqu'il inhibe la perfusion des cellules péri-infarctus et augmente la pression intracrânienne (35). L'arrêt des pompes ioniques entraîne aussi la perte du potentiel de membrane et donc la dépolarisation massive des neurones et des cellules gliales du cerveau (36). Cette
9
dépolarisation entraîne l'activation des canaux calciques (Ca2+)
voltage-dépendants présynaptiques qui vont libérer dans l'espace extracellulaire du glutamate, un acide aminé excitateur (37, 38). Le mécanisme de recapture de ce neurotransmetteur par le transporteur de glucose GLUT-1 étant aussi dérégulé, le glutamate atteint des niveaux très élevés dans le milieu extracellulaire et dans la fente synaptique (39). Cette concentration anormale de glutamate extracellulaire active les récepteurs perméables aux ions monovalents tels que le récepteur N-méthyl-D-aspartate (NMDA) et le récepteur α-amino-3-hydroxy-5-méthylisoazol-4-propionate (AMPA) et cause l'entrée de Ca2+ dans la cellule (40).
Stress oxydatif et nitrosatif
Le Ca2+ est un second messager universel important dans la cellule et coordonne
plusieurs processus métaboliques et signalétiques. Son augmentation intracellulaire à des niveaux anormaux entraînera une série d'évènements nucléaires et cytoplasmique qui influenceront profondément l'apparition et le développement des dommages. D'abord, il entraîne l'activation d'enzymes protéolytiques qui dégraderont les protéines du cytosquelette telles que l'actine et la spectrine ainsi que les protéines de la matrice extracellulaire comme la laminine, compromettant l'intégrité des cellules et de la vasculature (41, 42). Ensuite, l'augmentation de calcium intracellulaire induit la production de dérivés réactifs de l'oxygène (ROS) et d'oxyde nitrique (NO). Ces radicaux libres vont causer des dommages à la membrane via la peroxydation des lipides et vont promouvoir l'activation des mécanismes d'apoptose (43, 44). La phospholipase A2 sera aussi activée par les niveaux élevés de calcium, ce qui relâchera de l'acide arachidonique qui à son tour engendra la production d'anion superoxyde O2.- par la
voie du cyclo-oxygénase. La Ca2+/calmoduline-dépendante oxyde nitrique
synthase est aussi activée et synthétise de l'oxyde nitrique (NO) qui interagit avec le O2.- pour former du peroxynitrite (ONOO-), un oxydant hautement instable qui interagit et endommage l'ADN, les protéines et d'autres composants cellulaires (45). Des radicaux libres sont aussi produits par la mitochondrie lors de la reperfusion et entraînent un dommage supplémentaire appelé dommage de
10
reperfusion. Les ROS et le calcium intracellulaire promeuvent le gonflement de la mitochondrie et la rupture de son potentiel membranaire, la rendant incapable de générer de l'ATP. De même, cela cause la relâche additionnelle de radicaux libres dans l'espace intracellulaire et éventuellement la mort cellulaire (46). En somme, le stress oxydatif et nitrosatif induisent des processus fortement préjudiciables qui vont enclencher autant la formation de dommages structuraux que le déclenchement des processus de mort cellulaire.
Dépolarisations péri-infarctus
Une autre conséquence de l'augmentation de glutamate et de potassium dans l'espace extracellulaire est la formation de vagues de dépolarisation. En effet, au cœur de la lésion, les neurones se dépolarisent selon le mécanisme expliqué plus haut mais ne pourront jamais se repolariser en raison du manque crucial d'énergie. Cependant, dans la pénombre, les cellules parviennent lentement à rétablir leur potentiel de membrane et le tissu endure donc des vagues massives de dépolarisations qui peuvent durer des heures et qui vont se transmettre aux tissus environnants (47). Ces dépolarisations en vagues ne causent pas de dommages aux tissus sains puisqu'ils sont en mesure de se repolariser rapidement et de rétablir leurs gradients ioniques. Cette alternance polarisation-dépolarisation va cependant épuiser les réserves énergétiques des cellules qui sont déjà fortement éprouvées par les processus de la cascade ischémique et contribuer à augmenter le volume de la lésion, induisant la transition entre la région pénombre vers des régions du cœur (48). Les dépolarisations péri-infarctus peuvent aussi induire des dommages structuraux aux épines dendritiques qui sont seulement réversibles en présence d'un débit sanguin adéquat (49). Il a été démontré que des antagonistes des récepteurs NMDA, un récepteur ionotrope activé par le glutamate, administrés chez le rat ont pour effet de réduire les dépolarisations et diminuer le volume de la zone ischémique (50). En résumé, des évènements électriques normalement bien tolérés dans le parenchyme cérébral sain causent des effets dévastateurs dans la zone pénombre en raison du microenvironnement chimique et ionique altérés par la cascade ischémique.
