Impact du génotype de l'ACTN3 sur la conservation et
l'évolution de la force musculaire chez les individus
atteints de dystrophie myotonique de type 1
Mémoire
Xavier Allard-Chamard
Maîtrise en sciences cliniques et biomédicales - avec mémoire de l’Université
Laval
offert en extension à l’Université du Québec à Chicoutimi
Maître ès sciences (M. Sc.)
Université du Québec à Chicoutimi
Chicoutimi, Canada
Médecine
Université Laval
Québec, Canada
Impact du génotype de l’ACTN3 sur la conservation et
l’évolution de la force musculaire chez les individus
atteints de dystrophie myotonique de type 1
Mémoire
Xavier Allard-Chamard
Sous la direction de :
Résumé
Le but de cette investigation vise à analyser l’impact du génotype de l’ACTN3 sur la conservation et l’évolution de la force musculaire chez les individus atteints de DM1. Cette étude se veut une recherche longitudinale, les patients ayant été évalués sur deux périodes séparées de 9 ans (temps 1 et temps 2). L’analyse de l’impact du génotype de l’ACTN3 comporte deux volets. Le premier volet a pour objectif d’identifier si l’absence de protéine alpha-actinine 3 (génotype 577XX) provoque chez les personnes atteintes de DM1 un niveau de force musculaire global plus faible que chez l’individu des deux autres génotypes, en l’occurrence 577RX et 577RR. Le deuxième volet, quant à lui, a pour objet la comparaison de la perte de force musculaire entre le temps 1 et le temps 2 des trois génotypes.
Les patients ont été recrutés dans le registre de la clinique neuromusculaire du Saguenay. Les personnes invitées à participer à cette étude devaient avoir été testées par une analyse génétique confirmant la présence de la maladie (phénotype adulte et tardif) et être âgées de 18 ans ou plus. Un total de 113 participants, soit 42 hommes et 71 femmes, a été en mesure de compléter cette étude longitudinale. Ces derniers ont été évalués à l’aide de 17 tests musculaires.
Les résultats du temps 2 ont montré que l’absence de la protéine alpha-actinine 3 chez les hommes atteints de DM1 induisait un niveau de force musculaire global plus faible que chez ceux de génotype 577RX. En effet, le sexe masculin de génotype 577XX a vu sa force décroître plus rapidement au fil des années. Somme toute, si l’encadrement le permet, connaître le profil génotypique associé au gène ACTN3 chez les hommes atteints de DM1 est un élément important à prendre en considération afin de ralentir la progression de la maladie et d’optimiser les stratégies de réadaptation.
Abstract
The purpose of this investigation was to analyze the impact of the genotype of ACTN3 on the conservation and evolution of muscle strength in individuals with DM1. This study was a longitudinal study, the patients having been evaluated on two different occasions with 9 years in between (time 1 and time 2). The analysis of the impact of the ACTN3 genotype had two components. The first part aimed at identifying whether the absence of alpha-actinin 3 protein (genotype 577XX) caused a lower level of overall muscular strength in people with DM1 when compared to the other two genotypes, in this case 577RX and 577RR. The second part aimed at comparing the loss of muscle strength between time 1 and time 2 for the three genotypes.
Patients were recruited from the Saguenay Neuromuscular Clinic Registry. Those invited to participate in this study had been previously tested by a genetic analysis confirming the presence of the disease (adult and late phenotype) and be 18 years of age or older. A total of 113 participants, 42 men and 71 women, were able to complete this longitudinal study. These were assessed using 17 muscle tests.
At time 2, results showed that the absence of alpha-actinin 3 protein in men with DM1 induced a lower level of overall muscle strength than men with 577RX genotype. Indeed, the male genotype 577XX had a more rapid decrease in strength over the years. In light of these results, knowing the genotype profile associated with the ACTN3 gene in men with DM1 is an important element to consider in order to optimize rehabilitation strategies.
Table des matières
Résumé ... iii Abstract ... iv Liste des figures ... vii Liste des tableaux ... viii Liste des abréviations ... ix Remerciements ... x INTRODUCTION ... 1 CHAPITRE 1 ... 3 REVUE DE LITTÉRATURE ... 3 1.1 Mécanisme général musculaire ... 3 1.1.1 Structure microscopique du muscle ... 3 1.1.2 Typologie musculaire ... 4 1.1.3 Contraction musculaire ... 5 1.1.4 Adaptation à l'entraînement ... 6 1.2 Physiologie de l’alpha-actinine ... 6 1.2.1 La famille alpha-actinine ... 6 1.2.2 Structures du domaine ... 8 1.2.3 Interactions protéiques ... 11 1.2.4 Polymorphisme ... 12 1.3 Impacts du gène ACTN3 sur le vieillissement ... 14 1.3.1 Composition corporelle ... 14 1.3.2 Capacité fonctionnelle ... 15 1.3.3 Réponse à l’entraînement ... 16 1.4 Impacts du gène ACTN3 chez les sportifs ... 17 1.4.1 Performance musculaire ... 17 1.4.2 Dépense énergétique ... 18 1.4.3 Flexibilité ... 18 1.4.4 Blessures ... 19 1.4.5 Profils des athlètes ... 19 1.5 Impacts du gène ACTN3 dans différentes maladies ... 21 1.5.1 Maladies cardiovasculaires ... 21 1.5.2 Maladies neuromusculaires ... 22 1.6 Dystrophie myotonique de type 1 ... 24 1.6.1 Définition ... 24 1.6.2 Nombre de répétitions CTG ... 24 1.6.3 Prévalence ... 25 1.6.4 Signes et symptômes de la DM1 ... 26 1.6.5 Catégorisation de la DM1 (échelle de MIRS) ... 29 1.6.6 Réponse à l’entraînement chez les individus atteints de DM1 ... 30 1.7 Formulation des hypothèses de recherche ... 32 CHAPITRE 2 ... 34 MATÉRIELS ET MÉTHODES ... 342.1 Participants ... 34 2.2 Évaluation de la force musculaire ... 35 2.3 Caractéristiques sociodémographiques et cliniques des participants ... 35 2.4 Prises des mesures ... 36 2.5 Identification des génotypes ... 36 2.6 Statistiques ... 36 CHAPITRE 3 ... 38 RÉSULTATS ... 38 3.1 Mesures anthropométriques ... 38 3.2 Comparaison de la force musculaire entre les génotypes ... 40 3.3 Différence de perte de force musculaire selon les 3 génotypes ... 48 3.4 Taille d’effet du pourcentage de la force musculaire selon les 3 génotypes ... 52 3.5 Production de la force maximale en fonction du nombre de répétitions CTG pour tous les génotypes confondus ... 55 3.6 Production de la force maximale en fonction du nombre de répétitions CTG pour chacun des génotypes ... 60 3.7 Pourcentage de perte de la force maximale en fonction du nombre de répétitions CTG pour chacun des génotypes ... 73 CHAPITRE 4 ... 81 DISCUSSION ... 81 4.1 Hypothèses de recherche ... 81 4.2 Résultats inattendus ... 83 4.3 Nombre de répétitions CTG ... 85 4.4 Mesures anthropométriques ... 86 4.5 Mesure de la taille d’effet ... 88 4.5.1 Pourcentage de perte de la force musculaire ... 88 4.5.2 Nombre de répétitions CTG ... 89 4.6 Impact du génotype pour la prise en charge ... 90 4.7 Forces et limites de l’étude ... 91 CONCLUSION ... 92 PERSPECTIVES ... 93 BIBLIOGRAPHIE ... 94
Liste des figures
Figure 1 Localisation et structure des domaines du sarcomère
Figure 2 Représentation de l’interaction entre l’actine et la dystrophine
Figure 3 Structure de l’alpha-actinine : représentation du système mécanique des filaments d’actine.
