Analyse fonctionnelle de l’interaction entre les protéines
Fanconi et le corépresseur CtBP1 : l’antagoniste Wnt
Dickkopf-1 comme cible transcriptionnelle
Thèse
Caroline Huard
Doctorat en Biologie cellulaire et moléculaire
Philosophiae Doctor (Ph.D.)
Québec, Canada
Résumé
L’anémie de Fanconi (FA) est une maladie génétique caractérisée par une insuffisance médullaire, un risque augmenté de cancers et plusieurs types de malformations. FANCC constitue la seule des 15 protéines FA qui se localise principalement au cytoplasme. De ce fait, en plus de son rôle dans la réparation de l’ADN, FANCC pourrait intervenir dans d’autres mécanismes régulant la croissance des cellules hématopoïétiques. Pour mieux comprendre ses fonctions, nous avons cherché de nouveaux partenaires de FANCC. Le corépresseur de la transcription CtBP1 a été retenu pour analyse.
L’étude des interactions a confirmé le lien physique FANCC-CtBP1 et révélé que CtBP1 est un membre du complexe FA. Afin d’éclaircir la fonction des interactions, nous avons analysé le profil d’expression génique de cellules réprimées pour les gènes FA ou CtBP1. Les résultats ont montré une expression augmentée de l’antagoniste de la voie Wnt Dickkopf-1 (DKK1). Ainsi, les essais de gènes rapporteurs ont établi que CtBP1 et FANCC agissent comme répresseur transcriptionnel du promoteur DKK1. Nous avons aussi observé que FANCD2 réprime indirectement DKK1 en favorisant l’expression de l’oncogène c-Myc. Le mécanisme pourrait dépendre d’interactions entre les protéines FA, incluant FANCC, et les protéines de l’appareil transcriptionnel Wnt, CtBP1 et β-caténine. Par ailleurs, nous avons montré que FANCC s’accumule au noyau en réponse à la signalisation Wnt et qu’elle participe avec d’autres protéines FA à l’activation de la β-caténine. Ainsi, nous avons trouvé des niveaux élevés de DKK1 dans le surnageant des cellules appauvries en protéines FA et CtBP1, de même que dans les sérums de souris knockout FancA et FancC.
Notre étude a permis de montrer que FANCC et les protéines FA, de concert avec CtBP1, agissent dans la régulation transcriptionnelle de l’antagoniste DKK1. Ces résultats suggèrent que FANCC est une protéine clé impliquée dans la signalisation Wnt. Puisque DKK1 est impliqué dans des processus connus pour être perturbés dans la FA, la régulation de DKK1 par FANCC et CtBP1 représente un mécanisme pouvant expliquer la perte progressive des cellules souches hématopoïétiques et représente une étape cruciale pour la découverte de stratégies visant à prévenir l’insuffisance médullaire chez les patients FA.
Abstract
Fanconi anemia (FA) is a genetic disease characterized by bone marrow failure, excess cancer risk, as well as a broad array of malformations. FANCC is one of fifteen genes linked to the FA disease and encodes a protein that, unlike other FA proteins, is localized primarily to the cell cytoplasm. Because of this, in addition to its role in DNA crosslink repair, FANCC is proposed to function in other mechanisms that can regulate hematopoietic progenitor cell fate. To better understand its functions, we investigated for new partners of the FANCC protein. One candidate, the transcriptional corepressor CtBP1, was selected for further analyses.
Interatomic studies confirmed the physical link between FANCC and CtBP1 and revealed that CtBP1 is a member of the FA core complex. To investigate biological function of these interactions, we used a microarray strategy and found that the Wnt antagonist Dickkopf-1 (DKK1) is upregulated in FA- and CtBP1-depleted cells. Accordingly, CtBP1 and FANCC were found to act as transcriptional repressor on DKK1 promoter in reporter gene assays. We also observed that FANCD2 indirectly represses DKK1 in promoting the expression of c-Myc. Functional mechanism of these repressions may be explained on the observation that FANCC and FA core complex proteins interact with the Wnt transcriptional machinery proteins including CtBP1 and β-catenin. Furthermore, we showed that FANCC accumulates into the nucleus in response to Wnt signalisation and participates with other FA proteins in β-catenin activation. Therefore, we found increased levels of DKK1 in FA- and CtBP1-depleted cells supernatant as well as in sera from FancA and FancC knockout mice.
Functional interaction studies showed that FANCC and FA proteins with CtBP1 act in transcriptional regulation of the Wnt antagonist DKK1. These findings suggest that FANCC is a key protein involved in the Wnt signalling response. Because DKK1 is implicated in biological processes similar to those involved in FA pathogenesis, linking FANCC with CtBP1 to the regulation of DKK1 suggests a possible mechanism explaining the progressive loss of bone marrow cells and represents a crucial step for the development of novel strategies aimed at preventing bone marrow failure in FA patients.
Table des matières
RÉSUMÉ ... III
ABSTRACT ... V
TABLE DES MATIÈRES ... VII
LISTE DES FIGURES ... IX
LISTE DES TABLEAUX ... XI
LISTE DES ABRÉVIATIONS ... XIII
REMERCIEMENTS ... XXI
AVANT-PROPOS ... XXIII
CHAPITRE 1 INTRODUCTION ... 1
1.1 L’ANÉMIE DE FANCONI ... 1
1.1.1 ORIGINE ET CONTEXTE ACTUEL ... 1
1.1.2 CARACTÉRISTIQUES CLINIQUES ET CELLULAIRES ... 3
1.1.2.1 Aspects hématologiques de l’anémie de Fanconi ... 3
1.1.2.2 Anomalies caractéristiques de l’anémie de Fanconi et tumeurs ... 4
1.1.2.3 Aspects cellulaires et diagnostic de l’anémie de Fanconi ... 7
1.1.3 GÉNÉTIQUE MOLÉCULAIRE DE L’ANÉMIE DE FANCONI ... 8
1.1.3.1 Gènes associés à l’anémie de Fanconi ... 8
1.1.3.2 Protéines associées à l’anémie de Fanconi ... 10
1.1.3.3 La voie de signalisation de l’anémie de Fanconi ... 13
1.1.4 LES IMPLICATIONS DES PROTÉINES FANCONI ... 16
1.1.4.1 Réplication et réparation de l’ADN et cycle cellulaire dans l’anémie de Fanconi ... 16
1.1.4.2 Apoptose et détoxification des produits oxygénés dans l’anémie de Fanconi ... 19
1.1.4.3 Régulation transcriptionnelle dans l’anémie de Fanconi ... 21
1.1.4.4 Développement et hématopoïèse dans l’anémie de Fanconi ... 22
1.2 LA VOIE DE DÉVELOPPEMENT WNT DANS L’HÉMATOPOÏÈSE ... 25
1.2.1 L’ACTIVATION DE LA VOIE WNT STIMULE L’AUTORENOUVÈLEMENT DES CSHS ... 29
1.2.2 L’INHIBITION DE LA VOIE WNT DIMINUE L’AUTORENOUVÈLEMENT DES CSHS ... 29
1.3 LE CORÉPRESSEUR DE LA TRANSCRIPTION CTBP1 ... 32
1.3.1 STRUCTURE DU GÈNE ET DE LA PROTÉINE CTBP1 ... 32
1.3.2 RÉGULATION DE CTBP1 ... 34
1.3.3 COMPLEXE DE RÉPRESSION CTBP ET TRANSCRIPTION ... 35
1.3.4 CTBP1 DANS LE DÉVELOPPEMENT ... 37
1.3.5 CTBP1 DANS LA TUMORIGENÈSE ... 39
CHAPITRE 2 CONTEXTE, HYPOTHÈSES ET OBJECTIFS DE RECHERCHE ... 43
1. CARACTÉRISER LE RÔLE DU CORÉPRESSEUR CTBP1 DANS LA VOIE DE L’ANÉMIE DE FANCONI ... 44
2. CARACTÉRISER LE RÔLE DES PROTÉINES FA DANS LA VOIE DE SIGNALISATION WNT/CTBP1 ... 45
CHAPITRE 3 LES PROTÉINES FANCONI INTERAGISSENT AVEC CTBP1 ET MODULENT L’EXPRESSION DE L’ANTAGONISTE WNT DICKKOPF-1 ... 47
3.1 RÉSUMÉ ... 48
3.2 ABSTRACT ... 49
3.3 INTRODUCTION ... 50
3.4 MATERIALS AND METHODS ... 52
3.5 RESULTS ... 56
3.6 DISCUSSION ... 69
3.7 ACKNOWLEDGMENTS ... 72
3.8 AUTHORSHIP CONTRIBUTION AND CONFLICT OF INTEREST DISCLOSURE ... 72
3.9 REFERENCES ... 73
3.10 SUPPLEMENTAL METHODS ... 76
3.11 SUPPLEMENTAL FIGURES ... 77
CHAPITRE 4 LES PROTÉINES FANCONI EXERCENT UNE DOUBLE FONCTION DANS LA RÉPRESSION TRANSCRIPTIONNELLE DE DICKKOPF-1 ... 83
4.1 RÉSUMÉ ... 84
4.2 ABSTRACT ... 85
4.3 INTRODUCTION ... 86
4.4 RESULTS ... 88
4.5 DISCUSSION ... 99
4.6 MATERIALS AND METHODS ... 102
4.7 ACKNOWLEDGMENTS ... 105
4.8 AUTHORSHIP CONTRIBUTION AND CONFLICT OF INTEREST DISCLOSURE ... 105
4.9 REFERENCES ... 106
CHAPITRE 5 DISCUSSION ET PERSPECTIVES ... 109
Liste des figures
CHAPITRE 1 : INTRODUCTIONFigure 1.1 : Atteintes hématologiques de l’anémie de Fanconi……….……...4