11 L'inflammation
L'inflammation fait partie intégrante de la réponse suite à l'ischémie cérébrale et est caractérisée par l'activation des cellules gliales résidentes, par l'infiltration des leucocytes dans le cerveau et par la production de cytokines pro-inflammatoires. Le système immunitaire remplit normalement un rôle bénéfique dans le corps en éliminant les agents étrangers qui menacent l'intégrité des tissus. Cependant, en condition ischémique, le système immunitaire peut promouvoir une réponse inflammatoire chronique qui exacerbe les dommages cellulaires et compromet la viabilité de la pénombre ischémique en empêchant ou en favorisant le recrutement de certaines cellules à la lésion (51). La réponse neuroinflammatoire stimule les cellules gliales, plus précisément les microglies et les astrocytes, qui entourent les neurones endommagés. Ces cellules contribuent normalement à la santé neuronale. Les microglies rétractent leurs processus et adoptent alors une morphologie amiboïde qui est caractéristique de leur activation tandis que les astrocytes deviennent hypertrophiques (52). Une fois activées, les cellules gliales sécrètent des médiateurs pro et anti-inflammatoires qui vont promouvoir l'œdème, l'angiogenèse et la plasticité, dégrader des protéines, phagocyter les débris et compromettre la BHE (51, 53, 54). Ces médiateurs incluent des cytokines pro-inflammatoires telles que le facteur de nécrose tumorale (TNF-α), l'interleukine-1β (IL-1β), des médiateurs de l'inflammation comme le facteur d'activation plaquettaire (PAF), des chimiokines telles que la protéine chimiotactique des monocytes (MCP-1) et des métalloprotéinases matricielles (MMP) (54). En plus d'être libérées par les cellules immunitaires, certaines cytokines vont être libérées par les neurones endommagés. Ces médiateurs vont aggraver la lésion ischémique en favorisant encore plus la sécrétion de cytokines et de molécules d'adhésion, en activant les cellules gliales et en inhibant la recapture de glutamate dans les cellules (55). En parallèle, la réponse inflammatoire a aussi des effets bénéfiques sur la lésion. Lorsque des cellules deviennent ischémiques, les microglies migrent et entourent ces cellules de manière à ce qu'elles soient rapidement éliminées de l'environnement et ne puissent pas libérer le contenu toxique de leur cytoplasme (56). De plus, les astrocytes vont former ce qu'on appelle une cicatrice gliale au
12
pourtour de la lésion pour prévenir les infections et la création d'autres dommages cellulaires (57). Des cytokines anti-inflammatoires et des facteurs de croissance comme le facteur neurotrophique issu du cerveau (BDNF) et le facteur neurotrophe dérivé de la glie (GDNF) sont libérés et ont des effets neuroprotecteurs en promouvant la survie cellulaire (54). Par exemple, le facteur de croissance transformant (TGF-β) et IL-10 sont les deux médiateurs anti-inflammatoires majeurs retrouvés après un AVC qui sont capables de diminuer le dommage tissulaire. Ils sont capable de diminuer l'activation des cellules gliales et vont inhiber la sécrétion de IL-1β et TNF-α (51, 53). Certaines cytokines peuvent avoir des effets mixtes. Par exemple, TNF-α aura un effet pro-inflammatoire ou anti-inflammatoire dépendamment de sa liaison avec le récepteur TNFR1 ou TNFR2 (52). De plus, l'AVC induit la libération d'une autre catégorie de médiateurs inflammatoires, les chimiokines. Les chimiokines sont dans la famille des cytokines et sont impliquées dans la migration des cellules immunitaires et inflammatoires, telles que les neutrophiles et les monocytes. Les chimiokines sont généralement peu exprimées dans un cerveau en condition physiologique mais l'ischémie cérébrale en induit la production rapide et cause l'infiltration des cellules immunitaires de la circulation (58). En effet, les molécules d'adhésion exprimées à la surface des cellules endothéliales lésées comme la molécule d'adhésion intercellulaire (ICAM-1), VCAM-1 ou les P-sélectines et E-sélectines vont s'associer avec les récepteurs complémentaires spécifiques présents sur les leucocytes et les monocytes (59, 60). S'ensuit alors les étapes de la diapédèse, soit le mécanisme par lequel les cellules immunitaires traversent les cellules endothéliales d'un vaisseau sanguin ou d'un capillaire dans l'objectif de migrer à la zone lésée. La délétion de ICAM-1 ou P-sélectine induit des lésions ischémiques plus petites et une plus faible infiltration de neutrophiles, suggérant que ces molécules modulent le dommage ischémique durant la réponse inflammatoire (61). Chez l'humain toutefois, un essai clinique avec des anticorps anti-ICAM-1 s'est avéré inefficace et a même aggravé les signes cliniques des patients (62). Il semblerait donc que l'inflammation peut aggraver les lésions suite à un AVC selon différents mécanismes. Par exemple, les leucocytes recrutés dans les
micro-13
vaisseaux peuvent les obstruer et induire de l'hypoxie (ce qu'on appelle le phénomène de no-reflow) (63) ou les médiateurs toxiques sécrétés par les cellules inflammatoires activées comme l'iNOS qui induit l'oxyde nitrique et qui est un gaz toxique pour les cellules (64). Ainsi, le rôle néfaste ou bénéfique de l'inflammation dans la guérison des dommages liés à l'AVC reste ambigü et est encore aujourd'hui un sujet de débats dans la littérature.