Figure 4 Structure de l’alpha-actinine : représentation de l’ABD possédant deux domaines d’homologie à la calponine (CH1 et CH2), les quatre motifs SLR (SR) numérotés de 1 à 4 à partir de l’extrémité N ainsi que le domaine CaM comportant deux domaines EFh
Figure 5 Résultats de PCR sur un gel de polyacrylamide (8%) illustrant les génotypes 577XX, 577RX et 577RR
Liste des tableaux
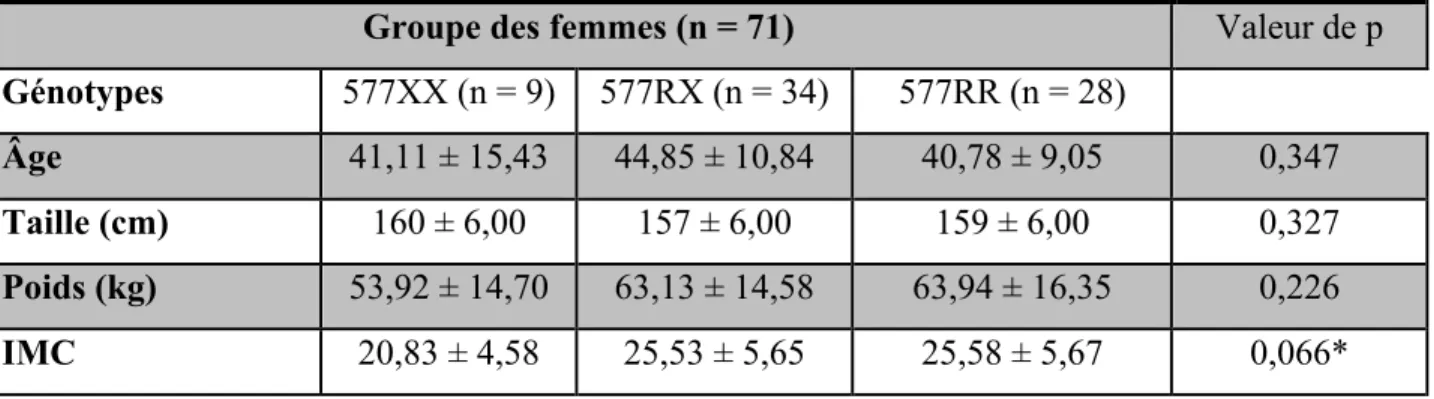
Tableau 1 Statistiques descriptives de l’âge et des mesures anthropométriques des hommes au temps 1
Tableau 2 Statistiques descriptives de l’âge et des mesures anthropométriques des femmes au temps 1
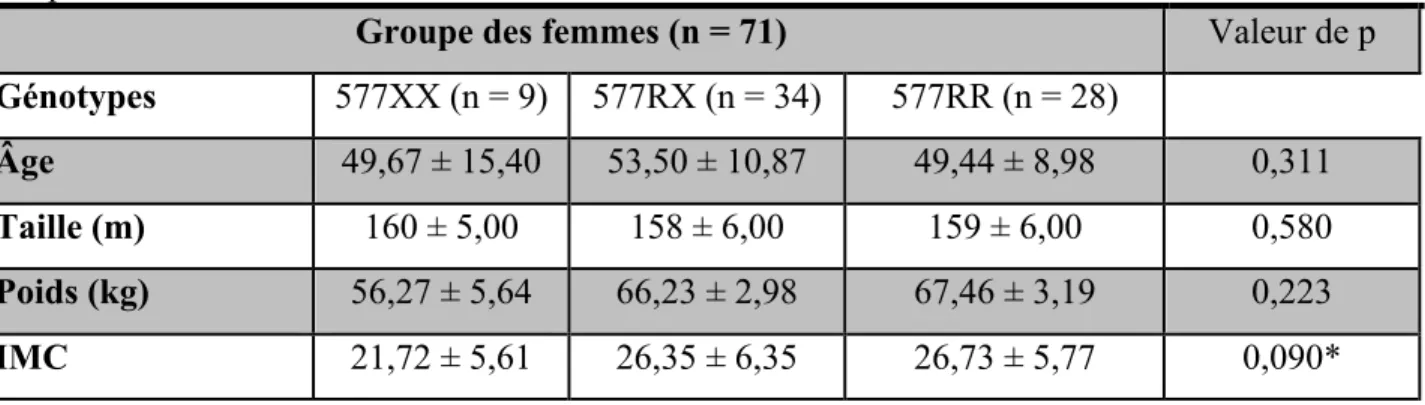
Tableau 3 Statistiques descriptives de l’âge et des mesures anthropométriques des hommes au temps 2
Tableau 4 Statistiques descriptives de l’âge et des mesures anthropométriques des femmes au temps 2
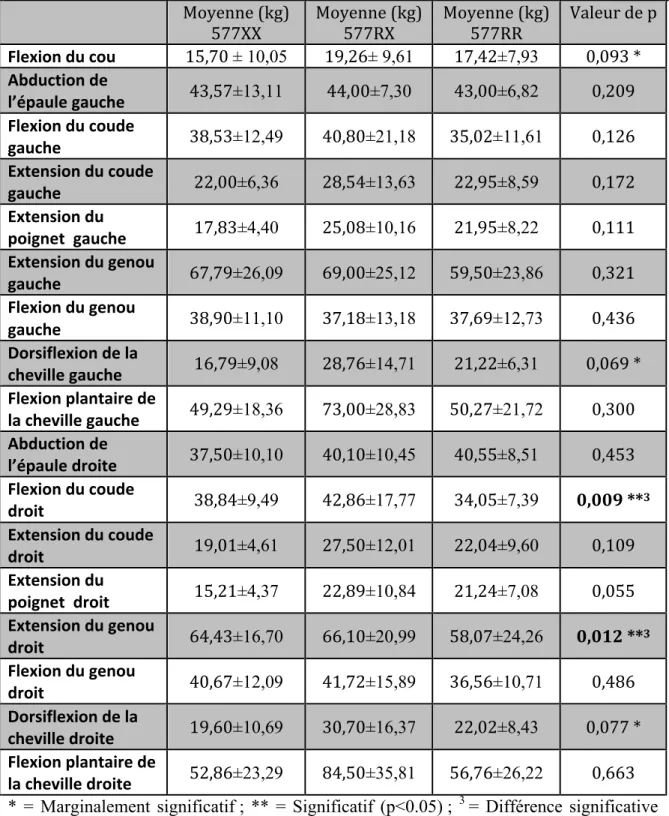
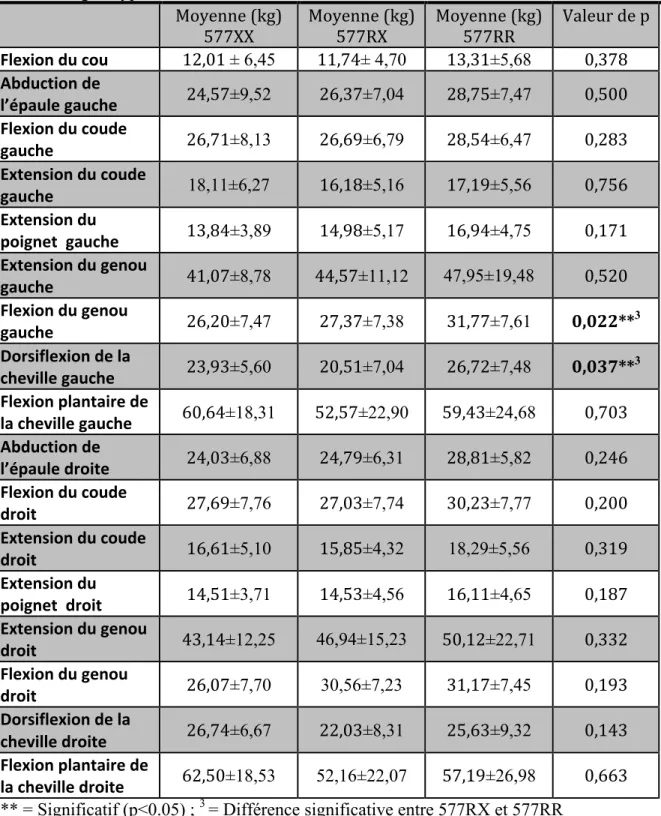
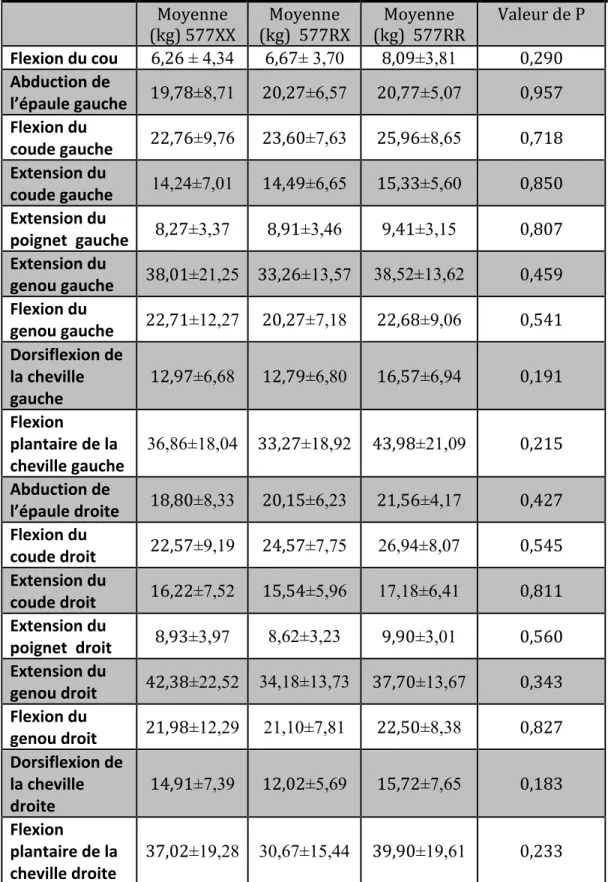
Tableau 5 Comparaison des moyennes de la force musculaire chez les hommes au temps 1 selon les génotypes
Tableau 6 Comparaison des moyennes de la force musculaire chez les femmes au temps 1 selon les génotypes
Tableau 7 Comparaison des moyennes de la force musculaire chez les hommes au temps 2 selon les génotypes
Tableau 8 Comparaison des moyennes de la force musculaire chez les femmes au temps 2 selon les génotypes
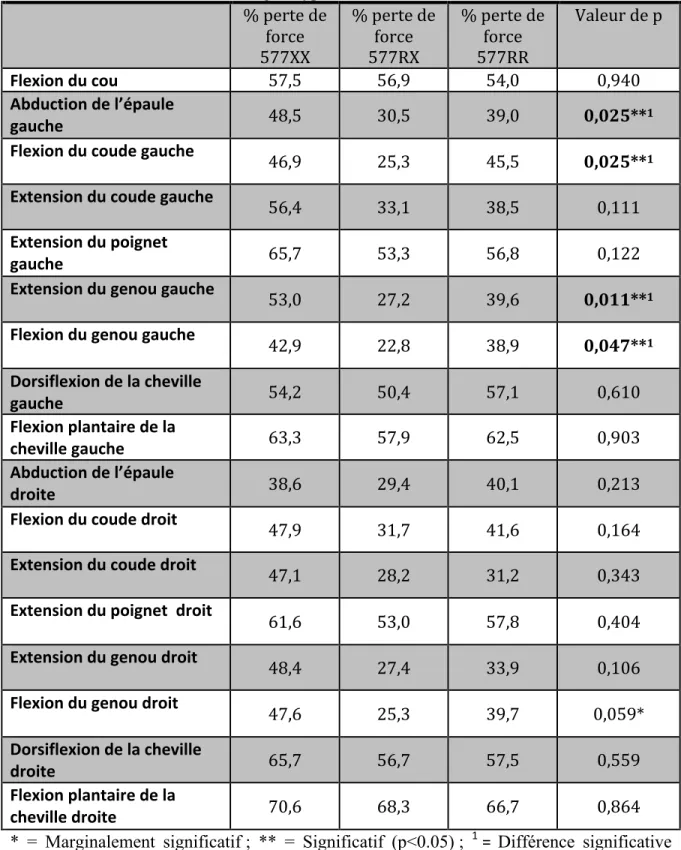
Tableau 9 Analyse de variance du pourcentage de perte de force musculaire chez les hommes selon les 3 génotypes
Tableau 10 Analyse de variance du pourcentage de perte de force musculaire chez les
femmes selon les 3 génotypes
Tableau 11 Taille d’effet du pourcentage de perte de force musculaire selon les 3
génotypes chez les hommes
Tableau 12 Taille d’effet du pourcentage de perte de force musculaire selon les 3
Liste des abréviations
ADN Acide désoxyribonucléique
DM1 Dystrophie myotonique de type 1 (maladie de Steinert)
ABD Domaine N-terminal de liaison à l’actine
SLR ou SR Domaine central en bâtonnets constitué de motifs spectrines répétés en série
CaM Domaine C-terminal d’homologie à la calmoduline
CH1 – CH2 Domaines d’homologie à la calponine EFh Domaines en main EF
SNPs Polymorphismes mononucléotidiques
ACTN3 Gène responsable de la protéine alpha-actinine 3
Pb Paire de bases
PROMM Myopathie myotonique proximale (dystrophie myotonique de type 2)
EMG Électromyogramme
MIRS Muscular impairment rating scale
Kg Kilogramme
PCR Polymerase chain reaction
Remerciements
Poursuivre ses études au deuxième cycle après avoir réalisé un baccalauréat n’est pas un choix qui se fait à la légère. Plusieurs questionnements et hésitations sont au rendez-vous. C’est pourquoi, le soutien de nos proches est un élément, quant à moi, essentiel à la réussite d’une maîtrise de recherche. Personnellement, je ne pouvais pas être mieux épaulé.