Figure 1.2 : Anomalies caractéristiques de l’anémie de Fanconi..………...……5
Figure 1.3 : Aspects cellulaires de l’anémie de Fanconi………...….7
Figure 1.4 : Représentation schématique des protéines de l’anémie de Fanconi………..…13
Figure 1.5 : La voie de signalisation Fanconi.………16
Figure 1.6 : Implication des protéines FA dans la réparation de l’ADN……….………18
Figure 1.7 : Développement des cellules souches hématopoïétiques.……….………26
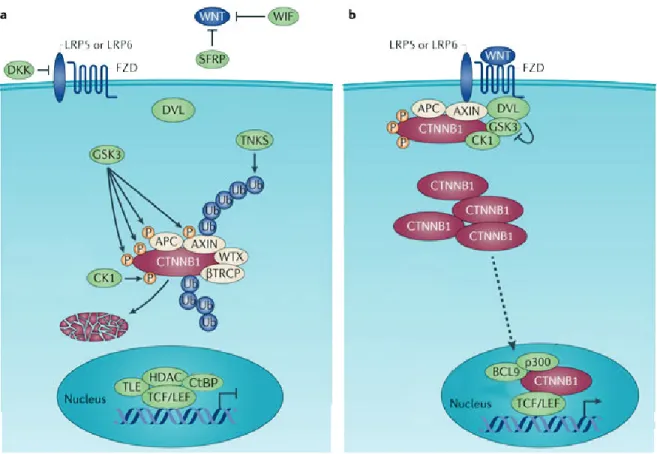
Figure 1.8 : La voie de signalisation Wnt………..….29
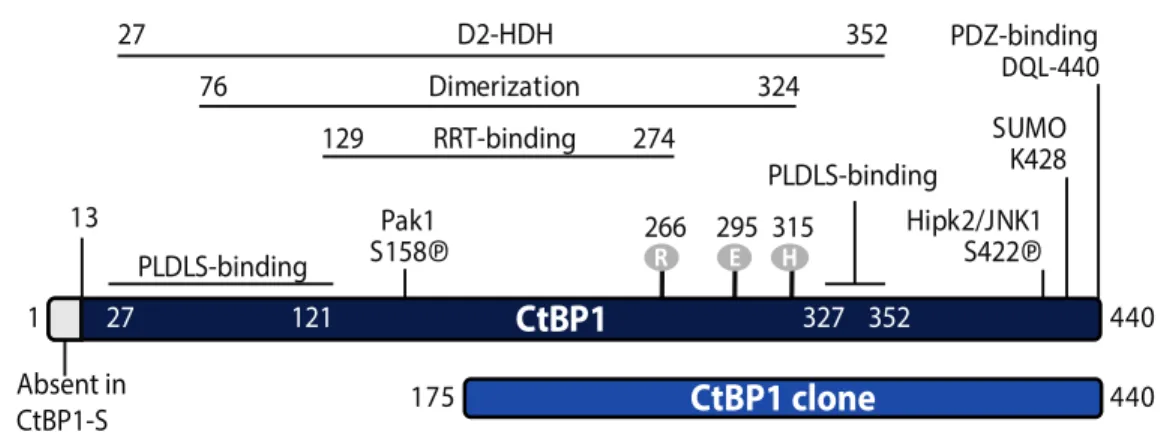
Figure 1.9 : Domaines structuraux et fonctionnels de CtBP1……….33
CHAPITRE 3 : LES PROTÉINES FANCONI INTERAGISSENT AVEC CTBP1 ET MODULENT L’EXPRESSION DE L’ANTAGONISTE WNT DICKKOPF-1 Figure 1. CtBP1 interacts with the N terminal region of FANCC in yeast……….63
Figure 2. CtBP1 interacts with FANCC in cells………...64
Figure 3. CtBP1 interacts with components of the FA core complex..………..65
Figure 4. CtBP1 is not essential for the stability or the activity of the FA core complex.………66
Figure 5. CtBP1 is essential for survival and proliferation.………..67
Figure 6. CtBP1 and FANCD2 regulate DKK1 expression.……….68
Supplemental Figure 1. Protein expression analysis following shRNA mediated gene repression….77 Supplemental Figure 2. CtBP1 interacts with FA core complex proteins………...…..78
Supplemental Figure 3. CtBP1 depleted cells die of apoptosis………..…79
Supplemental Figure 4. Expression profiling of CtBPi and FANCD2i cells………..……80
Supplemental Figure 5. CtBP1 is not required for the formation of RAD51 foci.……….……81
CHAPITRE 4 : LES PROTÉINES FANCONI EXERCENT UNE DOUBLE FONCTION DANS LA RÉPRESSION TRANSCRIPTIONNELLE DE DICKKOPF-1 Figure 1. FANCC forms a complex with CtBP1 and β-catenin………...92
Figure 2. β-catenin activation induces FANCC nuclear translocation.………..93
Figure 3. Nuclear entry of β-catenin requires FANCC.……….94
Figure 4. Nuclear translocation of β-catenin is dependent on a functional FA pathway.……….95
Figure 5. FANCC and CtBP1 act as transcriptional repressors of DKK1.………..96
Figure 6. FANCD2-deficient cells have reduced DKK1 expression………...97
Figure 7. Proposed model of the dual mechanism by which the FA pathway acts in transcriptional regulation of DKK1 gene.………98
Liste des tableaux
CHAPITRE 1 : INTRODUCTIONListe des abréviations
ng nanogramme µg microgramme mg milligramme µL microlitre mL millilitre nM nanomolaire µM micromolaire mM millimolaire α alpha ou anti β beta δ delta γ gamma ς zetaβTrCP beta-Transducin-repeat- Containing E3 ubiquitin Protein ligase δEF1 delta-crystallin Enhancer binding Factor 1
a. a. acide aminé
ACTH AdrenoCorticoTropic Hormone ADN Acide DésoxyriboNucléique
ADNc Acide DésoxyriboNucléique complémentaire Akt thymoma viral proto-oncogene
AML Acute Myeloid Leukemia - Leucémie myéloïde aigüe APC Adenomatous Polyposis Coli
ARF Alternative Reading Frame protein
ARM Armadillo repeat – Domaine de répétition Armadillo ATM Ataxia Telangiectasia Mutated protein kinase ATR Ataxia Telangiectasia and RAD3 related protein Bax BCL2-Associated X protein
BACH1 BRCA1-Associated C-terminal helicase 1
BCL9 B-cell CLL (Chronic Lymphocytic Leukemia) / Lymphoma 9 BCoR-L1 BCL6 corepressor-Like 1
BKLF Basic Krüppel-like Factor Bik BCL2-Interacting Killer BMP Bone Morphogenetic Protein
BRCA1/2 BReast CAncer susceptibility protein 1/2 BRG1 Brahma-related gene 1
BRIP1 BRCA1 Interacting Protein 1
BTB/POZ BR-C, Ttk and Bab / POx virus and Zinc finger
CBP CREB (cAMP responsive element binding protein) Binding Protein CD34 Cluster of Differentiation 34
CDKN2A/B Cyclin-Dependent Kinase inhibitor 2A/B CDYL ChromoDomain protein Y-Like
CHK1 Checkpoint kinase 1
Cip1 COP1 (COat Protein1)-Interacting Protein 1 CK1 Casein Kinase 1
CLP Common Lymphoid Progenitor CMP Common Myeloid Progenitor CMV Cytomegalovirus
CO2 Dioxyde de carbone
CRM1 Chromosome Region Maintenance 1 CSHs Cellules Souches Hématopoïétiques CSM Cellule Stromale Mésenchymateuse
CoREST REST (Repressor Element-1 Silencing Transcription factor) corepressor CtBP1/2 C-Terminal Binding Protein 1/2
C-terminal carboxy terminal
D2-HDH D-2-Hydroxyglutarate Dehydrogenase DEB Diépoxybutane
DKK1 Dickkopf-1
DMEM Dulbecco’s Modified Eagle’s Medium DSB Double Strand Break
Dsh Dishevelled
EGF Epidermic Growth Factor
EME1 Essential Meiotic Endonuclease 1 EMT Epithelial-Mesenchymal Transformation EpoR Erythropoietin receptor
ERCC1 Excision Repair Cross-Complementing group 1 FA Fanconi Anemia - Anémie de Fanconi
FAAP24/100 FA-Associated Protein 24/100 FAN1 FA-Associated Nuclease 1
FANC Fanconi Anemia protein - Protéine de l’anémie de Fanconi (groupe de complémentation A à M)
Fas Apoptosis Stimulating Fragment FAZF Fanconi Anemia Zinc Finger protein FBS Fetal Bovine Serum – Sérum de veau fœtal FOG Friend of GATA-1
G1/G2 Growth phase 1 / Growth phase 2 GATA-1 Globin transcription factor 1 GLP G9a-Like Protein
GM-CSF Granulocyte / Macrophage Colony Stimulating Factors GSK3β Glycogen Synthase Kinase 3 beta
GSTP1 Gluthathione-S-transferase pi 1
GVDH Graft Versus Host Disease - Réaction du greffon contre l’hôte HDAC Histone DeACetylases
HEF1 Human Enhancer of Filamentation 1
HEK293T Human Embryonic Kidney 293T cell – Cellules embryonnaires de rein humain 293T HeLa Henrietta Lacks human epithelial carcinoma cell line
HIPK2 Homeodomain-Interacting Protein Kinase 2 HPC2 Human Polycomb 2
Hsp70 Heat shock protein 70
ICAT Cell autonomous inhibitor of β-catenin and TCF
INF Interféron
IgG Immunoglobuline G
IL Interleukine
JAK Janus Kinase
kDa kilo Dalton
LcoR Ligand dependent nuclear receptor CORepressor LDL Low-Density Lipoprotein
LEF Lymphoid Enhancer Factor LRP5/6 LDL receptor-related protein 5/6 LSD1 Lysine-Specific Demethylase 1 M Phase de mitose cellulaire
MDS Myelodysplastic syndrome - Syndrome myélodysplasique
MEFs Mouse Embryonic Fibroblasts - Fibroblastes embryonnaires de souris MHF1/2 FANCM-Associated Histone Fold protein 1/2
MMC Mitomycine C
MO Moelle Osseuse
MRE11 Meiotic Recombination 11
mRNA messenger RiboNucleic Acid - Acide ribonucléique messager MTOB 4-Methythio-2-oxobutyric acid
MUS81 Methyl methansulfonate, UV Sensitive 81 NAD Nicotinamide Adenine Dinucleotide
NADPH Nicotinamide Adenine Dinucleotide Phosphate reduced form NES Nuclear Export Signal - Signal d’export nucléaire
NLS Nuclear Localization Signal - Signal de localisation nucléaire NBS1 Nijimegen Breakage Syndrome 1 protein
ng nanogramme
NHEJ Non-Homologuous End-Joining nNOS neuronal Nitric Oxide Synthase N-proximal amino proximal
p15Ink4a p15 Inhibitor of cyclin-dependent kinase 4a
p16Ink4b p16 inhibitor of cyclin-dependent kinase 4b
PAK1/6 p21-Activated Kinase-1/6 PALB2 Partner And Localizer of BRCA2
PBS Phosphate Buffered Saline - Tampon phosphate salin PCAF P300/CBP-Associated Factor
PCNA Proliferating Cell Nuclear Antigen
PDZ PSD95 (Post-synaptic density protein)-Dlg1 (Drosophila disc large tumor suppressor)-zo-1 (zonula occludens-1 protein)
PERP p53-Effector Related to PMP-22 (Peripheral Myelin Protein-22) PHD Plant Homeo Domain
PHF9 PHD Finger Protein 9
PIAS Protein Inhibitor of Activated STAT Pinin/DRS Pinin/Domain-Rich Serine protein
PKA Protein Kinase A PKB Protein Kinase B PKR Protein Kinase R PLDLS Pro-Leu-Asp-Leu-Pro Polζ Polymerase zeta PP2A Protein Phosphatase 2A
PTEN Phosphatase and TENsin homologue Puma p53 Upregulated Modulator of Apoptosis RAD50/51C RADiation sensitive 50/51C
REV1 REVersionless 1
RING Really Interesting New Gene RMN RAD50/MRE11/NBS1 RPA1 Replication Protein A1
RREB Ras Responsive Element Binding protein RRT Arg-Arg-Thr
RSV Rous Sarcoma Virus
S Phase de synthèse de l’ADN
SAE1/SAE2 SUMO1 Activating Enzyme subunit 1 / SUMO1 Activating Enzyme subunit 2 SDS Sodium Dodecyl Sulfate
SDS-PAGE SDS-PolyAcrylamide Gel Electrophoresis sFRP Secreted Frizzled Related Protein