L'apoptose
L'ischémie cérébrale, en causant l'activation excessive des récepteurs glutamates, la surchage de Ca2+, l'expression de radicaux libres et des dommages
mitochondriaux ou à l'ADN, va pousser les cellules à mourir soit par apoptose ou par nécrose (65). La nécrose est le processus de mort cellulaire principal qui se produit dans le cœur de la lésion et survient durant les premières phases de l'AVC tandis que l'apoptose survient plus tardivement et est plus observé dans la zone pénombre (3). Contrairement au démantèlement cellulaire de l'apoptose qui est un processus contrôlé et organisé, la nécrose implique des dommages à la membrane cellulaire et aux organelles. Les principales caractéristiques de la nécrose sont l'agglutination de la chromatine, le gonflement et la dégénérescence des organites, plus particulièrement de la mitochondrie, la destruction de l'intégrité membranaire pour finir avec la dissolution de la cellule et le déversement de son contenu dans l'espace extracellulaire. Puisque le déversement de ce contenu contient des débris intracellulaires antigéniques actifs, il est accompagné d'une réponse inflammatoire qui va alors causer une infiltration leucocytaire et de l'œdème tissulaire. Les enzymes protéolytiques relâchées sont aussi capables d'aller endommager les cellules environnantes, exacerbant le dommage (66). L'apoptose se manifeste dans un intervalle de temps pouvant aller à des jours ou des semaines après le début de l'AVC et est principalement confinée en dehors du cœur de la lésion. L'apoptose est un processus actif dépendant de l'expression de gènes spécifiques qui vont mener à l'autodestruction cellulaire et implique les caspases (67). Au niveau morphologique, il y a d'abord la condensation de la chromatine puis du cytoplasme qui est associé avec la formation de vacuoles
14
cytoplasmiques que l'on appelle les corps apoptotiques issus de la convolution de la membrane cytoplasmique et nucléaire. Puisque l'intégrité des membranes est conservée au cours du processus apoptotique, toute réaction inflammatoire est évitée (66). Le mécanisme d'apoptose caspase-dépendant est gouverné par deux voies principales d'activation: la voie extrinsèque et la voie intrinsèque. La voie intrinsèque implique la mitochondrie et peut être induite par plusieurs déclencheurs présents dans l'ischémie: radicaux libres, débalancement ionique, etc. Cela cause la formation de pores de transition de perméabilité de la membrane interne mitochondriale (MAC) et produit le relâchement du cytochrome c. Les MAC sont régulés par les protéines de la famille du gène Bcl2, qui englobent les protéines pro-apoptiques Bax et Bak ainsi que les protéines anti-apoptiques Bcl-2 et Bcl-xL qui vont respectivement favoriser ou inhiber la formation des pores. Le cytochrome c relâché dans le cytosol va s'associer à APAF1 et à la procaspase-9 pour former un complexe appelé apoptosome. L'apoptosome active par la suite des caspases effectrices comme la caspase-3 qui seront responsables du démantèlement systématique des structures cellulaires (66, 68).