Avant tout, je tiens à remercier et à exprimer ma profonde gratitude à mon Directeur de recherche le Dr Mario Leone. Sans sa supervision et son encadrement, rien de tout cela n’aurait pu être possible.
Merci à toute l’équipe du Groupe de Recherche sur les Maladie Neuromusculaire (GRIMN) et plus particulièrement au Dre Cynthia Gagnon qui m’a permis d’avoir accès à cette base de données unique. Leur professionnalisme ainsi que leur compétence ont permis l’avancée scientifique dans une des maladies neuromusculaires les plus répandues au Saguenay.
Bien évidemment, je ne peux passer sous silence le support inconditionnel que m’a apporté ma conjointe Andréanne. Son immense aide et sa bonne humeur m’ont permis de passer au travers les moments les plus difficiles de mon parcours scolaire. Je serai éternellement reconnaissant de son don de soi.
Merci à toute ma famille, tout particulièrement à ma mère Jocelyne et à mon frère Hugues qui ont cru en moi jusqu’à la fin. Vos encouragements et conseils m’ont été d’une importance majeure.
Finalement, un merci tout particulier à mon bon ami William. Nos journées et nuits de travail auront porté leurs fruits. Je meurs d’impatience de recommencer l’expérience avec toi pour… le doctorat !
INTRODUCTION
L’évolution des technologies géniques a été une avancée scientifique majeure à partir du 19ième siècle. La théorie de transmissions des gènes élaborée par Gregor Mendel ainsi que la découverte de la structure de l’acide désoxyribonucléique (ADN) réalisée par Rosalind Franklin, Francis Crick et James Watson (1953) ont amené les savants à accorder une importance particulière à notre patrimoine génétique (Carroll et al., 2013). Depuis, l’hérédité est un aspect que les individus n’ignorent plus. Une multitude de gènes, responsable de nos caractéristiques génotypiques et phénotypiques, est recensée dans le génome humain régulant, entre autres, les fonctions des protéines (Carroll et al., 2013). Parmi les 20 000 gènes que possède l’humain, approximativement 900 sont reconnus pour avoir un impact sur les fonctions musculaires (NCBI, 2018). L’ACTN3, responsable de la protéine alpha-actinine 3, est un de ces gènes découvert en 1992 (Yuruker & Niggli, 1992). Approximativement 16% des individus au niveau mondial souffrent d’une déficience en alpha-actinine 3 et 18% des caucasiens, soit environ une personne sur cinq, ne possède pas le gène ACTN3 (North et al., 1999). De plus, 25% des asiatiques ne semblent pas posséder ce gène contrairement aux Zoulous, un des peuples de l’Afrique du Sud, qui quant à lui, le possèdent à 99% (North, 2008).
Bien que l’ACTN3 ait fait l’objet d’études pour ses effets sur la fonction musculaire d’athlètes de haut niveau, de plus en plus de recherches se réalisent sur son impact dans certaines pathologies. En effet, des investigations sont effectuées sur le gène ACTN3 ainsi que sur sa protéine alpha-actinine 3 afin d’obtenir un portrait global de certaines maladies, la plupart d’origine musculaire (Pickering & Kiely, 2018). Bien qu’aucune pathologie précise ne soit associée à la présence ou à l’absence de ce gène, ce dernier pourrait toutefois influencer l’évolution des personnes atteintes de maladies spécifiques (Bernardez-Pereira et al., 2014). Malgré l’importance d’améliorer l’état fonctionnel des patients, plusieurs affections musculaires n’ont pas été étudiées résultant en un manque d’approfondissement de l’influence des génotypes de l’ACTN3. D’ailleurs, aucune recherche n’a été réalisée à ce jour sur la dystrophie myotonique de type 1 (DM1) et
le lien avec l’ACTN3. Selon le stade de la maladie, les capacités fonctionnelles des personnes atteintes de DM1, sont sévèrement touchées ce qui résulte en une diminution considérable de leur qualité de vie. Il s’avère donc digne d’intérêt de documenter l’influence des génotypes de l’ACTN3 sur cette maladie neuromusculaire étant donné les retombées thérapeutiques que cela pourrait engendrer.
CHAPITRE 1
REVUE DE LITTÉRATURE
1.1 Mécanisme général musculaire
1.1.1 Structure microscopique du muscle
Lorsqu'isolée, chaque fibre musculaire représente un long et fin cylindre subdivisé en plusieurs compartiments. Celui régissant les propriétés contractiles se nomme myofibrille. Les myofibrilles se retrouvent dans le cytoplasme musculaire et contiennent deux types de filaments protéiques (myofilaments) : la myosine et l'actine. Le myofilament nommé myosine est caractérisé par d'épais filaments tandis que celui de l’actine est composé d'une structure très fine. Ces deux types de filaments donnent une apparence striée aux muscles squelettiques (Wilmore et al., 2017).
Les myofibrilles sont divisées en segments nommés sarcomères, séparés par une zone appelée ligne Z. À l'intérieur des sarcomères, une portion sombre désignée bande A, comprend les filaments épais de myosine. Au contraire, la portion plutôt claire des sarcomères, la bande I, est constituée des filaments fins d'actine. De plus, les filaments d'actine recouvrent en partie les filaments de myosine. Conséquemment, les filaments d'actine sont localisés aussi au niveau de la bande sombre. L'endroit où aucun filament d'actine ne recouvre la myosine est nommé la zone H (Figure 1) (Wilmore et al., 2017).
Figure 1 : Localisation et structure des domaines du sarcomère. Représente (à l’aide des
flèches en pointillés) la direction des forces émises par les mécanismes musculaires. (Adapté selon McArthur, 2004)
1.1.2 Typologie musculaire
La notion de continuum (continuité) est à considérer lors de la classification des fibres musculaires. Bien que chacune des catégories des fibres musculaires soit associée à un système énergétique propre, le recrutement de tous les types de fibres est observé, quelle que soit l’ampleur de la contraction musculaire. En effet, selon le système énergétique sollicité, la différence essentielle réside dans la proportion des types de fibres recrutés (Pette, 1980).
1.1.2.1 Fibres à contraction rapide
Communément appelées fibres de type IIX ou encore fibres glycolytiques, ces dernières comprennent un faible nombre de mitochondries, organelles essentielles pour la production d'énergie. Ces fibres musculaires sont caractérisées par un métabolisme aérobie faible et disposent d'une moins grande résistance à la fatigue que les fibres à contraction lente. Contrairement aux fibres lentes, leurs avantages se situent au niveau de leur richesse en enzyme glycolytique et en glycogène, ressources qui leur confèrent une excellente
capacité à synthétiser des ATP rapidement tout en libérant du lactate. De surcroit, le nombre élevé d'enzymes ATPase optimise l'activité ATPasique ce qui engendre une contraction musculaire importante en raison d'une grande vitesse de constitution et de rupture des points d'union (Pette, 1980).
1.1.2.2 Fibres à contraction lente
Les fibres oxydatives, également nommées fibres de type I, sont constituées d’une kyrielle de mitochondries et de capillaires. La myoglobine, protéine musculaire utilisée comme navette sur lequel se fixe l'oxygène, lui confère une pigmentation rouge. Ce réseau de ramifications favorise l'apport de l'oxygène ainsi que son utilisation. Conséquemment, les fibres à contraction lente possèdent une excellente résistance à la fatigue musculaire, caractéristique prédominante du système énergétique dit aérobie (Pette, 1980).