SIP1 SNF1 (Sucrose Non Fermenting 1)-Interacting Protein 1 SLX1/4 Synthetic Lethal of unknown (X) function 1/4
Smad Mothers against DPP (dipeptide transport protein) homolog SNAI1/2 Snail homolog 1
STAT1 Signal Transduction and Activator of Transcription protein 1 SUMO1 Small Ubiquitin Modifier 1
SWI/SNF SWItch/Sucrose NonFermentable TALE Three–amino Acid Loop Extension TCF T-Cell Factor
TGF Transforming Growth Factor
TGIF TGFB (transforming growth factor beta) induced factor TLE Transducin-Like Enhancer
TLS Translesion synthesis – Synthèse translésionnelle TNF Tumor Necrosis Factor
TPR Tretratricopeptid repeat - Répétition tétratricopeptide UAF1 USP1-Associated Factor 1
UBC9 Ubiquitin-conjugating enzyme 9 UBE2T UBiquitin-conjugating enzyme E2T USP1 Ubiquitin Specific Peptidase 1
XIAP X-chromosome-linked Inhibitor of Apoptosis Protein XPF Xeroderma Pigmentosum group F
XRCC3 X-ray Repair Complementing defective repair in Chinese hamster cells 3 WAF1 Wild-type p53-Activated Fragment 1
WD Tryptophan-Aspartic acid dipeptide WIF1 Wnt Inhibitory Factor 1
WTX Wilms Tumor gene on the X chromosome ZEB1/2 Zinc finger E-box-Binding homeobox 1/2 ZNF217 Zinc finger protein 217
À Yan, Mathis et Maélie parce qu’ils sont ce que j’ai de plus cher
La vie, c’est comme une bicyclette, il faut avancer pour ne pas perdre l’équilibre.
Remerciements
Je tiens premièrement à remercier la Dre Madeleine Carreau de la confiance qu’elle m’a accordée en acceptant de me confier une place au sein de son équipe, mais surtout de son indulgence, de sa patience et de sa souplesse qui ont été rudement sollicitées tout au cours du déroulement de mon projet. Son expérience, sa rigueur scientifique et son entière disponibilité m’ont été des plus bénéfiques dans l’acquisition d’une solide expérience en laboratoire. Je tiens également à remercier le Dr Georges Lévesque pour sa disponibilité et ses explications généreuses.
Je désire remercier les membres de l’équipe de la Dre Madeleine Carreau et du Dr Georges Lévesque pour leur précieuse assistance et leur soutien moral tout au long de ce projet de recherche qui m’a sincèrement passionné et beaucoup appris. Merci particulièrement à Marie-Chantal Delisle, Valérie Bourdages et Chantal Godin, qui ont été mes complices pendant ces cinq dernières années, et dont sans elles je n’aurais pu garder le sourire. Merci d’avoir partagé mes bons moments et de m’avoir épaulé dans mes moments de faiblesse. Merci aussi à Cédric Tremblay, Audrey Magron, Feng Fei Huang, Tagrid Kaddar, Kevin Goggin, Carolina Koutras et Manel Ben Aissa d’avoir grandement participé à mon évolution, autant professionnelle que personnelle.
Je tiens spécialement à témoigner ma reconnaissance à mon conjoint, Yan, pour son amour, sa présence et son soutien tout au cours de ces longues années d’études, mais surtout pour m’avoir donné ce que j’ai de plus cher au monde, une famille. Merci à mon fils Mathis. Merci à ma fille Maélie. Merci de mettre le bonheur dans toutes mes journées. Merci d’être là mes amours.
De même, j’aimerais exprimer toute ma gratitude à mes parents et à ma sœur pour m’avoir inculqué les valeurs nécessaires à un cheminement de vie sain et surtout de m’avoir appris à persévérer et accepter. Merci à ma belle famille pour leur présence et leur soutien. Merci aussi à tous mes amis d’avoir rempli de joie et de rires d’innombrables soirées. Sans vous tous, je n’y serais pas parvenu.
Enfin, merci à mes organismes subventionnaires, les IRSC, le FRSQ, la fondation des étoiles et la fondation de l’anémie de Fanconi. Et, merci aux membres du jury d’avoir accepté d’évaluer mon travail, qui j’espère, sera à la hauteur de vos attentes.
Avant-propos
Cette thèse représente la synthèse et l’analyse des données que j’ai acquises au cours des cinq années de travail de mon doctorat sous la direction de la Dre Madeleine Carreau. Mes travaux ont porté sur la caractérisation de la fonction biologique de l’interaction entre les protéines Fanconi et le corépresseur de la transcription CtBP1. Ce projet a permis d’établir un lien étroit entre la voie de développement Wnt et la voie de signalisation Fanconi concernant la régulation génique de l’antagoniste Wnt Dickkopf-1 (DKK1). L’ensemble des résultats obtenus s’intègre dans un modèle éclaircissant la perte progressive des cellules souches hématopoïétiques observée chez les patients atteints de l’anémie de Fanconi. Cette thèse regroupe des concepts originaux et apporte des connaissances nouvelles qui pourront servir au développement d’outils thérapeutiques visant à réduire l’insuffisance médullaire observée chez les patients atteints de cette maladie et dans d’autres désordres métaboliques similaires.
Dans l’ensemble, cet ouvrage comporte cinq chapitres, incluant une introduction classique, la description du contexte du projet et des hypothèses, l’insertion de deux articles scientifiques et une discussion sur l’ensemble des résultats. Les articles scientifiques représentent la majeure partie des données récoltées et sont présentés sous forme manuscrite aux chapitres 3 et 4.
Le chapitre 3 a fait l’objet d’une publication récente dans le journal américain Blood.1 Pour
ce chapitre, je suis responsable de l’élaboration et de la réalisation de la plupart des manipulations. Le résultat de coimmunoprécipitation en surexpression (Figure 3b) a été obtenu par Cédric Tremblay, et Marie-Chantal Delisle a participé à la réalisation des expériences de détection par Élisa (Figure 6c et 6d). J’ai interprété les résultats, rédigé le manuscrit et préparé les figures. Dre Madeleine Carreau a dirigé la recherche, analysé les résultats et édité le manuscrit.
Le chapitre 4 fait l’objet d’une soumission dans le journal américain PNAS. Pour ce chapitre,
je suis responsable de l’élaboration et de la réalisation de la majorité des manipulations, à l’exception des immunoprécipitations de la figure 1a et des immunofluorescences dans les fibroblastes FA (Figure 2b et 4c) qui ont été réalisées par Cédric Tremblay. J’ai interprété les résultats, rédigé le manuscrit et préparé les figures. Dre Madeleine Carreau a dirigé la recherche, analysé les résultats et édité le manuscrit.
Chapitre 1
Introduction
1.1 L’anémie de Fanconi
1.1.1 Origine et contexte actuel
L’anémie de Fanconi (FA) a été rapportée pour la première fois en 1927 par le pédiatre suisse Guido Fanconi après la caractérisation des symptômes cliniques semblables dont souffraient trois enfants d’une même famille.2 Tous présentaient une anémie aplasique progressive ainsi qu’une
combinaison d’anomalies somatiques spécifiques, tels une petite stature, un hypogonadisme et une hyperpigmentation de la peau. Depuis cette première caractérisation, une recherche exhaustive de la littérature se rapportant à la FA a permis de rapporter plus de 2000 nouveaux cas.3-5
La FA apparait dans toutes les régions du globe et touche toutes les races et ethnies avec une incidence d’environ 3 naissances par millions chaque année.6 De façon inexpliquée, la maladie
apparait plus fréquemment chez le mâle que chez la femelle dans un ratio d’environ 1,2 : 1.7 Malgré
la faible prévalence mondiale de la FA et le nombre inconnu de patients atteints, les scientifiques estiment qu’en 2010 la population hétérozygote aux États-Unis pour l’un des gènes Fanconi se situait entre 1 : 156 et 1 : 209 avec un nombre de personnes atteintes de la maladie se situant entre 550 et 975.8 Des mutations fondatrices ont été trouvées chez les Africains de l’Afrique du Sud9,10, les juifs
de descendance ashkénaze et marocaine11-13, ainsi que chez les gitans espagnols14. Ces dernières
ethnies présentent une fréquence de mutations hétérozygotes de plus de 1 : 100 découlant possiblement d’un effet fondateur.