La voie extrinsèque est déclenchée lorsque des ligands comme FasL lient les récepteurs de mort cellulaire de la famille des récepteurs de nécrose tumorale (TNFR) situés à la surface de la cellule. FasL va activer son récepteur spécifique, FasR qui va libérer la protéine associée au domaine de mort Fas (FADD). FADD se lie à la procaspase 8 pour produire le complexe d'induction de mort (DISC) (67). Ce complexe va cliver la procaspase 8 et induire la formation de caspase 8 qui va activer une chaîne de caspases et initier le démantèlement des structures cellulaires comparablement à la voie intrinsèque.
La caspase 3 est un des régulateurs majeurs de l'apoptose suivant une ischémie et est fortement exprimée dans les stades précoces d'un AVC chez la souris et l'humain (69, 70). L'inactivation génétique de caspase 3 rend la souris plus résistante aux dommages ischémiques. Toutefois, selon le modèle d'ischémie utilisé, les méthodes d'inhibition des caspases ne sont pas toujours capables de réduire les lésions ischémiques. Les mécanismes d'apoptose et de nécrose sont redondants et complexes et il semblerait que cibler une seule caspase ne soit pas
15
suffisant en soi pour diminuer les dommages. Par exemple, des études tendent à montrer une activation précoce de l'apoptose indépendante de la mitochondrie et de la caspase 9 dans le cœur ischémique (71).
1.2.2.1 La BHE en condition ischémique
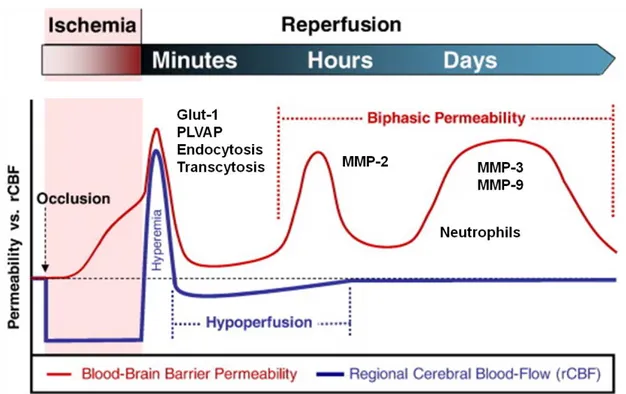
L'ischémie cérébrale va causer la rupture et le dysfonctionnement de la BHE. Plusieurs mécanismes sont impliqués, comme la dégradation de la matrice extracellulaire par des protéases, l'altération des propriétés du transport dans les cellules endothéliales et le remodelage des jonctions serrées (72). Des recherches intensives dans plusieurs modèles animaux utilisant différents modèles d'AVC suggèrent que l'ouverture de la BHE survient d'une manière biphasique: une phase précoce immédiate d'augmentation de la perméabilité observée environ 6h après l'ischémie suivie d'une ouverture retardée de la BHE notée 2-3 jours après l'AVC
(Fig 1.5) (73).
Figure 1.5. Schéma des événements phasiques de la barrière hémato-encéphalique associés à l'ischémie cérébrale et à
la reperfusion au cours du temps, tels que définis dans le texte. La lésion de reperfusion conduit à une ouverture biphasique de la BHE. L'ouverture précoce se produit plusieurs heures après le début de la reperfusion en raison de l'activation de MMP-2. Cette ouverture initiale est transitoire et réversible. De 24 à 72 heures plus tard, la réponse inflammatoire conduit à l'induction de MMP-3 et MMP-9, qui induisent des dommages plus intenses et irréversibles aux vaisseaux sanguins. [adapté de (26) et (74)]
16
Bien que le reperfusion du site de l'infarctus soit nécessaire pour soutenir les cellules en difficulté et leur faire parvenir l'oxygène et les nutriments dont elles ont besoin, le retour de la circulation sanguine va mener à un dommage secondaire entre autre causé par le stress oxydatif qui peut être responsable de la première phase de l'ouverture de la BHE. Les ROS (Section 1.2.2.1 Stress oxydatif et
nitrosatif) contribuent à la rupture de la BHE via plusieurs façons: dommage
oxydatif aux molécules cellulaires (protéines, lipides et ADN), activation des protéases; réorganisation du cytosquelette; modulation des protéines des jonctions serrées; et la régulation à la hausse des médiateurs inflammatoires (75). Les antioxydants comme la superoxyde dismutase recombinante (SOD) ou le polyéthylèneglycol-SOD sont capables de réduire la lésion de la BHE induite par l'ischémie et l'œdème vasogénique.