1.1.2.3 Fibres à contraction intermédiaire
Ces faisceaux hybrides sont appelés fibres rapides glycolytiques et oxydatives ou fibres de type IIa. Cette dénomination réfère à des caractéristiques mitoyennes entre les fibres à contraction rapide et lente. De ce fait, les fibres intermédiaires servent de transition entre les fibres oxydatives et les fibres glycolytiques en réponse à une nouvelle charge d'exercice. Ce phénomène porte le nom de phase d'adaptation (Pette, 1980).
1.1.3 Contraction musculaire
Les motoneurones, cellules nerveuses innervant les fibres musculaires, sont responsables de stimuler le muscle squelettique afin que ce dernier puisse développer une tension et générer une force mécanique sur les os. Cette contraction musculaire est un enchaînement ordonné engageant la myosine et l'actine par le biais du métabolisme de résynthèse de l'ATP. Le raccourcissement musculaire se produit par le glissement des
myofilaments d'actine sur les myofilaments de myosine résultant en une diminution de la distance entre deux lignes Z utilisés comme ancrage. En outre, le sarcomère perd en longueur par la diminution des bandes H et I. Un nombre élevé en myofibrilles signifie que la cellule contient une quantité importante de ponts d'union actomyosine ce qui engendrera un grand déploiement de force. Le nombre de ponts d'union qui entre en contact à chaque instant indique la quantité de force générée par le muscle. Ce phénomène permet la réalisation des tâches quotidiennes, notamment la locomotion (Billat, 2017).
1.1.4 Adaptation à l'entraînement
Les fonctions musculaires, assujetties à plusieurs conditions complexes, sont malléables par leur plasticité musculaire. L'adaptation du muscle à l'entraînement se réalise grâce à divers mécanismes tels qu'un accroissement de la surface des fibres musculaires, une modification des tissus conjonctifs et capillaires et une augmentation de l'ultrastructure du muscle. De surcroit, des adaptations morphologiques et biochimiques du muscle surviennent à court et à long terme. Ces adaptations permettent d'accroître les performances physiques à des fins sportives, mais aussi pour des fins fonctionnelles (Billat, 2017).
1.2 Physiologie de l’alpha-actinine
1.2.1 La famille alpha-actinine
L’alpha-actinine est une protéine globulaire appartenant à la superfamille des actines. En physiologie, elle est responsable de lier plusieurs protéines, dont la dystrophine, les B-spectrines et l’utrophine. Ces protéines sont caractérisées par la présence d’un domaine central d’unités répétitives en « bâtonnets » bordé, de part et d’autre, par un domaine de liaison au calcium en C-terminal et d’un domaine de liaison à l’actine en N-terminal (Beggs et al., 1992). L’actine fait partie d’une famille de protéine ubiquitaire retrouvée chez les mammifères (Beggs et al., 1992), les oiseaux (Arimura et al., 1988) ainsi que chez les invertébrés (Barstead, Kleiman & Waterston, 1991). Au cours de l’évolution,
l’addition de site d’épissage alternatif combiné à la duplication du gène de l’actine a engendré la génération de plusieurs variantes chez les mammifères. En tout, on dénote quatre gènes exprimés produisant un total de six isoformes distinctes. Par contre, chez l’humain seulement 4 sont activement transcrites. Chacune de ses protéines a un profil d’expression tissulaire propre (Beggs et al., 1992). En se basant sur leur dépendance au calcium, leurs propriétés physico-chimiques, leurs dynamiques d’expression et leurs localisations subcellulaires, ces protéines peuvent être a priori classifiées en deux catégories distinctes : les isoformes cytosquelettiques sensibles au calcium et les isoformes sarcomériques insensibles au calcium. La fonctionnalité de ces isoformes est influencée par le type cellulaire dans lequel elles sont exprimées (Blanchard, Ohanian & Critchley, 1989).
Chez les vertébrés, l’alpha-actinine est présente dans les muscles striés squelettiques, muscles cardiaques et muscles lisses (Beggs et al., 1992). L’alpha-actinine 1 et 4 est cytosquelettique tandis que conjointement l’alpha-actinine 2 et 3 forment le groupe sarcomérique. Leur expression est gouvernée respectivement par les gènes ACTN1, ACTN2, ACTN3 et ACTN4. Contrairement à d’autres familles de gènes concentrés en un seul locus, leurs positionnements chromosomiques sont épars ; ils sont situés au niveau du chromosome 14 (14q-22-24), 1 (1Q-42-43), 11 (11Q-13-14) et 19 (19Q-13-2) (Vainzof et al., 1997).
Dans le muscle squelettique, ces actinines sont concentrées dans une région sarcomérique nommée : « lignes Z ». Ces lignes forment une structure ancrant l’ensemble des filaments musculaires. De surcroît, malgré un rôle mécanique bien documenté (Billat, 2003), les actines représentent un groupe de joueurs prépondérants dans l’intégration des cascades de signalisation et des voies métaboliques gouvernant l’activité musculaire. Durant l’évolution des mammifères, l’alpha-actinine 2 a acquis la capacité de contrôler les signaux toniques du système oxydatif des fibres des muscles squelettiques. Elle est d’abord présente dans les types de fibres musculaires de type I dites lentes et en moindre quantité dans les fibres de type IIA, soit une sous-catégorie des types de fibres rapides, où elle ne représente que 50% des alpha-actinines présentent. En ce qui a trait à l’alpha-actinine 3, cette dernière est uniquement recrutée lorsque le système glycolytique s’active
secondairement à une utilisation accrue des fibres musculaires rapides (100% dans les fibres de type IIX et 50% dans les fibres de type IIA) (MacArthur & North, 2004). Cette protéine est essentielle afin d’obtenir des contractions musculaires optimales et rapides (Yang et al., 2009).
Malgré une forte homologie de structure et le fait qu’il serait en théorie possible que l’alpha-actinine 2 puisse compenser l’absence d’alpha-actinine 3, il existe des preuves solides suggérant que le gène ACTN3 ait été maintenu dans le génome conséquemment à des fonctions indépendantes de l’ACTN2. En effet, depuis sa divergence d’avec l’ACTN2, il y a 1300 millions d’années, la séquence ACTN3 est restée hautement conservée. De plus, ces gènes sont exprimés différemment, autant au niveau temporel que spatial, lors du développement embryonnaire. En effet, dans les modèles développementaux murins, l’expression de l’ACTN2 ne chevauche pas complètement celle de l’ACTN3 dans le muscle squelettique (Mills et al., 2001).
1.2.2 Structures du domaine
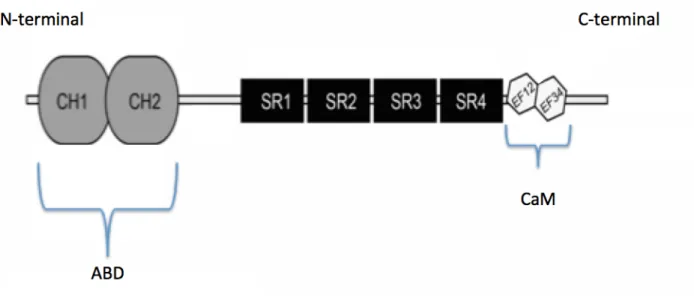
Les membres de la famille alpha-actinine sont définis par une structure en 3 domaines : (1) un domaine N-terminal de liaison à l’actine (ABD), (2) un domaine central en bâtonnets constitué de motifs spectrines répétés en série (SLR ou SR) et (3) un domaine C-terminal d’homologie à la calmoduline (CaM) (Figure 3 et Figure 4) (Sheterline, 1998).
Structuralement, l’ABD possède deux domaines d’homologie à la calponine nommés CH1 et CH2 disposés en tandem (Figure 4). Ces régions sont les principaux sites conservées dans la famille des alpha-actinines. Cette conservation reflète le maintien évolutif particulièrement fort au niveau de leur actine cible (Sheterline, 1998). L’architecture de ces ABDs se retrouve également dans de nombreuses autres protéines, y compris dans la famille des spectrines. Dans toutes ces protéines, les deux domaines CH interagissent de manière conjointe pour maximiser une liaison de haute affinité à l’actine (Gimona, 2002).
Le domaine central en bâtonnets est constitué d’une série répétitive de quatre motifs SLR numérotés de 1 à 4 à partir de l’extrémité N (Figure 3). Ce domaine est nettement moins conservé que l’ABD. En effet, le nombre exact de répétitions des motifs SLR dans le domaine des bâtonnets centraux a varié au cours de l’évolution. À titre d’exemple, les protéines de type alpha-actinine du protozoaire Entamoeba histolytica, du champignon
Schizosaccharomyces pombe et du parasite Trichomonas vaginalis semblent en posséder un
(Vire A, 2004), deux (Wu J-Q, 2001) et cinq respectivement (Bricheux, 1998). Ce phénomène est contrasté avec toutes les autres isoformes de vertébrés connus qui en comprennent quatre classiquement. De façon intéressante, on peut noter que certains membres de la famille comportent jusqu’à 30 motifs SLR (Thomas, 1997). La fonction la mieux définie des motifs SLR semble être la facilitation d’interactions protéine-protéine afin de permettre une homo/hétérodimérisation par contact entre les domaines SLR de deux molécules distinctes. Les partenaires d’interactions renferment un amalgame de protéines d’échafaudage et de signalisation (Thomas, 1997).
Le domaine C-terminal CaM comporte deux domaines en main EF (EFh pour «EF hands») (Figure 2). Évolutivement, ce site est le plus distinct d’un point de vue fonctionnel. Dans l’isoforme cytosquelettique, donc non sarcolemmique, les domaines EFh lient le calcium cytoplasmique et régulent le mouvement de la liaison à l’actine et à d’autres protéines (Tang, 2001). Au contraire, les isoformes sarcomériques (muscles striés et lisses) souffrent d’une délétion de plusieurs acides aminés ainsi que de plusieurs substitutions ce qui engendre une incapacité de liaison calcique (Noegel, 1987).