L’âge médian lors du diagnostic de la maladie de Fanconi est de 6,5 ans avec une répartition de 0 à 50 ans. L’observation d’une pancytopénie (diminution de toutes les lignées cellulaires
hématopoïétiques) lors d’un hémogramme, accompagnée d’une augmentation du volume des globules rouges et de l’hémoglobine fœtale, en plus d’une diminution du nombre de cellules dans la moelle osseuse causée par la diminution des précurseurs hématopoïétiques, suggère dans un premier temps le diagnostic d’anémie de Fanconi.15
Le traitement de la FA est similaire à ceux des formes d’anémies aplasiques acquises.15 Les
patients Fanconi sont généralement traités avec des soins de support pour le déficit hématopoïétique, comme des transfusions sanguines et de l’antibiothérapie, mais ces traitements se révèlent efficaces uniquement pour une courte durée. Les traitements à plus long terme se rangent en quatre catégories : greffe de la moelle osseuse, traitement aux androgènes et aux facteurs de croissance synthétiques et thérapie génique. Actuellement, le seul traitement pour les complications hématologiques de l’anémie de Fanconi reste la greffe de sang de cordon ou de cellules souches hématopoïétiques (CSHs) par une transplantation allogénique. Le moment optimal de la greffe constitue un défi puisque les meilleures réponses aux greffes s’obtiennent lorsqu’elles sont effectuées avant l’apparition de complications. De plus, la greffe de CSHs ne corrige pas les manifestations non hématologiques de l’anémie de Fanconi, et le risque de développer des tumeurs solides est augmenté après la transplantation, en particulier chez les patients Fanconi qui développent une grave réaction du greffon contre l’hôte (GVHD).
L’espérance de vie moyenne pour les patients atteints de la FA est d’environ 29 ans avec une répartition de 0 à 50 ans.7 La sévérité du phénotype clinique et la vitesse d’évolution de la
maladie dépendent des éléments génétiques de fond et des facteurs environnementaux, en plus de la spécificité mutationnelle du gène FA.16,17 Les patients présentent un large éventail de
caractéristiques cliniques, chacun ayant une pénétrance incomplète. Les principales caractéristiques cliniques observées chez les patients Fanconi peuvent être divisées en trois catégories : les défauts des cellules souches, les anomalies du développement et les néoplasies.
1.1.2 Caractéristiques cliniques et cellulaires
1.1.2.1 Aspects hématologiques de l’anémie de FanconiLa FA est une maladie génétique récessive qui se caractérise principalement par une anémie aplasique causée par un défaut dans le renouvèlement des cellules souches hématopoïétiques de la moelle osseuse.18,19 L’épuisement des CSHs mène à la diminution des
lignées cellulaires périphériques reliées à ces premières, soit les érothrocytes, les plaquettes et les leucocytes. Par conséquent, les patients atteints de la maladie présentent une fatigue chronique, des problèmes d’hémorragies spontanées et des difficultés à combattre les infections, ceci amenés respectivement par les déficits en érythrocytes, en plaquettes et en leucocytes. Cette atteinte hématologique constitue la cause principale de la grande morbidité et de la mortalité des patients homozygotes Fanconi. À la naissance, le compte sanguin des patients Fanconi est généralement normal et la macrocytose (augmentation de la taille des globules rouges) est le premier signe d’anomalie détecté. Il s’ensuit une thrombopénie (diminution des plaquettes circulantes), une neutropénie (diminution des neutrophiles) et une anémie (diminution des globules rouges). Finalement, une pancytopénie apparait typiquement entre l’âge de 5 et 10 ans avec un âge médian de 7 ans (Figure 1.1B).11,20,21 Plus de 85 % des patients présentent une aplasie médullaire avant
l’âge de 20 ans (Figure 1.1E).18,22,23
Il existe d’autre part pour les patients Fanconi une incidence de développement cumulative de leucémies myéloïdes aiguës (AML) d’environ 30 % à l’âge de 40 ans, ainsi qu’un risque de développement d’un syndrome myélodysplasique (MDS) de 5 % tous âges confondus.19,24-26 Les
MDS sont des altérations clonales des cellules souches pluripotentes ou myéloïdes, caractérisées par une hématopoïèse inefficace et par une probabilité marquée de transformations leucémiques (Figure 1.1C). L’évolution des MDS en AML se produit à la suite d’une transformation maligne d’un progéniteur myéloïde, générant une descendance de cellules myéloïdes bloquées à un stade précoce de la différenciation cellulaire incapable de maturation terminale (Figure 1.1F).27 Cette
population cellulaire clonale envahit la moelle osseuse et le système sanguin, perturbe le fonctionnement des progéniteurs normaux et entraine une insuffisance médullaire. Le mécanisme de l’inhibition de l’hématopoïèse polyclonale normale, qui mène à la synthèse des globules rouges, des plaquettes et des leucocytes, est soit un encombrement physique de l’espace médullaire, soit une
inhibition de la différenciation des progéniteurs hématopoïétiques normaux, ou bien l’association de ces deux mécanismes.
L’incidence cumulative des anomalies hématologiques, incluant l’aplasie médullaire, les AML et les MDS, est supérieure à 90 % au cours de la vie des patients Fanconi.28 Bien que le déficit
hématologique constitue le principal signe clinique de la maladie, la plupart des patients sont diagnostiqués à la suite d’observation de malformations congénitales à la naissance.
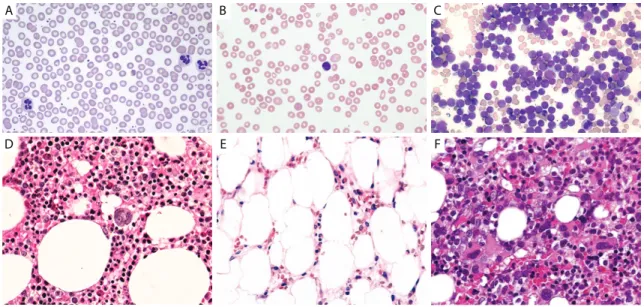
Figure 1.1 : Atteintes hématologiques de l’anémie de Fanconi. Les patients souffrant d’anémie de Fanconi présentent une pancytopénie lors d’un hémogramme (B) et une aplasie médullaire caractérisée par une hypocellularité de la moelle osseuse et une diminution des cellules hématopoïétiques à l’origine des précurseurs myéloïdes et lymphoïdes (E) comparativement aux patients sains (A et D). Les patients Fanconi présentent également une forte prédisposition au développement de syndromes myélodysplasiques caractérisés par une hypercellularité de la moelle osseuse (F) et un encombrement physique nuisant au développement normal des cellules hématopoïétiques dégénérant souvent en leucémies myéloïdes aiguës (C). Photos : 29-33
1.1.2.2 Anomalies caractéristiques de l’anémie de Fanconi et tumeurs
Un nombre important de patients FA, soit environ 60 à 70 %, souffrent de malformations congénitales diverses qui sont de sévérité variable.7 Parmi les anomalies les plus fréquentes, on
A B C
D E F
retrouve une petite stature, l’absence ou la duplication de pouce, l’absence de radius, de la dyschromie (tache café au lait, hypo ou hyper pigmentation de la peau), et une microphtalmie (diminution de la taille de l’œil) (Figure 1.2). Également, d’autres malformations peuvent toucher le système squelettique, rénal, auditif, cardio-pulmonaire, gastro-intestinal ou nerveux central.
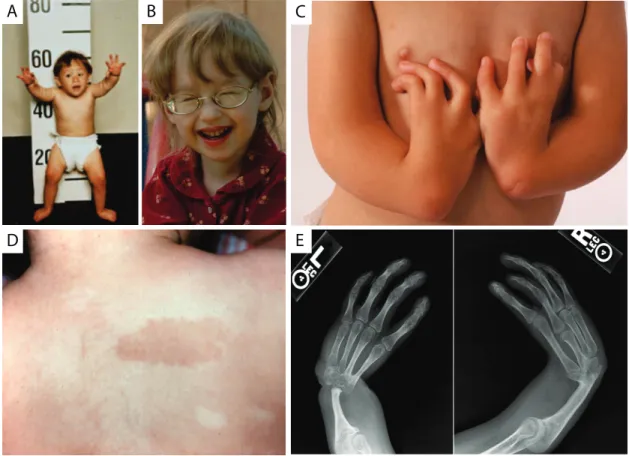
Figure 1.2 : Anomalies caractéristiques de l’anémie de Fanconi. Les patients souffrant d’anémie de Fanconi présentent communément comme caractéristiques une petite taille (A), une microphtalmie (B), une absence de pouce et de radius (C et E) et des tâches de type café au lait (D). Photos : 34-37
Une diminution notable de la fertilité, autant chez l’homme que chez la femme, est par surcroit observée, causée d’une part par des cycles menstruels anovulatoires et irréguliers, et d’autre part, par un hypogonadisme et une spermatogenèse anormale.38,39 Une fréquence élevée
d’anomalies endocrines est par ailleurs observée chez les patients Fanconi pouvant entrainer une déficience en hormones de croissance, de l’hypothyroïdisme, un déséquilibre du métabolisme du glucose et de l’insuline, de l’obésité, de la dyslipidémie ainsi que d’autres syndromes
A C
D E
métaboliques.40,41 Le retard de développement entrainé par les anomalies endocrines est de plus
associé à un retard de développement mental. L’étendue et la gravité des défauts développementaux chez les patients Fanconi sont associées à l’apparition précoce d’une pancytopénie.4 Cette
association est cohérente avec la notion que l’hématopoïèse embryonnaire peut être compromise.42
Ces importantes observations cliniques suggèrent que le fœtus Fanconi peut être exposé à des dommages à l’ADN in utero, qui peuvent conduire à des anomalies du développement et l’épuisement des réserves des CSHs embryonnaires. L’épuisement des réserves hématopoïétiques au cours du développement peut ultérieurement promouvoir une progression plus rapide vers l’anémie aplasique dans la première décennie de la vie.