Les protéases sont normalement présentes sous une forme latente dans le cerveau. Certaines protéases sont exprimées de manière constitutive et participent aux processus cellulaires normaux, telles que MMP-2 qui est retrouvé dans le liquide céphalorachidien et est produit par les astrocytes (74). L'activation de MMP-2 requiert la présence de l'inhibiteur tissulaire de la métalloprotéinase-MMP-2 et la métalloprotéinase membranaire de type 1, ce qui restreint son activité enzymatique à la proximité immédiate de la membrane. Un autre groupe de protéases activées suivant l'AVC sont les MMP inductibles telles que MMP-3 et MMP-9. Lorsqu'elles sont secrétées, elles se diffusent rapidement et peuvent agir sur plusieurs substrats de la matrice extracellulaire. Les MMP vont dégrader les protéines de la membrane basale telles que la fibronectine, la laminine et l'héparane sulfate après une agression ischémique, ce qui contribue à la dégradation de la BHE (76). Les cytokines induisent l'expression de ces MMP via l'action du facteur de transcription Кβ (NF-Кβ). Les neutrophiles sont aussi capables d'induire le relâchement de MMP-9. Ainsi, dans la lésion secondaire causée par la reperfusion, les protéases participent à l'ouverture biphasique de la BHE. Une phase initiale réversible liée à l'activation de la MMP-2 précède une phase ultérieure de 24 à 48 heures associée à l'induction de MMP-3 et de MMP-9. Les MMP sont fortement activées suite à un AVC et les niveaux plasmiques de MMP-9 corrèlent avec la sévérité de la lésion
17
chez les patients atteints d'AVC (77). Les souris surexprimant la SOD-1 ont aussi une activation réduite des MMP-9 et présentent des lésions et de l'œdème réduit causé par la prévention de l'ouverture de la BHE (78). Toutefois, les protéases semblent avoir un rôle protecteur plus tard après l'ischémie, jouant un rôle dans la plasticité, l'angiogenèse et la réparation tissulaire. Leur inhibition 7 jours après un AVC élimine le remodelage neurovasculaire et augmente les dommages fonctionnels (79). L'ischémie cérébrale entraîne aussi l'activation d'autres protéases telles que l'activateur tissulaire du plasminogène (tPA) qui sera discuté plus loin (Section 1.3. Le système de l'activateur tissulaire du plasminogène). La dérégulation des protéines des jonctions serrées fait partie intégrante du remodelage de la BHE suite à un AVC. Dans une étude utilisant des souris taguées fluorescentes pour claudine-5 afin de visualiser la dynamique des jonctions serrées, des changements ultrastructuraux sont observés 48h après un AVC chez la souris: présence de lacunes (jonctions serrées discontinues) et de saillies (extensions linéaires des brins des jonctions serrées) et diminution de l'expression d'occludine et de ZO-1 corrélée avec une extravasation augmentée de IgG (80). Plusieurs autres études ont démontré que la modulation des protéines des jonctions en conditions ischémiques est corrélée avec une augmentation de la perméabilité de la BHE (26, 81, 82). Toutefois, dans les premiers stades suivant un AVC, les altérations structurelles des jonctions serrées semblent minimes tandis que l'on note la présence accrue d'endocytose et de transcytose (mesurées par l'absorption et le transport d'albumine fluorescent) dans les cellules endothéliales 6h après l'ischémie (80). Il semblerait que ce soit la protéine cavéoline-1 qui médie cette ouverture initiale de la BHE (83). Les cellules endothéliales acquièrent un phénotype de cellules endothéliales fenestrées ressemblant à ce qu'on retrouve dans le reste du corps, notamment en augmentant l'expression de PLVAP (7), mais aussi en augmentant l'abondance des transporteurs GLUT-1 du côté luminal (84) et en diminuant l'expression du transporteur de résistance multi-drogue ABCB1 du côté abluminal (85). Ces changements permettent de stabiliser le micro-environnement cérébral sous les conditions changeantes d'une ischémie en éliminant les métabolites toxiques du site de l'injure, en limitant l'entrée de
18
molécules issues de la circulation sanguine et en augmentant l'accès au glucose du parenchyme cérébral (17, 84, 85). Fait intéressant, ce concept d'extravasation transendothéliale a déjà été décrit dans les premières études d'ischémie et a été plus tard négligé dans la littérature (86, 87). Une analyse comparative récente de trois modèles différents d'AVC révèle quatre étapes dans la dégradation aiguë de la BHE: gonflement endothélial, rupture de la membrane endothéliale, rupture des jonctions serrées entre les cellules endothéliales et les cellules gliales adjacentes et finalement la rupture vasculaire complète (88). Cette séquence distincte d'événements à la BHE (transcytose et endocytose accrues suivie par le remodelage des jonctions serrées) met en évidence l'implication dynamique des deux voies transcellulaires et paracellulaires modulant la perméabilité de la BHE dans les heures et les jours après un AVC. L'évaluation spatio-temporelle des modifications de la barrière suggère que l'apparition du dysfonctionnement de la BHE correspond à la sévérité de l'ischémie (89).