Figure 2 : Représentation de l’interaction entre l’actine et la dystrophine (Adapté selon
Henderson, 2012).
Figure 3 : Structure de l’alpha-actinine. Représente le système mécanique des filaments
d’actine. Les domaines ABD, EFh, SR ainsi que la vinculine régularisant les forces mécaniques de l’alpha-actinine (Adapté selon Shimin, 2017).
Figure 4 : Structure de l’alpha-actinine. Représente l’ABD possédant deux domaines
d’homologie à la calponine (CH1 et CH2), les quatre motifs SLR (SR) numérotés de 1 à 4 à partir de l’extrémité N ainsi que le domaine CaM comportant deux domaines EFh (Adapté selon Haywood et al., 2016).
1.2.3 Interactions protéiques
Compte tenu de leur propension à former des interactions homotypiques par la voie de leur domaine SLR, les partenaires principaux de liaison des alpha-actinines sarcomériques sont, en effet, elles-mêmes (Blanchard, 1989 et Flood, 1997). Celles-ci se juxtaposent les unes aux autres à contresens pour former des dimères. Les deux alpha-actinines sarcomériques humaines ont la propriété de s’assembler dynamiquement en homodimères ou hétérodimères avec une stabilité et affinité comparables. Ce phénomène suggère une importante conservation structure-fonction entre les deux isoformes (Chan, 1998). La dimérisation a deux retombées importantes sur la fonctionnalité des alpha-actinines : (1) elle donne naissance à une molécule bivalente comportant un ABD à chaque extrémité provoquant la réticulation des filaments d’actine et (2) elle amène le domaine CaM C-terminale d’une molécule d’alpha-actinine à proximité de l’ABD N-terminal de son partenaire de liaison. La conséquence immédiate de l’approximation de ces deux domaines est de permettre une corégulation entre les deux zones (Chan, 1998).
Bien que l’alpha-actinine sarcomérique ait une tendance naturelle à former des homodimères, plusieurs de ses fonctions sont gouvernées par des interactions hétérotypiques avec d’autres macromolécules. Pour n’en nommer que quelques-unes, on note une kyrielle de protéines structurantes, dont l’actine filamentaire (Kuhlman, 1994), la myotiline, la titine (Ohtsuka, 1997) de même que la nébuline (Nave, 1990) et la vinculine (Shimin, 2017). En outre l’alpha-actinine sarcomérique intervient au niveau de deux complexes protéiques majeurs en liant simultanément la dystrophine (Hance, 1999) et les intégrines (Otey, 1990). Cette double fonction permet la transduction de signaux extracellulaires vers des fibres musculaires au sarcomère (Hance, 1999).
Également, il a été démontré que les deux actines du muscle squelettique interagissent avec la famille des calsarcins, se situant dans les lignes Z des muscles striés. Ces groupes d’acides aminés entrent en contact avec la Ca2+ et la calmoduline influençant la calcineurine (Frey, 2002). Celle-ci est une protéine de signalisation à l’origine de l’hypertrophie musculaire ainsi que du type de fibre musculaire (Olson, 2000). Enfin, des enzymes impliquées dans le cycle de Krebs, soit le fructose-1,6-bisphosphatase et l’aldolase, interagissent avec les deux actines du muscle squelettique. Il est à préciser que leur influence spécifique reste encore incomprise (Masters, 1984).
1.2.4 Polymorphisme
Les polymorphismes mononucléotidiques (SNPs) ont retenu l’attention de la communauté scientifique étant donné leur capacité de prédire la susceptibilité de la morbidité associée à plusieurs pathologies (Cargill et al., 1999). En effet pour l’alpha-actinine 3, il a été observé qu’un changement de nucléotide, soit cytosine (C) à une thymine (T), à la position nucléotidique 1747 (C1747T) dans l’exon 16 du gène ACTN3 induirait une conversion de l’arginine (CGA) en un codon-stop (TGA) au 577e acide aminé (R577X). Ce changement physiologique résulte en une protéine tronquée qui est, par le fait même, non fonctionnelle et engendre sa dégradation prématurée. Chez les homozygotes affectés par ce SNP, la protéine alpha-actinine 3 est déficiente. On réfère à ces individus en parlant du génotype 577XX. La présence de la protéine alpha-actinine 3 dans les fibres
musculaires de type II témoignerait chez les homozygotes des allèles sauvages, correspondant à une personne de génotype 577RR. Le génotype davantage présent dans la nature est celui communément appelé l’hybride, soit 577RX (se référer à la figure 4) (North, 1999).
Figure 5 : Résultats de PCR sur un gel de polyacrylamide (8%) illustrant les génotypes
577XX, 577RX et 577RR. Le génotype 577RR démontre deux bandes (205 pb* et 85 pb), le génotype 577XX affiche trois bandes (85 pb, 97 pb et 108 pb), le génotype 577RX exprime 4 bandes (85 pb, 97 pb, 108 pb et 205 pb)
*pb = paire de bases
Finalement, la mutation créant une carence en alpha-actinine 3 (577XX) n’a démontré aucune association pathologique (Vainzof & Costa, 1997) et n’a pas été recensée chez d’autres mammifères (Mills et al, 2001). Néanmoins, la carence ou la présence de cette protéine influe sur d’autres aspects dont le vieillissement, les performances sportives
et la progression de certaines maladies (Barakat-Haddad et al., 2017; Delmonico et al., 2008; Ma et al., 2013).
1.3 Impacts du gène ACTN3 sur le vieillissement
1.3.1 Composition corporelle
La dégradation musculaire, appelée sarcopénie, est une manifestation courante chez les personnes âgées (Morley et al., 2001). Ce phénomène débute dès le début de l’âge adulte, soit vers 25 ans (Lexell, Taylor & Sjöström, 1988) et est caractérisé par une perte de masse maigre de 10% vers 40 ans et de 40% vers 70 ans. Ce changement physiologique se justifie, entre autres, par une altération graduelle du volume des fibres de type IIA et IIB (Porter, Vandervoort & Lexell, 1995). De surcroît, une masse adipeuse plus importante ainsi qu’une certaine fragilité osseuse témoigne d’un âge avancé (Bonnefoy, 2010; Breuil & Euller-Ziegler, 2004). Conséquemment, l’ACTN3 est un gène qui a intrigué les chercheurs chez les aînés par sa nature à influencer certaines fibres musculaires et par son implication au niveau des systèmes énergétiques (Olson & Williams, 2000).
L’influence de l’ACTN3 dans l’évolution de la masse maigre, indice de force musculaire (Kraemer & Ratamess, 2004), se révèle encore incomprise chez les aînés. Les individus de génotype 577XX, donc qui présentent une déficience totale de la protéine alpha-actinine 3, ont manifesté une différence significative au niveau de la masse musculaire dans trois études (Cho, Lee & Kang, 2017; Lima et al., 2011; Walsh et al., 2008). Ces dernières démontrent un risque accru de développer de la sarcopénie dès le jeune âge lorsqu’un individu est caractérisé par les deux allèles mutés. Une seule étude, ciblant les hommes caucasiens, n’a détecté aucune différence de la masse maigre entre les trois génotypes (McCauley, Mastana & Folland, 2010).
Le tissu adipeux chez les individus d’âge avancé semble être davantage important chez les personnes de génotype 577XX comparativement à celles des génotypes 577RR et
577RX (Walsh et al., 2008). Présenter le génotype 577XX semble donc constituer un désavantage pour la masse adipeuse chez les aînés. Il en serait de même pour la densité osseuse, qui elle, décroît d’une manière plus importante lorsqu’un allèle est muté (Cho et al., 2017). Au contraire, les individus du génotype 577RR bénéficieraient d’un effet protecteur diminuant ainsi les risques de fractures ostéoporotiques (Min, Lim & Kim, 2016).
1.3.2 Capacité fonctionnelle
Certaines évidences tendent à démontrer que la mutation R577X peut contribuer à la variation des fonctions musculaires chez les aînés. À ce jour, la plus grande recherche réalisée sur l’alpha-actinine 3 auprès des personnes âgées est celle de 2008 comportant 2568 caucasiens (Delmonico et al., 2008). L’objectif étant de mettre en lumière l’influence qu’a l’ACTN3 sur le processus du vieillissement, les participants devaient avoir l’âge minimal de 65 ans. Sur une période de 5 ans, les aînés de génotype 577XX démontrèrent une détérioration plus rapide au test de 400 mètres marche que leurs confrères et consœurs caractérisés par un ou aucun allèle muté. Les autres tests n’ont pas révélé de différences majeures entre les génotypes (Delmonico et al., 2008).
En plus de prédire la performance aux 400 mètres chez les personnes âgées, l’alpha-actinine 3 influencerait aussi l’équilibre (Masud & Morris, 2001). Considérant que le tiers des personnes de 65 ans et plus trébuche durant l’année, il est intéressant de dresser un profil des personnes plus sujettes à ce type d’accident (Masud & Morris, 2001). Selon Judson (2010), les individus de génotype 577RX ainsi que 577XX sont plus à risque de tomber comparativement à ceux de génotype 577RR. En effet, le génotype influence la capacité de rétablir l’équilibre après le commencement d’une chute. L’augmentation du risque de chute est de 33% chez les personnes de génotype 577XX. Il est à noter que la population de cette recherche n’était constituée que de femmes (Judson et al., 2010).