Les causes de mortalité des patients Fanconi sont surtout associées au déficit sévère du système hématopoïétique et à diverses malformations congénitales. De même, le pronostic des patients est assombri par une susceptibilité accrue au développement de différents cancers, incluant principalement les leucémies myéloïdes aiguës et les tumeurs solides de type carcinomes des cellules épithéliales.18,21 Parmi les tumeurs solides les plus développées chez les patients Fanconi,
on retrouve celles des régions de la tête et du cou, des voies aériennes et digestives supérieures, ainsi que celles des voies uro-gynécologiques.24-26,43 L’incidence de développement de tumeurs
solides pour les patients Fanconi n’ayant pas été soumis à une transplantation de la moelle osseuse est de 5 à 10 %.26 L’impact des transplantations entraine une augmentation du risque d’apparition de
tumeurs solides pouvant aller jusqu’à 42 %.26 Cette augmentation du risque serait reliée aux
différents traitements d’irradiation, d’immunosuppression et de myélosuppression administrés en préparation de la transplantation, ainsi qu’aux complications associées à cette transplantation comme les infections et les réactions de rejet.44
L’hétérogénéité et la diversité des malformations congénitales observables chez les patients Fanconi ainsi que l’apparition des anomalies hématologiques ou cancéreuses au cours de leur vie rendent le diagnostic des patients difficile à partir des simples observations cliniques. Une analyse du comportement cellulaire des lymphocytes en culture des patients est nécessaire pour s’assurer de l’identification exacte de la maladie et éviter les diagnostics différentiels de la FA.
1.1.2.3 Aspects cellulaires et diagnostic de l’anémie de Fanconi
La forte prédisposition aux cancers des patients Fanconi découlerait de l’incapacité des cellules à maintenir l’intégrité génomique provoquant une accélération de l’accumulation des changements génétiques.45 En effet, les cellules lymphocytaires provenant de patients atteints de la
maladie montrent une instabilité chromosomique et une hypersensibilité aux agents pontant l’ADN, tels la mitomycine C (MMC) et le diépoxybutane (DEB), lorsqu’elles sont mises en culture.46-48 Le
traitement avec ces agents clastogéniques entraine des points de cassures aléatoires à une fréquence anormalement élevée sur les chromatides des chromosomes dans les cellules FA en comparaison avec les cellules saines. Ces chromosomes altérés s’apparient de façon imparfaite pour générer des chromosomes quadrilatéraux qui peuvent être visualisés et quantifiés en microscopie au cours de la métaphase (Figure 1.3A).48-51 La formation de ces chromosomes instables provoque
l’arrêt spontané du cycle cellulaire au cours de la phase S ou G2/M pouvant être mesuré en cytométrie de flux (Figure 1.3B).52
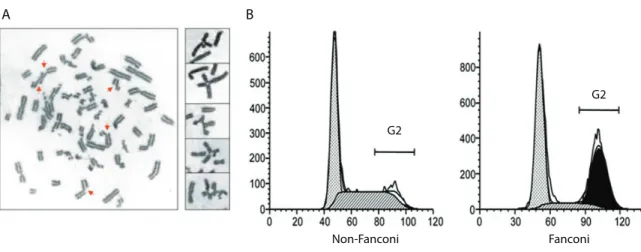
Figure 1.3 : Aspects cellulaires de l’anémie de Fanconi. Les cellules des patients Fanconi soumises à un traitement au DEB présentent l’apparition de formes chromosomiques quadrilatérales au cours de la métaphase (A) et un blocage du cycle cellulaire en phase G2 (B). Tirée de 53 et modifiée de 54
L’outil diagnostique le plus utilisé pour confirmer l’atteinte du patient repose sur l’observation de l’hypersensibilité des cellules FA aux agents pontant l’ADN.55 Un test au DEB est effectué sur des
G2
G2
Non-Fanconi Fanconi
cultures de lymphocytes provenant du sang périphérique du patient. L’apparition de cassures et la formation de structures quadrilatérales observables sur les chromosomes témoignent de la positivité ou de la négativité du test.6,56 Par contre, des résultats faux négatifs peuvent être obtenus pour
certains patients qui possèdent à la fois des cellules d’apparence normale et des cellules marquées du phénotype FA.57,58 Cette mosaïque s’explique par le mécanisme de réversion génique menant à
la correction du gène dans certaines cellules souches, qui à la base étaient mutées différemment sur chacun des allèles d’un même gène Fanconi.59,60
La gravité des aberrations chromosomiques et de l’arrêt du cycle cellulaire en phase G2, à la suite d’un traitement des cellules FA avec des agents pontant l’ADN, varie largement d’un patient à l’autre. En somme, le tableau clinique présenté par les patients Fanconi est extrêmement variable autant pour les anomalies hématologiques et physiques que pour les caractéristiques des cellules FA. Cette hétérogénéité phénotypique s’explique possiblement par la présence des nombreuses protéines Fanconi différentes pouvant être impliquée dans la maladie.
1.1.3 Génétique moléculaire de l’anémie de Fanconi
1.1.3.1 Gènes associés à l’anémie de FanconiLes patients ayant reçu un diagnostic d’anémie de Fanconi présentant un phénotype et des caractéristiques cellulaires similaires sont réunis en différents groupes de complémentation. L’analyse de la survie cellulaire des lignées lymphoblastoïdes des patients Fanconi à la suite d’un traitement aux agents pontant l’ADN après hybridations somatiques et complémentations a conduit à l’identification de 15 groupes FA. Chacun des groupes correspond à un gène muté responsable de l’apparition de la maladie (Tableau 1.1). Les gènes Fanconi identifiés jusqu’à maintenant sont répartis sur l’ensemble du génome, et sont appelés FANCA, FANCB, FANCC, FANCD1/BRCA2, FANCD2, FANCE, FANCF, FANCG, FANCI, FANCJ/BRIP1/BACH1, FANCL/PHF9, FANCM/HEF1, FANCN/PALB2, FANCO/RAD51C et FANCP/SLX4.61-67 Aujourd’hui, l’assignation des patients aux
différents groupes de complémentation s’effectue par la correction du phénotype cellulaire après la transfection virale des cellules du patient en culture avec les différents gènes Fanconi connus. Les
mutations génétiques permettant d’identifier le groupe auquel appartient le patient peuvent aussi être déterminées à la suite d’un séquençage de l’ADN.68
Environ 85 % des patients Fanconi présentent des mutations dans les gènes FANCA, FANCC ou FANCG, tandis que les autres gènes comptent pour moins de 5 % des mutations trouvées chez les patients FA. À ce jour, certains patients restent sans affectation indiquant la possibilité d’identifier de nouveaux gènes FA.6,56 Des mutations dans le gène RAD51C
(provisoirement appelé FANCO) ont été associées à un trouble s’apparentant à l’anémie de Fanconi, ce qui suggère que ce gène pourrait représenter un autre gène FA, mais la confirmation de ce groupe de complémentation reste à effectuer.69,70 Plus récemment, le séquençage d’exome par la
méthode de Sanger de l’ADN des patients n’appartenant à aucun groupe a permis d’identifier les gènes ERCC4 (XPF) et XRCC2 comme des gènes potentiellement associés à la FA.71,72
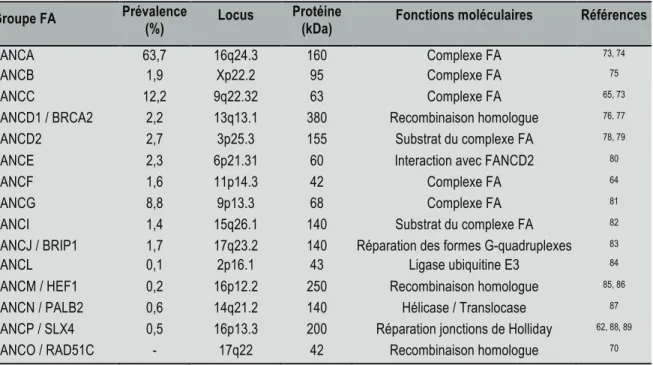
Tableau 1.1 : Gènes importants impliqués dans l’anémie de Fanconi.