1.3 Le système de l'activateur tissulaire du plasminogène
Trois approches ont été investiguées pour le traitement des patients ayant un AVC ischémique: la neuroprotection, la thrombolyse et le retrait mécanique du caillot sanguin. En contraste avec la neuroprotection qui s'est avéré majoritairement infructueuse, la thrombolyse par le tPA et le retrait mécanique du caillot sont les seuls traitements approuvés par la Food and Drug Administration (FDA) à ce jour (90, 91). Le tPA est en fait une sérine protéase produite de manière endogène par les cellules endothéliales du corps suite à un stress vasculaire, comme la formation d'un thrombus occludant un vaisseau, afin de lyser le caillot (92). Un thrombus consiste en des cellules sanguines piégées dans une matrice de fibrine. La dissolution enzymatique de ce caillot de fibrine est induite par la sérine protéase de type trypsine, la plasmine. Cette forme active de plasmine est générée par un précurseur qui se nomme le plasminogène. La conversion du plasminogène en plasmine requiert le clivage protéolytique par un activateur du plasminogène, qui chez les mammifères peut être soit le tPA ou l'activateur urokinase du plasminogène (93). Cette cascade de conversion est finement régulée par
19
différents inhibiteurs qui touchent à l'activité protéolytique du tPA - l'inhibiteur 1 et 2 des activateurs du plasminogène (PAI-1et PAI-2), la neuroserpine et la protéase nexine-1 (PN-1) — ou qui inhibent directement la plasmine — l’α2-anti-plasmine et l’α2macroglobuline (94, 95). Le tPA est normalement internalisé via une interaction avec la protéine apparentée aux récepteurs des lipoprotéines à faible densité 1 (LRP1) puis dégradé (96). Le tPA possède une demie-vie très courte d'environ 5 min après quoi il est dégradé au foie par LRP1 (97). Le tPA est disponible pour les cliniciens afin de dissoudre le caillot sanguin d'un AVC par la technologie recombinante (rtPA) (98, 99). Cependant, de plus en plus de preuves suggèrent que, dans le contexte d'un AVC ischémique aigu, le tPA affecte l'intégrité de l'unité neurovasculaire et pourrait avoir à la fois des effets protecteurs ou délétères, selon l'endroit de son action et son temps d'administration.
1.3.1 Fonction du système de l’activateur tissulaire du plasminogène
Le tPA est une glycoprotéine qui fait partie de la superfamille des sérines protéases et est composé de 5 domaines (100). La partie N-terminale s'appelle le domaine fibronectine ou en doigts et est impliqué dans la liaison du tPA à la fibrine (101). Ce domaine peut aussi interagir avec plusieurs récepteurs membranaires comme le LRP1 ou l'annexine 2 (102, 103). Ce sont ses différents domaines qui lui confèrent un effet pléiotropique au sein du système nerveux central. Le tPA a été identifié pour la première fois dans la circulation sanguine (104). Il y est principalement produit par les cellules endothéliales (105) mais, dans le parenchyme cérébral, il peut aussi être sécrété par plusieurs cellules: astrocytes, neurones, oligodendrocytes et microglies (106). Pour ce qui est du tPA exogène, il peut traverser la BHE via transcytose médié par LRP1 (107). Le tPA exerce plusieurs rôles dans le cerveau juvénile, adulte et âgé. Le tPA est nécessaire lors de l'embryogenèse dans les régions en cours de migration cellulaire et de remodelage tissulaire (108). Les souris déficiente en tPA démontrent une migration neuronale retardée comparé à leur équivalent sauvage (109). Le tPA possède aussi des propriétés neuroprotectrices et joue un rôle dans les processus de potentialisation à long terme (LTP) (110) et de dépression à long terme (LTD), qui sont considérés comme étant les supports cellulaires et moléculaires des
20
processus d'apprentissage et de mémoire (111). L'administration d'un inhibiteur de tPA est capable de stopper la phase terminale de la LTP (112). Le tPA participe aussi à la plasticité synaptique et au remodelage neuronal via ses actions de dégradation de la matrice extracellulaire (113) et son habilité à activer les pro-neurotrophines (BDNF, NGF) (114). Ainsi, il a été démontré que le tPA est un facteur important dans la mémoire spatiale (115), la réponse au stress, l'anxiété et l'apprentissage de la peur (116). Toutefois, le tPA exogène peut induire la mort neuronale par excitoxicité médiée par la suractivation des récepteurs NMDA (117). Les récepteurs LRP pourraient agir à titre de co-récepteurs pour le tPA, qui à leur tour augmenteraient l'influx de Ca2+ via les récepteurs NMDA (118). De manière