Deux études supplémentaires ont été réalisées sur les capacités fonctionnelles concernant l’importance du polymorphisme R577X chez les aînés. Contrairement aux
résultats significatifs de Delmonico (2008) et de Judson (2010), aucune association n’a été trouvée entre les génotypes de l’ACTN3 et la capacité fonctionnelle (Bustamante-Ara et al., 2010; McCauley et al., 2010). En effet, ces deux dernières études stipulent que les personnes âgées ont la même force et la même capacité d’ambulation quel que soit la nature des allèles. Nonobstant ces résultats, leurs nombres de participants étant beaucoup plus bas que les études de Delmonico (2008) et de Judson (2010), leur envergure apparaît moins importante.
1.3.3 Réponse à l’entraînement
Pour bien déterminer l’influence de l’expression de l’ACTN3 sur l’activité physique, Delmonico (2005) a évalué la réponse à un programme d’entraînement d’une durée de 10 semaines chez les aînés. Les individus devaient réaliser une extension du genou de la jambe droite trois fois par semaine. Suite à ce programme d’entraînement, tous les individus ont vu leur force s’accroître. Aussi, l’ACTN3 a vraisemblablement influencé la progression de la puissance chez les hommes. En effet, une plus grande expression du gène, soit chez les hommes de génotype 577RR et 577RX, a occasionné un gain en puissance plus notable (Delmonico, 2005). Selon cet auteur, davantage d’études doivent être réalisées afin de déterminer l’influence de ce gène puisque dans son étude la progression de la puissance n’a pas révélé de différence significative chez les femmes.
Lors du vieillissement, une déficience de la protéine alpha-actinine 3 se traduit donc par une accélération de la sarcopénie, une masse adipeuse plus importante ainsi que par une fragilité osseuse accentuée (Cho et al., 2017; Min et al., 2016; Walsh et al., 2008). De surcroît, les capacités fonctionnelles tendent à diminuer plus rapidement et, chez les aînés masculins possédant la protéine alpha-actinine 3, il serait plus facile d’accroître leur force musculaire (Delmonico et al., 2008).
1.4 Impacts du gène ACTN3 chez les sportifs
1.4.1 Performance musculaire
Une déficience de la protéine alpha-actinine 3 engendre une substitution par la protéine alpha-actinine 2 (MacArthur & North, 2004). Ce phénomène a pour conséquence de ralentir les propriétés métaboliques et physiologiques du système musculaire résultant en un amoindrissement des fibres rapides par une augmentation des fibres oxydatives. Ce changement physiologique pénalise le corps quant à la génération de force et de puissance. Conséquemment, la performance dans les sports qui exigent de la force et de la puissance musculaire en est affectée (MacArthur & North, 2004).
Ce phénomène a été avéré par Moran (2006) avec une population de 1084 grecs ayant effectué les tests physiques suivants : la force de préhension, le lancer au basketball, le saut vertical, le 40 mètres sprint, une course d’agilité ainsi qu’un test de VO2max. Les résultats de cette recherche ont démontré que parmi l’ensemble de ces tests, les personnes de génotype 577RR obtiennent un meilleur résultat dans l’épreuve du 40 mètres sprint, soit un test qui requiert une grande puissance musculaire (Moran et al., 2006).
De surcroît, les dommages musculaires induits par l’exercice ont été identifiées comme l’une des principales causes du ralentissement progressif chez une personne réalisant de la course à pied (Del Coso et al., 2013). De ce fait, une recherche a été menée afin de déterminer l’influence du gène ACTN3 sur les altérations musculaires (Del Coso, Valero, et al., 2017). Jusqu’à maintenant, la littérature stipule qu’un allèle X est plus enclin à créer un dommage musculaire en raison d’une déficience de la production de l’alpha-actinine 3 dans les fibres musculaires de type II (Clarkson, Hoffman, et al., 2005; Pimenta et al., 2012; Venckunas et al., 2012; Vincent et al., 2010). Dans le but de confirmer ce phénomène, 71 coureurs avec une expérience d’au moins trois marathons depuis les trois dernières années ont été ciblés pour participer à une étude. Avant et après le marathon, une épreuve de saut vertical a été réalisée par chacun des participants afin de mesurer la puissance de leurs jambes. Après l’accomplissement de cette compétition, les coureurs dont
les génotypes possédaient minimalement un allèle X ont rapporté un plus grand niveau de fatigue et des douleurs accrues à la région lombaire durant la course que les coureurs de génotype 577RR. De plus, les coureurs de génotype 577XX et 577RX affichaient un taux plus élevé en créatine kinase et en myoglobine, confirmant ainsi des dommages musculaires accrus. Par conséquent, ces derniers présentaient une réduction de la puissance des jambes dans leurs sauts verticaux nettement inférieur à celle des coureurs de génotype 577RR (Del Coso, Valero, et al., 2017). Toutefois, contrairement à une autre étude (Del Coso, Salinero, et al., 2017), les participants n’ont pas diminué leurs cadences de course malgré leur concentration plasmatique élevée en créatine kinase et en myoglobine. L’auteur note que l’âge, l’alimentation, le volume d’entraînement ainsi que l’intensité de la course peuvent avoir occasionné des répercussions dans cette recherche, mais qu’aucune n’a été contrôlée (Del Coso, Valero, et al., 2017).
1.4.2 Dépense énergétique
Toujours au niveau de la course, Pasqua et al (2016) a analysé la dépense énergétique requise chez les individus des trois différents génotypes. Un total de 150 hommes sains a participé à cette étude. La tâche consistait à courir à 10 et à 12 km/h à une vitesse constante. En analysant la biomécanique des mouvements de course, il est ressorti que les hommes de génotype 577RX nécessitaient un coût énergétique moindre comparativement aux participants de génotypes 577XX et 577RR. Ces résultats supportent le fait que le génotype 577RX est un profil intermédiaire pour les qualités musculaires de la force et de l’endurance. Par conséquent, cette combinaison permet à l’appareil locomoteur d’utiliser, de façon plus optimale, l’énergie exigée (Pasqua et al., 2016).
1.4.3 Flexibilité
La flexibilité est un autre paramètre analysé en lien avec l’ACTN3 (Kikuchi et al., 2017). Dans cette étude, les chercheurs ont réalisé l’évaluation de deux cohortes d’individus d’origine japonaise. La première comprenait 776 individus et la deuxième, 1257 personnes. La flexibilité a été évaluée grâce à un test de flexion du tronc en position
assise (sit-and-reach). Dans les deux groupes, les individus de génotype 577RR ont présenté nettement moins de mobilité articulaire que ceux des deux autres génotypes. L’auteur émet donc l’hypothèse que les personnes possédant des génotypes 577RX et 577XX sont vraisemblablement plus performantes dans les sports qui nécessitent une grande amplitude de mouvements comme par exemple la gymnastique (Kikuchi et al., 2017).
1.4.4 Blessures
Les chercheurs ont également tenté d’approfondir les liens entre l’alpha-actinine 3 et les lésions musculaires. Dans le cadre d’une étude menée par Myosotis et al. en 2007, un total de 257 joueurs de soccers professionnels ont été évalués. Après analyse, les joueurs de soccer possédant le profil 577XX ont été plus susceptibles de se blesser. Comparativement aux deux autres génotypes, ces derniers affichaient une incidence de blessures 2 fois et demie plus élevée. Cependant, aucune différence n’a été détectée pour les risques de se blesser chez les joueurs de génotype 577RX et ceux de génotype 577RR. Par ailleurs, la sévérité des blessures a également été un élément corrélé avec l’alpha-actinine 3. Les blessures enregistrées chez les joueurs de génotype 577XX étaient plus sévères que celles pour les personnes affichant les génotypes 577RX et 577RR. Finalement, les blessures des joueurs de génotype 577RX étaient plus graves que celles pour les joueurs de génotype 577RR (Myosotis et al., 2017).
1.4.5 Profils des athlètes
Considérant l’avantage physiologique important que peut procurer la protéine alpha-actinine 3, un profil des athlètes a été dressé afin de cerner la prépondérance ou non de génotypes spécifiques selon les sports.
En 2005, Niemi, durant son étude, s’est concentré sur le génome mitochondrial ainsi que sur l’alpha-actinine 3 auprès d’athlètes finlandais d’endurance et de sprint. Sa recherche a précisé que le génotype 577XX se retrouve en moins grand nombre chez les
sprinters que chez les athlètes d’endurance. L’auteur révèle qu’un sprinteur de compétition est plus sujet à posséder le gène ACTN3 lié à un génotype 577RR (Niemi & Majamaa, 2005).
Bien que l’étude de Niemi (2005) démontre l’avantage d’un individu de génotype 577RR dans les sports de puissance, présenter un génotype 577RX aurait tout de même un avantage. Dans ce sens, une recherche a été effectué chez 486 athlètes russes dans une variété de sports afin d’évaluer plusieurs qualités musculaires telles que l’endurance, la résistance et la puissance. Le nombre nettement supérieur de personnes de génotype 577RR mais également 577RX suggère une prédisposition aux sports de puissance (Druzhevskaya et al., 2008).
Les individus de génotype 577XX, bien que désavantagés dans les sports de puissance démontrent, quant à eux, un certain avantage dans les sports d’endurance comparativement aux individus de génotype 577RR. Dans un groupe composé de 118 femmes et 132 hommes pratiquant des sports d’endurance de compétition au niveau provincial ou national (aviron, athlétisme, marathon, cycliste de longue distance, natation (≥400 mètres)), les résultats obtenus supportent le fait que le génotype 577RR est grandement moins représentés chez ces athlètes qu’au sein d’une population sédentaire. De plus, les individus de génotype 577XX sont associés aux athlètes d’endurance. Il est à noter toutefois que ce phénomène n’a pas été observée chez les hommes mais seulement chez les femmes athlètes (Shang et al., 2010). De surcroît, chez 115 athlètes pratiquant du patinage de vitesse, une biopsie prélevée dans le quadriceps des athlètes de génotype 577XX a révélé une proportion plus élevée de fibres lentes et ces sportifs professionnels se retrouvaient dans le bassin des individus performant les épreuves des plus longues distances (4000 à 7500 mètres) (Ahmetov et al., 2011). Ces deux recherches (Ahmetov et al., 2011; Shang et al., 2010) établissent une association entre les athlètes d’endurance et le génotype 577XX.