Source : Fanconi Anemia Mutation Database at Rockefeller University http://www.rockefeller.edu/fanconi/mutate/. Groupe FA Prévalence (%) Locus Protéine (kDa) Fonctions moléculaires Références
FANCA 63,7 16q24.3 160 Complexe FA 73, 74
FANCB 1,9 Xp22.2 95 Complexe FA 75
FANCC 12,2 9q22.32 63 Complexe FA 65, 73
FANCD1 / BRCA2 2,2 13q13.1 380 Recombinaison homologue 76, 77
FANCD2 2,7 3p25.3 155 Substrat du complexe FA 78, 79
FANCE 2,3 6p21.31 60 Interaction avec FANCD2 80
FANCF 1,6 11p14.3 42 Complexe FA 64
FANCG 8,8 9p13.3 68 Complexe FA 81
FANCI 1,4 15q26.1 140 Substrat du complexe FA 82
FANCJ / BRIP1 1,7 17q23.2 140 Réparation des formes G-quadruplexes 83
FANCL 0,1 2p16.1 43 Ligase ubiquitine E3 84
FANCM / HEF1 0,2 16p12.2 250 Recombinaison homologue 85, 86
FANCN / PALB2 0,6 14q21.2 140 Hélicase / Translocase 87
FANCP / SLX4 0,5 16p13.3 200 Réparation jonctions de Holliday 62, 88, 89
Plusieurs études ont tenté en vain de mettre en évidence une corrélation claire entre les groupes de complémentation et la sévérité des phénotypes FA observés chez l’humain.90,91 Les
analyses corrélatives sont complexifiées par un nombre insuffisant de patients pour certains groupes de complémentation et par l’existence de plusieurs mutations différentes pour d’autres groupes.92,93
Les études de concordance chez les jumeaux monozygotiques ainsi que chez les familles consanguines avec plusieurs membres atteints de la FA ont montré qu’une seule et même mutation présente une pénétrance incomplète du phénotype.94-96 Bien qu’il existe peu de corrélations entre le
génotype et le phénotype des patients, les patients porteurs d’une mutation hypomorphique présentent généralement un phénotype moins agressif de la maladie avec des complications plus tardives. En revanche, les patients porteurs de mutations dans les gènes FANCD2 et FANCD1/BRCA2 présentent habituellement un phénotype plus sévère avec une progression plus précoce de la maladie, ainsi qu’un développement plus rapide des complications tumorales.97-100
1.1.3.2 Protéines associées à l’anémie de Fanconi
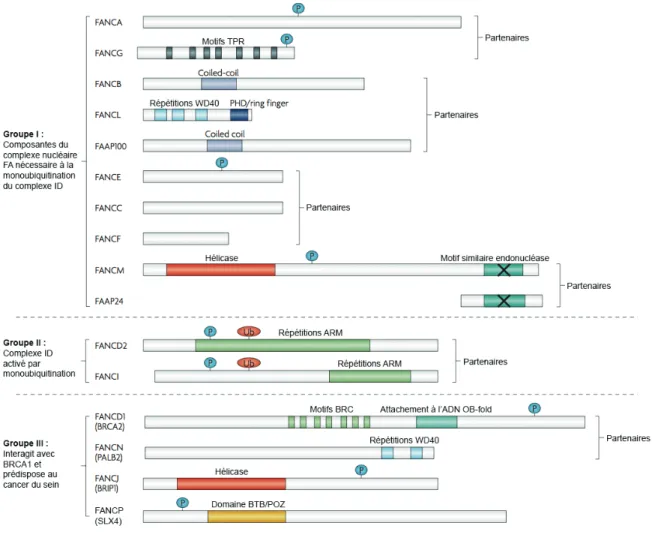
Les protéines correspondant à chaque groupe de complémentation présentent très peu d’homologie entre elles et plusieurs protéines ne possèdent aucun motif semblable à d’autres protéines répertoriées qui pourrait les relier à une fonction précise. Le rôle exact de chacune des protéines Fanconi demeure inconnu à ce jour. Par ailleurs, la présence de domaines conservés pour plusieurs d’entre elles permet d’émettre une hypothèse sur leur fonction potentielle (Figure 1.4).101
La distribution des protéines dans la cellule, les modifications post-traductionnelles ainsi que les partenaires des protéines Fanconi identifiés jusqu’à maintenant renseignent également sur les mécanismes dans lesquels les protéines Fanconi sont impliquées.102
Les protéines FANCA, FANCB, FANCD1/BRCA2, FANCE et FANCJ possèdent des séquences de localisation nucléaire (NLS) suggérant que ces protéines agissent à l’intérieur du noyau cellulaire.103 FANCA possède de plus cinq domaines d’export nucléaire (NES) nécessaire à
sa translocation cytoplasmique dépendante de CRM1.104 Par conséquent, la localisation cellulaire
des protéines FA intervient dans le bon fonctionnement des processus moléculaires. La protéine FANCC ne possède aucun domaine de localisation cellulaire et contrairement aux autres protéines Fanconi, elle se retrouve majoritairement au cytoplasme.105-108 Puisque la majorité des protéines FA
du noyau représente une étape essentielle à son activité fonctionnelle.108,109 Il est d’ailleurs connu
qu’une mutation dans le gène FANCC, entrainant la substitution de la proline 554 en une leucine, empêche la translocation nucléaire de FANCC et perturbe l’activité fonctionnelle de la protéine.110
Figure 1.4 : Représentation schématique des protéines de l’anémie de Fanconi. Plusieurs protéines Fanconi possèdent des domaines d’interaction protéines-protéines, tels des motifs TPR (FANCG) ou BRC (FANCD1), des répétitions WD40 (FANCL et FANCN) et Armadillo (ARM) (FANCD2 et FANCI) ou un domaine BTB/POZ (FANCP). Certaines protéines Fanconi sont connues pour avoir une activité enzymatique de type hélicase (FANCM et FANCJ) ou d’ubiquitine ligase (FANCL). Tirée et adaptée de 101
Certaines protéines Fanconi possèdent des domaines d’interactions protéiques favorisant le regroupement de partenaires nécessaires à leur fonction moléculaire. Par exemple, FANCD1/BRCA2 possède plusieurs répétitions BRC essentielles à son interaction directe avec RAD51, une protéine connue pour son rôle dans la recombinaison homologue.111,112 De la même manière, FANCG est
capable de rassembler plusieurs partenaires Fanconi à l’aide de sept domaines répétés TPR, participant d’une part aux interactions protéine-protéine nécessaire à la formation du complexe FA, et d’autre part à l’assemblage d’un sous-complexe comprenant FANCD2 ainsi que les protéines de réparation par recombinaison homologue FANCD1/BRCA2 et XRCC3.113,114 FANCN/PALB2 et
FANCL présentent des domaines répétés d’interactions protéiques de type WD leur permettant de s’associer respectivement avec BRCA2 et d’autres membres du complexe FA.84,86,115 Finalement,
FANCD2 et FANCI possèdent des séquences répétitives de type Armadillo (ARM) également responsables d’interactions protéiques.82,116,117 Ainsi, les protéines Fanconi peuvent jouer un rôle
dans différents processus moléculaires à la suite d’interactions avec des protéines associées à un mécanisme particulier.
Plusieurs protéines Fanconi ont été identifiées comme des gènes de susceptibilité au cancer impliqués dans la réparation de l’ADN.101,118 Comme mentionné précédemment, FANCD1/BRCA2 est
impliqué dans la recombinaison homologue après s’être lié à RAD51. De même, FANCG participe à ce processus par l’assemblage du sous-complexe FANCD2, FANCD1/BRCA2 et XRCC3. FANCJ et FANCM possèdent un domaine hélicase nécessaire à la résolution de structures secondaires pouvant survenir au cours de la réplication de l’ADN.119,120 FANCN/PALB2 est un partenaire
important pour la stabilisation et la localisation de l’interaction entre BRCA1 et BRCA2, et sa présence est nécessaire au processus de recombinaison homologue.121 De façon plus spécifique,
FANCL possède un domaine d’ubiquitine ligase responsable de la monoubiquitination des protéines FANCD2 et FANCI suggérant une implication de ces protéines dans des mécanismes de régulation, telle la réparation de l’ADN.78,82,122-124 FANCP/SLX4 représente une des protéines Fanconi
contenant le plus de domaines favorisant les interactions protéiques suggérant que FANCP/SLX4 agit comme une plateforme permettant le regroupement de plusieurs partenaires impliqués dans la réparation de l’ADN et la formation d’un complexe protéique fonctionnel.125
Les protéines Fanconi possèdent toutes des caractéristiques structurales et fonctionnelles diverses qui peuvent les relier à plusieurs mécanismes de régulation cellulaire. L’hétérogénéité
phénotypique des patients renforce l’idée de multifonctionnalité des protéines FA. L’absence de fonction commune précise suggère que les différentes protéines Fanconi agissent en tant qu’intermédiaires dans une voie métabolique canonique ou sous la forme d’un complexe multiprotéique.84,120,126-128
1.1.3.3 La voie de signalisation de l’anémie de Fanconi
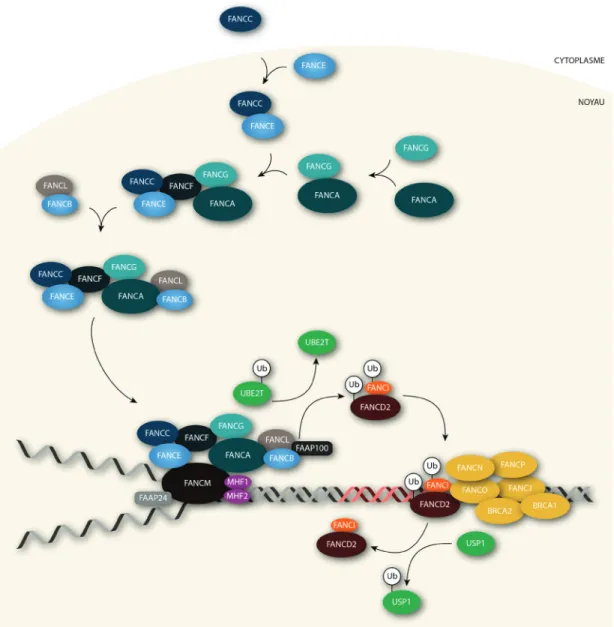
Il a été montré que la majorité des protéines Fanconi s’accumulent à l’intérieur du compartiment nucléaire pour former différents complexes et activer la voie métabolique Fanconi. (Figure 1.5) Dans la voie de signalisation FA, les protéines sont subdivisées en trois complexes.