intéressante, en réponse au tPA, LRP1 a été décrit comme capable d'assembler un système de co-récepteurs pour initier la signalisation cellulaire (119). Cependant, en plus de son rôle dans la mort neuronale excitotoxique, les études in vivo, in vitro et ex vivo suggèrent également que le tPA peut avoir des effets pro-survie et anti-apoptotique sur les neurones et les oligodendrocytes (120-122). La plupart de ces études suggèrent que les effets trophiques du tPA se produisent indépendamment de son activité protéolytique, avec l'activation des voies de signalisation PI3K/Akt-, AMPK- ou mTor-HIF-1alpha-dépendantes (123). L'interaction du domaine fibronectine du tPA avec les récepteurs LRP1 (124) et annexine II (125) sont en partie responsables de son effet sur la modulation inflammatoire en favorisant l'activation microgliale, en induisant la production de NO et en induisant le recrutement de macrophages en plus de la production de cytokines (126-128). Il semblerait aussi que le tPA en soi puisse exercer une action de cytokine anti-inflammatoire (129).
Dans le cerveau âgé, l'activité protéolytique du tPA diminue, ce qui entraîne des conséquences fonctionnelles (130). Par exemple, la diminution des niveaux de tPA dans l'hippocampe entraîne une déficience dans la mémoire spatiale (131). Il existe aussi de nombreux liens dans la littérature entre le système tPA/plasmine et la maladie d'Alzheimer, incluant sur l'activité de la clairance du peptide A-β (132). Il est connu que la plasmine est capable de promouvoir la dégradation du peptide A-β (133). Dans la maladie d'Alzheimer, on note une augmentation sévère des
21
niveaux de PAI-1, induisant alors une diminution de l'activité protéolytique du tPA (130).
1.3.2 L'action de l'activateur tissulaire du plasminogène sur la barrière hémato-encéphalique
Le rtPA circulant exerce des effets modulateurs sur la perméabilité de la BHE et, suite à un stimulus comme l'ischémie, peut causer une toxicité vasculaire et neuronale menant à la rupture de la BHE et à la formation de transformations hémorragiques (134). Les effets délétères du rtPA à la BHE sont attribués à: l'activation des MMP, la liaison aux récepteurs LRP, la dégranulation des mastocytes et l'expression du facteur de croissance de l'endothélium vasculaire (VEGF).
De nombreuses études suggèrent que le tPA peut réguler à la hausse et activer différents membres de la famille MMP (en particulier MMP-3 et MMP-9). En fait, l'hypothèse de «tPA-induced MMP-9» est généralement acceptée et appuyée par de nombreuses preuves expérimentales (135). Les MMP dégradent les protéines des jonctions serrées et de la lame basale, entraînant une rupture de la BHE, un œdème cérébral, la mort des cellules neuronales et des hémorragies (136, 137). Des expériences in vivo ont montré que le traitement thrombolytique au tPA après un AVC embolique augmentait les taux de MMP-9 dans le cerveau et que l'administration concomitante d'inhibiteurs de MMP réduisait les transformations hémorragiques et les lésions cérébrales (138, 139). La claudine-5 et l'occludine, des composantes des jonctions serrées, contiennent des sites de clivage de MMP extracellulaires et constituent un substrat direct des MMP (140, 141), suggérant que les MMP peuvent dégrader les jonctions serrées directement. Il est connu que les perturbations structurelles des liens entre l'occludine et les filaments d'actine peuvent conduire à la modification de la perméabilité paracellulaire (142) et, ainsi, la dégradation de l'occludine par les MMP est donc susceptible de déclencher l'ouverture de la BHE (143). Bien que les mécanismes précis derrière cette activation restent à élucider, des études récentes suggèrent un rôle des LRP, auxquels le tPA a une forte affinité (144).