Les résultats d’une seule étude vont à l’encontre des propos ci-dessus. En effet, elle suppose que le génotype 577RR est davantage présent dans une équipe de natation
performant des épreuves de moyenne distance (≥ 400 mètres, ≤1500 mètres) que dans la population en général. De plus, ce même génotype est davantage représenté dans cette équipe de nageurs que le génotype 577XX et 577RX. Nonobstant que le phénomène n’apparaisse que chez les femmes, l’auteur affirme que ses résultats sont en contradiction avec les études précédentes réalisées sur ce même sujet (Li et al., 2017).
Bien que les personnes n’ayant pas de déficience en alpha-actinine 3 soient plus sujettes à pratiquer un sport de force et de puissance et qu’au contraire, les athlètes avec une grande déficience de cette protéine soient plus enclines à pratiquer un sport d’endurance, le haut niveau sportif rend plus visible la distribution de ces génotypes (Yang et al., 2003).
La présence d’alpha-actinine 3 a une influence indéniable sur ces performances sportives. Bien que la littérature scientifique soit davantage en mesure de démontrer l’impact du génotype 577RR sur les performances nécessitant de la force musculaires, les génotypes 577RX et 577XX tendent à être avantagés pour d’autres aspects, dont la dépense énergétique aérobie et la flexibilité (Kikuchi et al., 2017; Pasqua et al., 2016). Néanmoins, la fréquence et la gravité des blessures demeurent amoindries chez les personnes de génotype 577RR (Myosotis et al., 2017).
1.5 Impacts du gène ACTN3 dans différentes maladies
La littérature scientifique recense peu d’études en ce qui concerne l’impact de l’ACTN3 sur l’évolution des différentes maladies. Les recherches essayant d’établir un tel lien ciblent des pathologies cardiovasculaires et majoritairement des maladies neuromusculaires en raison des fonctions de la protéine alpha-actinine 3.
1.5.1 Maladies cardiovasculaires
Dans le cœur, l’isoforme prédominant est l’alpha-actinine 2. Les études suggèrent qu’une déficience en alpha-actinine 3 engendre une substitution par l’alpha-actinine 2
(Mills et al., 2001). Conséquemment, les chercheurs ont analysé l’impact du polymorphisme R577X sur la mortalité chez les patients souffrant d’insuffisance cardiaque. Afficher un ou deux allèles mutés du gène ACTN3 ne constituerait pas un bon pronostic pour les individus atteints d’insuffisance cardiaque. En effet, les patients de génotype 577XX et 577RX présentent une durée de survie significativement inférieure par rapport aux personnes de génotype 577RR. De ce fait, le gène ACTN3 pourrait concourir à un effet protecteur chez les personnes souffrant d’une maladie du cœur qui résulterait en un accroissement de leur espérance de vie (Bernardez-Pereira et al., 2014).
1.5.2 Maladies neuromusculaires
Les maladies neuromusculaires, pathologies dites dégénératives, comprennent entre autres les différentes formes de dystrophie musculaire. Ces dernières sont caractérisées par un groupe hétérogène de troubles musculaires. Chaque trouble musculaire est associé à des gènes spécifiques. Par exemple, la dystrophie musculaire de Duchenne et la dystrophie musculaire de Becker ont en commun une absence ou une défectuosité de la protéine dystrophine (Hoffman et al., 1987). Ces anomalies, liées à une mutation du gène, n’ont pas toutes été détectées pour tous les types de dystrophies musculaires (Passos-Bueno et al., 1996). Néanmoins, connaître davantage l’aspect génique des maladies neuromusculaires peut vraisemblablement orienter vers des pistes de solutions. La défaillance en alpha-actinine 3 et son impact sur la force et les capacités fonctionnelles sont les deux aspects qui ont été survolés dans certains types de dystrophie musculaire.
1.5.2.1 Défaillance en alpha-actinine 3
Une recherche a été réalisée afin de constater la déficience en alpha-actinine 3 chez certaines formes de dystrophie musculaire, notamment la dystrophie musculaire de Duchenne, la dystrophie musculaire congénitale avec déficit primaire en mérosine, et la sarcoglycanopathie. L’hypothèse suggérait que l’atteinte d’une dystrophie musculaire est associée à une absence importante en alpha-actinine 3. Bien qu’aucune classification par
génotype des individus n’ait été effectuée, la présence d’alpha-actinine 3 a été mesurée à l’aide d’une réaction en chaîne par polymérase. L’analyse a démontré, comme émise dans l’hypothèse, que de souffrir d’une forme de dystrophie engendre une absence importante en alpha-actinine 3. Néanmoins, cette absence a été justifiée par un manque d’expression génique plutôt que par une dégradation secondaire de cette protéine (Vainzof et al., 1997).
1.5.2.2 Force et capacité fonctionnelle
L’impact de l’ACTN3 sur la force et la capacité fonctionnelle de patients atteints de dystrophie musculaire de Duchenne a également fait l’objet d’une étude (Hogarth et al., 2017). Il apparaît que le génotype de l’ACTN3 influence ces deux éléments lorsqu’il y a présence de cette maladie. Au total, 272 patients ont été analysés avec des tests physiques mesurant ces deux aspects. Autant la force que les capacités fonctionnelles sont affectées par le génotype. En effet, les individus associés au génotype 577XX, donc présentant une déficience en alpha-actinine 3, ont réussi des performances moindres, et ce, dans toutes les évaluations. Les tests physiques effectués ont consisté à réaliser un 10 mètres marche, une extension du genou, une extension du coude ainsi qu’une prise de force de préhension. L’étude a conclu que les individus de génotype 577RR se sont révélés significativement plus forts dans tous les tests musculaires et ont parcouru le 10 mètres marche plus rapidement (Hogarth et al., 2017).
Après analyse, l’expression ou non du gène ACTN3 influe dans l’évolution de différentes maladies. La présence d’alpha-actinine 3 engendre un effet protecteur chez les personnes atteintes d’une maladie cardiovasculaire en plus d’accroître le niveau global de force chez les individus souffrant de la dystrophie musculaire de Duchenne (Bernardez-Pereira et al., 2014; Hogarth et al., 2017).
1.6 Dystrophie myotonique de type 1
1.6.1 Définition
Une pathologie neuromusculaire est une maladie qui affecte le système nerveux central ainsi que le système nerveux périphérique. Les motoneurones qui sont touchés sont à l’origine des troubles de transmissions de l’influx nerveux engendrant un dysfonctionnement moteur et sensoriel au niveau des cellules musculaires (Li & Hondzinski, 2012). Au total, pas moins de 600 pathologies neuromusculaires ont été répertoriées. Leurs manifestations ainsi que leurs atteintes sont très variées (Cup et al., 2007). En effet, le patrimoine génétique influence grandement les probabilités qu’un individu souffre ou non d’une telle pathologie. L’effet fondateur qu’a subi le territoire du Saguenay-Lac-Saint-Jean est un bon exemple à l’appui. L’immigration d’une population sur ce territoire autrefois vierge est responsable d’une douzaine de maladies héréditaires (Moreau, Vézina & Labuda, 2007). La faible variété génétique de cette population a engendré une augmentation du nombre de personnes souffrant d’une maladie neuromusculaire nommée dystrophie myotonique de type 1 (DM1) (Beffy et al., 2010). Il est essentiel de bien distinguer la DM1 de la DM2. Lorsqu’il s’agit de la DM1, la littérature scientifique utilise également le terme maladie de Steinert tandis que pour la DM2, le terme employé est myopathie myotonique proximale (PROMM). La DM1 ainsi que la DM2 font partie d’un groupe de maladies qui comprend également la dystrophie musculaire de Duchenne, de Becker, oculopharyngée, pour n’en nommer que quelques-unes (Emery, 2002).
1.6.2 Nombre de répétitions CTG
La DM1 est caractérisée par un nombre important de répétitions CTG (bases nucléiques cytosine, thymine, guanine), qui est supérieur à 38, allant même au-delà de 1000 répétitions dans certains cas. Cette expansion instable est issue du gène DMPK qui code pour une protéine kinase sérine/thréonine, située sur le chromosome 19. Cette mutation est
exceptionnelle puisque cette dernière se localise sur une partie du gène DMPK qui ne code en aucun cas pour la protéine kinase sérine/thréonine (Ansved, 2001).
De surcroît, le nombre de répétitions de ces bases nucléiques est à l’origine de la sévérité de la maladie (Tsilfidis et al., 1992). Conséquemment, il existe une corrélation inversement proportionnelle entre l’âge du début de la manifestation de la maladie et le nombre de répétitions CTG. Autrement dit, lorsque le diagnostic se fait en bas âge, le nombre de mutations est élevé et, au contraire, lorsque le diagnostic est fait à un âge avancé, le nombre de mutations est faible. Chez les familles atteintes de DM1, un phénomène d’anticipation génétique est noté. En effet, les symptômes cliniques apparaissent plus tôt et la gravité de la maladie s’accroît avec la transmission aux générations successives. Par ailleurs, l’augmentation des mutations est plus couramment associée à la transmission chez la femme que chez l’homme (Huang et al., 2005).