Le premier complexe, appelé complexe nucléaire ou complexe I, agit en amont de la voie et se compose de 7 protéines FA.81,84,120,129-136 Les protéines FANCA et FANCG s’associent dans un
premier temps au cytoplasme puis migrent ensuite au noyau.137 De la même façon, les protéines
FANCC et FANCE interagissent dans le compartiment cytoplasmique avant de s’accumuler dans le compartiment nucléaire.137,138 Les sous-complexes FANCA/FANCG et FANCC/FANCE sont ensuite
liés ensemble par la protéine FANCF, et les protéines FANCB et FANCL viennent finalement compléter le complexe nucléaire FA.75,84,120,137 La protéine FANCM a déjà été considérée comme un
membre du complexe I, mais des études ultérieures dans les cellules mutantes FA-M ont rapporté des résultats suggérant que FANCM agit en amont et en aval de la voie FA.139 D’autres protéines
ont été identifiées comme des partenaires essentiels du complexe nucléaire, incluant FAAP20, FAAP24 et FAAP100, MHF1 et MHF2, FAN1 ainsi que HES1, mais aucune mutation pour ces protéines n’a encore été associée à l’anémie de Fanconi.140-142 Après un dommage à l’ADN, FANCM
en association avec d’autres protéines reconnait et modifie les fourches de réplication bloquées par le biais de son activité hélicase et translocase, et réunis le complexe nucléaire à la chromatine à la suite de son hétérodimérisation avec FAAP24.45,120,129,143,144
Figure 1.5 : Voie de signalisation de l’anémie de Fanconi. (A) Assemblage du complexe nucléaire. Les hététodimères FANCA-FANCG et FANCC-FANCE sont stabilisés par une interaction avec FANCF. L'accumulation de FANCC au noyau nécessite la protéine FANCE. Le sous-complexe FANCA-FANCG-FANCF-FANCC-FANCE se lie ensuite à FANCB et FANCL. Le complexe nucléaire est ensuite chargé sur la chromatine suite à son interaction avec FANCM. (B) Ubiquitination du complexe ID. Le complexe nucléaire interagit avec le conjugateur d’ubiquitine UBE2T par le biais de FANCL, ce qui provoque le chargement de l'ubiquitine sur le complexe FA. Le complexe ID (FANCD2-FANCI) est alors ubiquitiné par le complexe FA, ce qui permet son assemblage sur la chromatine avec les constituants du complexe III (C). (D) Le complexe ID est reconnu par la peptidase d'ubiquitine USP1, qui retire les ubiquitines présentes sur FANCD2 et FANCI. Ceci provoque le relargage du complexe ID, le rendant de nouveau disponible pour être activé par le complexe FA. Ub : ubiquitine.
La fonction principale du complexe nucléaire après sa formation est de modifier par monoubiquitination les protéines du deuxième complexe, appelé complexe ID, constitué des protéines FANCD2 et FANCI.82,84,122,123 FANCL, par le biais de son domaine PHD caractéristique
des ligases ubiquitine de type RING, est la sous-unité catalytique du complexe nucléaire responsable de la modification post-traductionnelle de FANCD2 et FANCI.78,82,122 La participation des autres
sous-unités du complexe I à l’ubiquitination des membres du complexe ID est indirecte, et n’est nécessaire qu’à l’assemblage adéquat et au maintien de la stabilité du complexe.75,134 L’interaction
entre FANCE et FANCD2 confère une spécificité du substrat pour le complexe et contribue à l’orientation exacte des protéines nécessaires au transfert de l’ubiquitine sur la lysine cible.80 La
formation du complexe ID par hétérodimérisation est une étape essentielle et préalable à la reconnaissance et à la monoubiquitination de FANCD2 et FANCI par le complexe I. Le transfert d’ubiquitine sur une protéine implique la participation de plusieurs molécules, chacune étant responsable d’une action précise.145,146 Dans la voie FA, la protéine UBE2T, un conjugateur
d’ubiquitine E2, a été identifiée comme étant le partenaire de FANCL responsable de l’apport de l’ubiquitine jusqu’au complexe nucléaire.147 Et, à ce jour, l’activateur d’ubiquitine E1 reste inconnu.
La monoubiquitination de FANCD2 ainsi que de FANCI modifie leur distribution cellulaire, et favorise l’interaction entre le complexe ID et les protéines Fanconi du complexe III agissant en aval de la voie, soit FANCD1, FANCJ, FANCN et FANCP ainsi que RAD51C (provisoirement FANCO).69,70 Finalement, la déubiquitination de FANCD2 et FANCI par le complexe enzymatique
UAF1-USP1 permet le relargage de FANCD2 et FANCI et le retour à un niveau basal de la voie FA.148,149
La cascade signalétique, comprenant la formation du complexe nucléaire, la monoubiquitination des protéines du complexe ID et le chargement de ces protéines sur la chromatine de façon concomitante aux protéines du complexe III, constitue le sentier canonique de l’anémie de Fanconi. La voie de signalisation FA est impliquée dans divers mécanismes cellulaires, dont le développement hématopoïétique 150, la réparation de l’ADN 151, la régulation du cycle
1.1.4 Les implications des protéines Fanconi
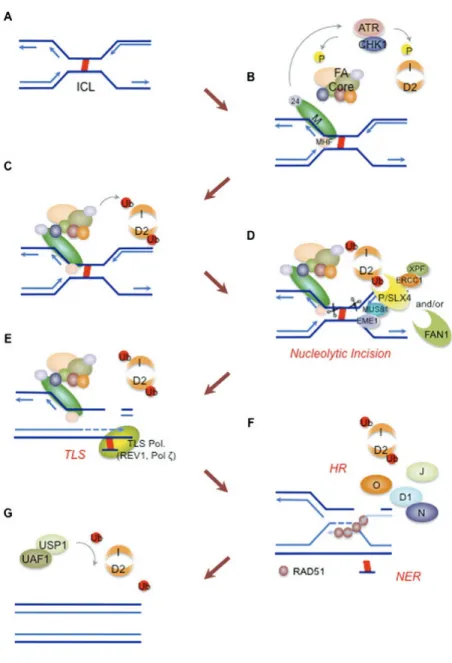
1.1.4.1 Réplication et réparation de l’ADN et cycle cellulaire dans l’anémie de Fanconi La régulation de la réparation de l’ADN après un dommage à l’ADN représente la fonction la mieux caractérisée de la voie de signalisation FA (Figure 1.6).151 La FA a été classée comme une
maladie héréditaire affectant le système de réparation de l’ADN après l’observation d’une hypersensibilité des cellules de patients Fanconi, lorsque mises en culture avec des agents pontant l’ADN.117,120,153,154 L’apparition d’aberrations chromosomiques dans ces conditions reflète un
dysfonctionnement dans la voie de réparation de l’ADN spécialisée dans la résolution des ponts double brins sur l’ADN, une lésion bloquant la réplication de l’ADN et la transcription des gènes.155
La sous-unité FANCM est responsable de l’initiation de la voie. Elle forme avec FAAP24 un hétérodimère capable de reconnaitre les lésions à l’ADN, d’amorcer le point de contrôle signalétique effectué par ATR-CHK1 et de rassembler le complexe nucléaire FA pour stabiliser la fourche de réplication bloquée.129,156,157 Au cours de l’initiation, plusieurs protéines Fanconi sont phosphorylées
par le complexe ATR-CHK1 renforçant le lien entre la voie FA et la réparation de l’ADN.158 Les
protéines MHF1 et MHF2 maintiennent la stabilité de l’interaction entre FANCM et la chromatine et contribuent à l’activation de la voie de signalisation FA.140,142 Le complexe FA activé modifie par
monoubiquitination FANCD2 (FANCD2-Ub) et FANCI (FANCI-Ub). La modification de FANCD2 par monoubiquitination est possible uniquement après que cette dernière ait été phosphorylée par la kinase ATR, elle-même recrutée par la protéine RPA1 qui aura reconnu le dommage à l’ADN.159
FANCD2-ub agira ensuite comme intermédiaire dans le regroupement de nucléases au niveau des lésions de l’ADN, telles FAN1 et FANCP/SLX4, pour l’initiation de l’incision nucléotidique.160-165 SLX1
et les nucléases MUS81-EME1 et XPF-ERCC1 sont ensuite rassemblées au site du dommage par l’intermédiaire d’une interaction avec FANCP/SLX4. Elles participent ensuite au décrochage du pont interbrin transformant la fourche de réplication bloquée en bris de l’ADN doubles brins.166-170
Figure 1.6 : Implication des protéines FA dans la réparation de l’ADN. (A) Deux fourches de réplication convergent vers un pont liant les deux brins d’ADN. (B) Le complexe FANCM-FAAP24-MHF1/2 reconnait les fourches bloquées, déclenche la réponse du point de contrôle dépendant de ATR-CHK1 entrainant la phosphorylation de FANCA-E-D2-I et rassemble le complexe FA. (C) Le complexe FA modifie par ubiquitination FANCD2 et FANCI, et le complexe ID est mobilisé vers la lésion de l’ADN. (D) FANCD2-Ub, FANCP/SLX4, ERCC1-XPF, MUS81-Eme1 et FAN1 se mobilisent à la lésion et coordonnent l’incision nucléotidique. (E) Le décrochage laisse des nucléotides attachés au brin complémentaire, qui seront contournés par la synthèse d’ADN translésionnelle effectuée par les polymérases REV1 et Polζ. (F) L’incision crée une rupture de l’ADN sur les deux brins qui est réparée par recombinaison homologue. En aval, les protéines FA stimulent l’invasion des brins d’ADN par RAD51 et la résolution des intermédiaires de recombinaison. La réparation par excision de nucléotides supprime les adduits restants et comble le vide. (G) Le complexe USP1-UAF1 supprime les ubiquitines de FANCD2-I et achève la réparation. Tiré de : 151
Le complexe nucléaire FA ne régule pas uniquement les étapes d’incision de l’ADN; il contribue également à la synthèse d’ADN translésionnelle (TLS) en favorisant le regroupement du complexe polymérase PCNA-REV1-Polζ à la fourche de réplication bloquée permettant la reprise de la réplication en aval des intermédiaires du pont interbrin.171,172 Cette fonction du complexe est
indépendante de la monoubiquitination de FANCD2, mais nécessite la présence de la sous-unité FAAP20 pour établir un lien entre le complexe nucléaire FA et le complexe PCNA-REV1.172,173
L’incision nucléotidique laisse comme intermédiaire de réparation des cassures de l’ADN double brin (DSB). La recombinaison homologue constitue le mécanisme principal utilisé par la cellule pour réparer les DSB et elle implique l’utilisation du nouveau gabarit d’ADN produit par TLS. La voie de signalisation Fanconi promeut ce mécanisme, mais aussi supprime la voie de réparation des lésions de l’ADN par liaison des bouts sans homologie (NHEJ) afin de prévenir la réparation infidèle de l’ADN.79,174 L’interaction entre les protéines FANCD1/BRCA2, FANCN/PALB2 et BRCA1
en aval de la voie signalétique permet le chargement de la protéine RAD51 sur le nouveau brin d’ADN, une étape clé dans la recombinaison homologue.76,86,121,175,176 FANCJ/BRIP1, après sa
dimérisation, agit en aval de la voie pour compléter la recombinaison homologue en dégageant les polymères RAD51 des filaments d’ADN simple brin. Et BRCA1 régule l’activité hélicase de FANCJ/BRIP1 en empêchant le retrait prématuré de RAD51.177-179
La phosphorylation de la protéine FANCD2, de même que NBS1, par la kinase ATM est par ailleurs observable, à la suite d’un dommage à l’ADN causé par des radiations ionisantes.180,181
L’arrêt au point de contrôle de la phase S du cycle cellulaire est dépendant de ces étapes de phosphorylation.180 La protéine NBS1 phosphorylée forme un complexe avec les protéines RAD50
et MRE11. Une fois phosphorylée, FANCD2 migre aux foyers de réparation de l’ADN avec le complexe MRN (MRE11-RAD50-NBS1) pour permettre la réparation par recombinaison homologue.182,183 Le complexe MRN agit sur FANCD2 en favorisant sa stabilité et sa localisation aux
sites de dommages.184 Par conséquent, FANCD2 assume une fonction charnière à l’intersection de
deux voies signalétiques suivant deux modifications post-traductionnelles de FANCD2 soit, la réparation de l’ADN et le contrôle du cycle cellulaire.