22
Le LRP est un des sites de liaison principal du tPA à la surface cellulaire (102). Le LRP est un membre de la famille des récepteurs des lipoprotéines et lie une variété de ligands associés à la matrice extracellulaire et serait principalement impliqué dans le métabolisme des lipoprotéines et dans la clairance des protéases (102). En outre, l'augmentation de la perméabilité de la BHE par le rtPA se produit via l'activation de LRP (107, 145, 146). En effet, le tPA injecté dans le liquide céphalorachidien en l'absence d'ischémie induit une augmentation dose-dépendante de la perméabilité vasculaire, effet qui n'est pas diminué chez les souris déficientes en plasminogène, suggérant l'implication de d'autres acteurs que le plasminogène et les MMP (145). Des antagonistes des LRP sont capables d'abolir les effets délétères du tPA, indiquant que ce dernier agit bel et bien via les LRP (145). L'expression des LRP à la surface cellulaire est augmentée dans les cellules endothéliales sous stress ischémique, et l'activation du LRP par la liaison à la rtPA stimule les voies de signalisation telles que la voie NF-κB (147).
Le tPA peut aussi avoir un effet sur les mastocytes, ce qui aggraverait le dommage de reperfusion après une ischémie cérébrale. Des expériences in vitro ont montré que le tPA stimule directement la dégranulation des mastocytes, libérant ainsi des substances vasoactives préformées, des enzymes protéolytiques, des anticoagulants et des facteurs chimiotactiques qui vont pouvoir causer ou aggraver l'hémorragie cérébrale et la lésion de reperfusion (148).
Des études récentes suggèrent aussi que les effets du tPA à la BHE pourraient être attribués par l'augmentation de l'expression du facteur de croissance de l'endothélium vasculaire (VEGF) par le récepteur LRP (149). Le VEGF sécrété se lie au récepteur 2 (VEGFR-2) à la surface des cellules endothéliales via un mécanisme autocrine et induit sa phosphorylation. L'activation du récepteur conduit à une augmentation de l'endocytose, entraînant une augmentation de la perméabilité de la BHE (143). Bien que le tPA possède de nombreux effets délétères sur la BHE, il est encore aujourd'hui le seul traitement disponible suite à un AVC ischémique.
23
1.3.3 L'activateur tissulaire du plasminogène dans les accidents vasculaires cérébraux et ses limitations
L'utilisation du tPA comme thérapie thrombolytique pour recanaliser une artère obstruée prend sa source dans deux essais randomisés contrôlés par placebo qui ont montré un bénéfice substantiel d'un traitement précoce par le rtPA chez des patients avec AVC ischémique aigu (90, 150) et plusieurs méta-analyses d'essais randomisés appuient l'utilisation du rtPA dans les 4,5 heures suivant le début de l'AVC ischémique (151, 152). Cependant, l'adoption généralisée de la thérapie thrombolytique par le rtPA a progressée lentement. Parmi les différents obstacles limitant l'utilisation de rtPA, sa fenêtre thérapeutique étroite a été la plus importante (153). L'étude européenne sur les accidents vasculaires cérébraux a montré que le rtPA administré entre 3 et 4,5 h après l'apparition des symptômes améliorait significativement les résultats cliniques des patients avec un AVC ischémique aigu, mais augmentait par 10 le risque d'hémorragies intracrâniennes symptomatiques (150). Il a été aussi confirmé qu'un traitement donné au-delà de 4,5 heures n'était pas associé à un avantage statistiquement significatif, mais plutôt à une augmentation des dommages au cerveau (154). Ainsi, certains médecins sont réticents à prescrire le rtPA suite à un AVC, de peur d'engendrer des transformations hémorragiques. Pour répondre à cette crainte, il existe des directives strictes concernant les critères d'inclusion et d'exclusion pour le traitement de l'AVC aigu avec le rtPA conçus pour minimiser le risque de complications hémorragiques. Ces critères stricts, combinés à une fenêtre thérapeutique étroite de 4,5 heures, ont pour conséquence que moins de 5% des patients ayant eu un AVC et qui sont éligibles à la thérapie thrombolytique peuvent en bénéficier (155). Administré en dehors de la fenêtre thérapeutique, le rtPA devient alors neurotoxique, entraîne des complications hémorragiques et cause une augmentation de la morbidité et de la mortalité chez les patients (156, 157) .
1.4 La voie de signalisation Wnt/ β-caténine
La voie de signalisation Wnt/β-caténine (de wingless et integration site) régule plusieurs processus biologiques et cellulaires dans le tissu en homéostasie comme la prolifération, l'apoptose, la polarité, la différentiation et la pluripotence des