1.6.3 Prévalence
Au Saguenay-Lac-Saint-Jean, la maladie neuromusculaire la plus répandue est la DM1. La prévalence de cette pathologie dans cette région est de 189 personnes sur 100 000 (Mathieu et al., 1990). Cela équivaut à plus de dix fois la prévalence au niveau mondial qui est de 2,1 à 14,3 individus par 100 000 habitants (Mathieu et al., 2003). Au total, quatre phénotypes sont observés : la forme adulte, la forme juvénile, la forme tardive ainsi que la forme congénitale (Fokstuen et al., 2001). Généralement, bien que la maladie puisse apparaître à tout âge, la plupart des personnes reçoivent le diagnostic avant la vingtaine. Les signes sont surtout détectables en raison de son caractère génétique qui engendre une certaine anticipation de la maladie. Le phénotype de la DM1 considéré comme étant le plus grave est la forme dite congénitale. Dès la naissance, le poupon démontrera plusieurs signes liés à cette maladie. Pour donner naissance à un enfant symptomatique, il est à noter que la mère doit souffrir elle-même de la maladie. Par ailleurs, avant la naissance, il est possible de détecter des signes de la maladie. Notamment, il semble qu’un mouvement plus lent du fœtus soit remarqué au second trimestre de grossesse (Koch et al., 1991).
1.6.4 Signes et symptômes de la DM1
1.6.4.1 Atteintes des muscles squelettiques
L’altération des muscles squelettiques est un des symptômes les plus manifestes chez les individus atteints de DM1. Une faiblesse musculaire distale apparaîtra de prime abord, pour par la suite s’étendre vers les régions proximales. Cette dysfonction a pour conséquence, entre autres, d’occasionner une fragilité au niveau des chevilles. Les muscles stabilisateurs des chevilles étant affectés, le phénomène des pieds tombants est un handicap que subissent les individus atteints de la maladie. La diminution de la force au niveau des chevilles oblige les personnes atteintes de DM1 à traîner leurs pieds et provoque ainsi des pertes d’équilibre et des chutes (Kroksmark et al.2005). L’incapacité fonctionnelle s’instaure donc progressivement en raison de la démarche de l’individu qui est chancelante. Plusieurs études ont été menées afin de déterminer de manière quantitative la force musculaire des individus souffrant de DM1. En analysant les recherches effectuées sur le sujet, il apparaît que l’affaiblissement musculaire des chevilles se produit 10 ans après que la maladie ait été diagnostiquée. Évaluer la force des chevilles des patients atteints de DM1 est donc essentiel pour apprécier l’évolution de la pathologie. Bien qu’une faiblesse musculaire soit aussi détectée au niveau de la musculature proximale, la diminution de la force est plus prononcée au niveau de celle distale (Ashton-Miller et al., 1996).
La DM1 engendre également de la myotonie. Ce handicap oblige les muscles à ne se relâcher qu’après une période de temps donnée. Autrement dit, un individu qui agrippe un objet va éprouver de la difficulté à relâcher sa prise en raison de la faible vitesse de relâchement des muscles impliqués (Wheeler et al., 2007). Les capacités fonctionnelles sont donc grandement affectées étant donné que l’usage des mains fait partie intégrante des tâches de la vie quotidienne (Harper, 2004). De surcroît, il est possible que les muscles du pharynx et de la langue soient affectés engendrant des troubles de la parole, de la mastication ainsi que de la dysphagie (Leonard et al. 2001). Chez les adultes présentant le phénotype de la DM1, près de 100% souffre de myotonie. Cette problématique est causée par l’hyperexcitabilité des myocytes en raison d’un dysfonctionnement des canaux sodiques
responsables de contrôler l’influx intracellulaire du sodium (Cannon & Bean, 2010). C’est pourquoi, lorsqu’un individu souffrant de DM1 effectue un électromyogramme (EMG), celui-ci révèle des potentiels d’actions (ondes M) inhabituels. Cette anormalité cause un délai dans le relâchement des muscles suite à une contraction (Arsenault et al., 2006). La température ambiante influence également la gravité de la myotonie. Un individu qui est soumis au froid verra sa myotonie s’aggraver tandis qu’au contraire, une personne qui s’échauffe souffrira moins de celle-ci (Emre & Henn, 1985).
1.6.4.2 Dégradation du système visuel
Au niveau du système visuel, le globe oculaire verra ses fonctions altérées. La présence de cataractes, de ptôsis, soit une chute de la paupière supérieure et de la diplopie, communément appelée double vision, sont sujets à survenir. Ces effets auront pour conséquence d’embrouiller la vue engendrant donc inévitablement une altération des fonctions visuelles. Néanmoins, de telles conditions sont davantage présentes chez les individus plus âgés (Ekström et al., 2010).
1.6.4.3 Débalancement du système endocrinien
La plupart des problèmes du système endocrinien sont habituellement traitables. Chez les hommes, la fertilité peut être perturbée en raison d’une réduction du niveau de testostérone associée à une atrophie testiculaire très courante après la puberté. Chez les femmes, la fertilité est également amenuisée bien que leurs ovaires ne soient pas affectés. D’autre part, le risque de fausse couche peut être accru en raison d’un dérèglement hormonal. Chez certaines personnes atteintes de DM1, le pancréas sécrète de l’insuline en excès ce qui s’apparente au diabète sucré. De plus, l’individu malade est davantage sujet à réagir anormalement à certains médicaments. L’hypersensibilité aux anesthésiques et aux relaxants musculaires sont des conséquences reliées à la condition des patients. Une forte somnolence après la prise d’un médicament est donc souvent observée. Cette sensibilité accrue est détectée autant chez l’individu légèrement atteint que gravement atteint de DM1 (Andoin et al., 2005; Matsumura et al., 2009).
1.6.4.4 Anomalies cardiaques
Les troubles cardiaques connus qui peuvent s’installer chez les personnes atteintes de DM1 sont les palpitations et l’arythmie. La gravité de ces symptômes peut varier selon les individus. Ces troubles cardiaques sont dus, pour la grande majorité des cas, aux troubles de conduction nerveuse entre les atriums et les ventricules (Harper, 2001). Ces troubles de conduction se manifestent à l’électrocardiogramme par des blocs atrioventriculaires et par des blocs de branche. Un tel défaut de conduction fera dévier l’influx nerveux qui emprunte alors un trajet non souhaité. Dans ce cas-ci, le nouveau chemin ralentit l’efficacité ainsi que la vitesse de la pompe du myocarde (Gosling et al., 2003). Les individus atteints de DM1 voient donc leur seuil de tolérance à l’exercice diminuer (Moore et al., 2016).
1.6.4.5 Dysfonctionnements du système respiratoire
Un individu souffrant de DM1 voit ses voies respiratoires se détériorer (Benhayon et al., 2015). En effet, il est commun de détecter chez ces personnes des problèmes d’infection et de toux. Les muscles utilisés lors de la respiration, davantage faibles, entraînent une difficulté respiratoire ainsi que de l’essoufflement. Les individus peuvent ressentir de l’essoufflement au repos même lorsqu’ils sont en position couchée. Lorsque l’insuffisance respiratoire est importante, le recours à une ventilation nocturne est suggéré afin d’atténuer les symptômes de somnolence et de fatigue (Cuvelier et al., 2012). Dépendamment de la sévérité de l’insuffisance respiratoire, les vaccins antigrippaux et contre le pneumocoque sont recommandés (Gagnon et al., 2010).
1.6.4.6 Altérations du système capillaire
La santé des cheveux est affectée autant chez l’homme que chez la femme. Pour ce qui est de l’homme, l’alopécie est fréquente tandis que chez la femme, les cheveux deviennent ténus. Si aucun traitement n’est effectué, il y aura progression jusqu’à la calvitie (Finsterer & Fellinger, 2011).
Bref, bien que les signes et symptômes de la maladie de la DM1 soient principalement musculaires, cette dernière est définie comme une pathologie multisystémique de par ces atteintes au niveau des systèmes visuel, endocrinien, cardiaque, respiratoire et capillaire.
1.6.5 Catégorisation de la DM1 (échelle de MIRS)
Lors du diagnostic, la DM1 comprend 5 classes établies selon l’atteinte et la sévérité des problèmes musculaires (Figure 5) (Mathieu et al., 2001).
Classe 1 : Aucune déficience musculaire clinique n’est détectée.
Classe 2 : L’atteinte musculaire est minimale. Il y a présence de myotonie, d’hypotrophie des muscles temporaux et maxillaires, d’une faiblesse faciale ainsi que d’une faiblesse au niveau des fléchisseurs du cou. Excepté pour les fléchisseurs des doigts, aucune faiblesse distale n’est détectée.
Classe 3 : Aucune faiblesse proximale n’est détectée excepté au niveau des extenseurs des coudes. De plus, il y a une fatigabilité accrue au niveau de la musculature distale.
Classe 4 : Faiblesse sévère au niveau de la musculature distale ainsi qu’une fatigabilité modérée des muscles proximaux sont présents.
Classe 5 : L’atteinte musculaire est générale. L’individu a perdu son autonomie et doit avoir recours à un fauteuil roulant.
Figure 6 : Progression des altérations musculaires selon les classes de la DM1 (Mathieu et
al., 2001).
1.6.6 Réponse à l’entraînement chez les individus atteints de DM1
L’activité physique au sein d’une population générale est un facteur clé quant au maintien de l’autonomie physique et psychologique (McKinlay, 1995). La DM1 étant une maladie dégénérative incurable, les effets de l’entraînement pourraient permettre une possible réduction, voire même une amélioration des symptômes chez ces patients. Les entraînements en résistance ainsi que les activités aérobies sont deux paramètres ayant été les plus évalués chez les individus qui présentent le phénotype de la DM1.
1.6.6.1 Entraînement en résistance
Cinq recherches ont approfondi les impacts d’un entraînement en résistance chez les personnes atteintes de DM1. L’investigation scientifique la plus ancienne est celle de