La détection des dommages causés à l’ADN s’avère défectueuse dans les cellules FA puisque des altérations évidentes du point de restriction de la phase réplicative sont observables.185
et être plutôt reconnus au cours du point de restriction G2 après la terminaison de la réplication.186,187
Une compensation au point de restriction de la phase G2/M est alors opérée en allongeant le temps de cette phase pour permettre la réparation de l’ADN.186,188 Des études récentes ont montré que la
voie de signalisation FA est contrôlée par ATR, et que le complexe FANCM-FAAP24 a un rôle à jouer dans l’activation de la voie de signalisation ATR.156 L’activité translocase de FANCM, non
nécessaire à l’activation de la voie FA, est requise pour son rôle dans la signalisation ATR-CHK1.156
D’autre part, la protéine FANCC est aussi connue pour remplir un rôle dans la régulation du cycle cellulaire après son association avec la kinase mitotique dépendante de la cycline Cdc2, laquelle régule la transition de la phase G2 vers M.189 Conséquemment, les cellules FA-C montrent un arrêt
du cycle cellulaire au cours de la phase G2/M suggérant un rôle direct de la protéine FANCC dans la signalisation du cycle cellulaire.190
Même s’il est évident que la voie de signalisation FA joue un rôle primordial lors de la réplication et la réparation du matériel génétique ainsi que dans la surveillance des points de contrôle au cours du cycle cellulaire, ces mécanismes ne peuvent expliquer toutes les manifestations cliniques de la maladie liées au développement ou à l’hématopoïèse.
1.1.4.2 Apoptose et détoxification des produits oxygénés dans l’anémie de Fanconi
Les cellules FA sont sujettes à une apoptose prématurée. En effet, les études sur les cellules de patients et de souris mutantes pour les gènes Fanconi ont clairement montré que les protéines FA sont impliquées dans la régulation de la survie et de la mort cellulaire.191-196 Les cellules des patients
atteints de la FA présentent une expression élevée des récepteurs de mort cellulaire Fas dans les cellules CD34+ et montrent une apoptose constitutive.197,198 Plus particulièrement, les cellules FA-C
entrent en apoptose plus rapidement que les cellules normales en réponse à divers signaux apoptotiques provenant de l’environnement extérieur.194,199-201 En effet, les cellules FA-C sont
hypersensibles à plusieurs signaux apoptotiques, tels le TNF-α et l’INF-γ, et ce phénotype est réversible par l’expression de la protéine FANCC sauvage.199 Des études ont montré qu’en plus de
sa présence dans le complexe multiprotéique nucléaire, la protéine FANCC est impliquée dans diverses voies apoptotiques de façon indépendante des autres protéines FA. Certaines mutations dans le gène FANCC empêchent la protéine de jouer son rôle au cours du processus d’apoptose sans affecter sa capacité à participer aux activités de réparation de l’ADN avec les autres protéines
Fanconi du complexe nucléaire.202 Par exemple, FANCC assure la modulation de la voie
apoptotique par la suppression de l’activation de la kinase PKR après s’être liée à Hsp70, une molécule chaperonne connue pour protéger les cellules contre les stress oxydatifs, les agents chimiothérapeutiques, les radiations et les facteurs tumoraux INF-γ et TNF-α.199-201,203 De la même
façon, l’inactivation de la caspase 3, également provoquée par l’interaction de FANCC avec Hsp70, rend compte de la fonction anti-apoptotique de FANCC.199-201,203 De ces faits, il a été conclu que
FANCC agit de concert avec Hsp70 pour prévenir l’apoptose dans les cellules exposées à l’INF-γ et TNF-α. Ensuite, FANCC promeut la survie cellulaire par le sentier de signalisation JAK/STAT en réponse à divers cytokines et facteurs de croissance à la suite de son association avec le facteur de la transcription STAT1.204 Dans les cellules FA-C, cette interaction est abolie et empêche la liaison
de STAT1 à la chaine α du récepteur de l’IFN et réduit la phosphorylation de STAT1.204 STAT1 ne
peut alors se mobiliser au noyau, induisant un message dans la cellule de privation de facteur menant finalement à l’apoptose cellulaire.204 Durant le processus d’apoptose, FANCC subit une
modification protéolytique donnant naissance à une protéine FANCC tronquée.205 L’analyse de la
séquence protéique de FANCC a révélé la présence de trois sites putatifs pouvant être reconnus par des protéases de type caspase. L’inhibition de la protéolyse de FANCC par la mutagenèse d’un de ces sites n’entraine pas d’hypersensibilité aux agents pontant l’ADN, mais retarde l’induction de l’apoptose. Ces résultats suggèrent que la protéine FANCC est régulée par une modification protéolytique durant le processus d’apoptose et que cette modification est pro apoptotique. Les cellules FA montrent aussi une accélération du raccourcissement des télomères, qui pourrait être causée par le nombre de divisions cellulaires anormalement élevé dans les cellules hématopoïétiques pour générer des cellules matures ou par l’apparition de bris dans les séquences télomériques.206-209 Finalement, la suractivité du processus d’apoptose dans les cellules des patients
Fanconi peut découler de l’incapacité des cellules FA à réparer les dommages à l’ADN, des interactions incorrectes avec les autres protéines du sentier apoptotique et de la prématurité du raccourcissement des télomères.199,210
Les cellules des patients atteints de la FA présentent de plus une défaillance dans la réponse aux stress oxydatifs. En effet, des radicaux oxygénés pouvant entrainer des dommages à l’ADN s’accumulent à la suite de réactions d’oxydoréduction et amènent un retard de croissance cellulaire et un blocage du cycle cellulaire en phase G2.211-217 Cette affirmation est supportée par
l’observation d’une interaction entre FANCC et la NADPH-cytochrome p450 réductase, laquelle est impliquée dans l’activation oxydoréductrice de nombreux xénobiotiques.218 L’augmentation de
l’activité de détoxification des composés toxiques et carcinogènes de GSTP1 à la suite de son interaction avec FANCC soutient également une implication des protéines FA dans la réponse aux stress oxydatifs.210 D’autres protéines Fanconi sont aussi impliquées dans le processus de
détoxification : FANCD2, par son interaction avec les protéines BRCA et ATM impliquées dans la régulation des dommages oxydatifs 211; FANCE, par son interaction avec les protéines FANCC et
FANCD2 connues pour leurs réponses aux stress oxydatifs 80,135,219; FANCG, par ses interactions
avec le cytochrome P450 2E1 et la protéine mitochondriale peroxyredoxine-3 220-222; et enfin,
FANCA, par sa régulation négative dépendante de la kinase Akt/PKB impliquée dans la signalisation cellulaire dépendante de l’oxydoréduction 223.
Le rôle des protéines Fanconi dans la signalisation liée au stress oxydatif a été montré dans plusieurs lignées cellulaires, dont les CSHs.224,225 Cette observation suggère une fonction potentielle
de la voie de signalisation Fanconi dans l’hématopoïèse.
1.1.4.3 Régulation transcriptionnelle dans l’anémie de Fanconi
Un rôle moins considéré des protéines Fanconi est la régulation de la transcription des gènes au cours du cycle cellulaire. L’interaction entre certaines protéines Fanconi et divers facteurs de la transcription suggère un rôle potentiel de la voie FA dans la régulation de l’expression de certains gènes. Le premier partenaire des protéines Fanconi impliqué dans la transcription à avoir été identifié est FAZF, et il a été identifié comme interagissant spécifiquement avec FANCC.226
FAZF est un répresseur transcriptionnel appartenant à la famille des BTB/POZ jouant un rôle important dans la régulation de différents processus développementaux incluant la prolifération cellulaire, la différenciation et la formation tumorale.227 La surexpression de FAZF dans des cellules
myéloïdes mène à son accumulation au cours de la phase G1 du cycle cellulaire et à l’apoptose des cellules. Ceci suggère que FAZF exerce un rôle essentiel au cours de la prolifération cellulaire et de l’induction de l’apoptose.228,229 Puisque les mutations dans la protéine FANCC interfèrent avec la
liaison de son partenaire FAZF, et que les CSHs provenant de souris FancC mutantes présentent un cycle cellulaire accéléré; 187 une hypothèse plausible est que l’interaction entre FANCC et FAZF