Architecture et essentialité des complexes de biosynthèse des acides mycoliques de la bactérie pathogène Mycobacterium tuberculosis
Texte intégral
Figure


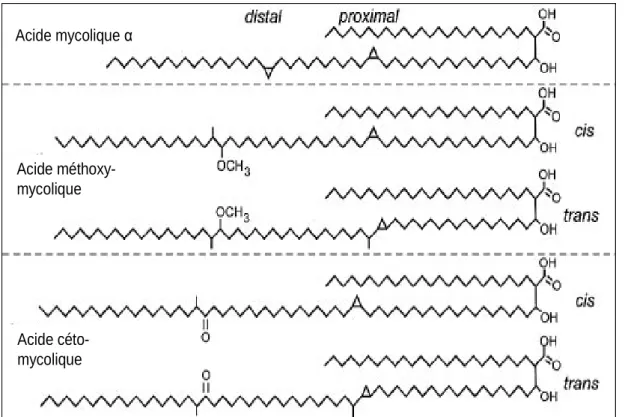
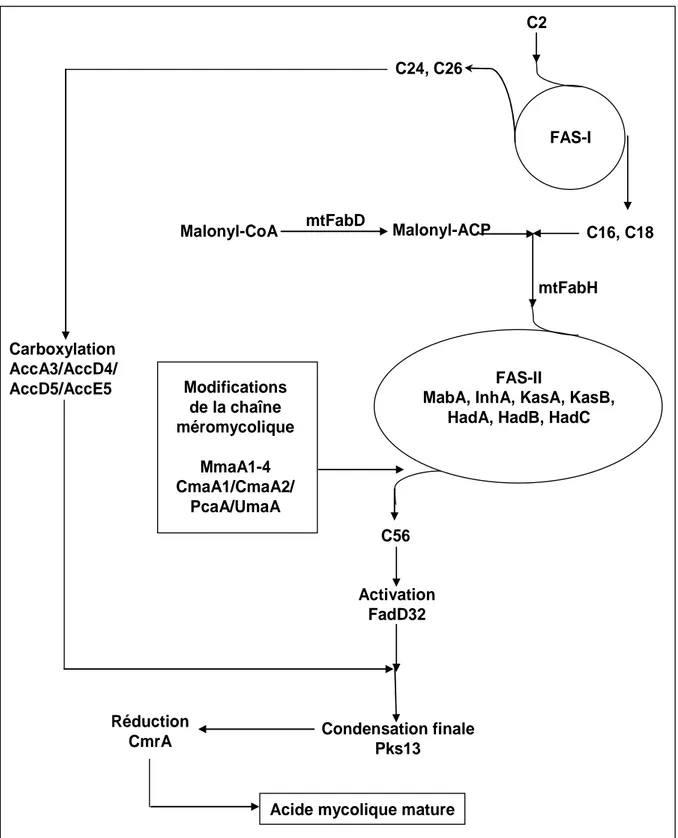
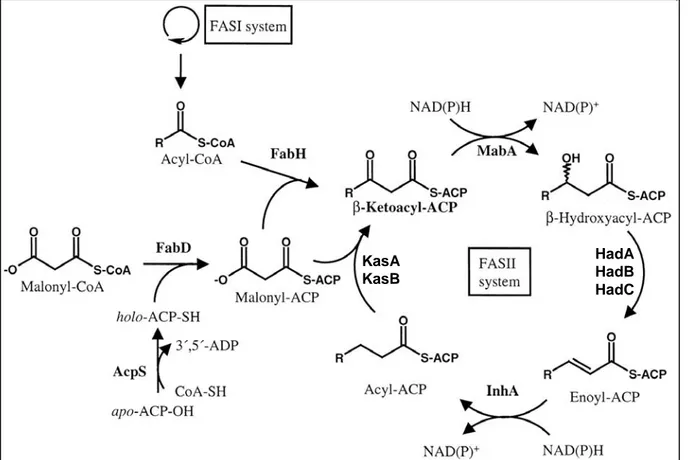
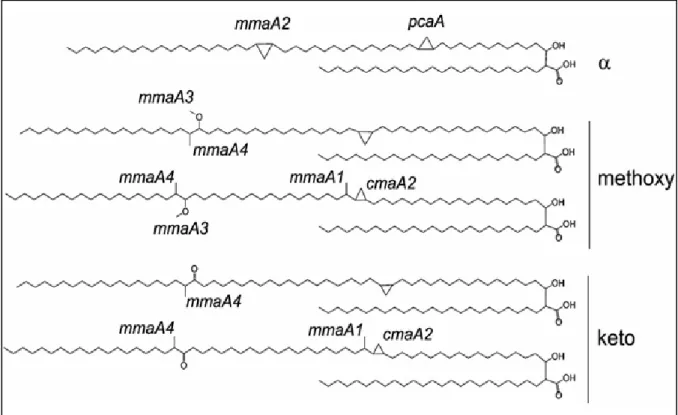

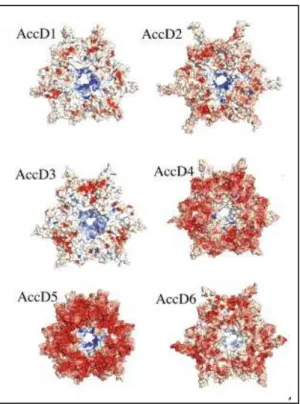
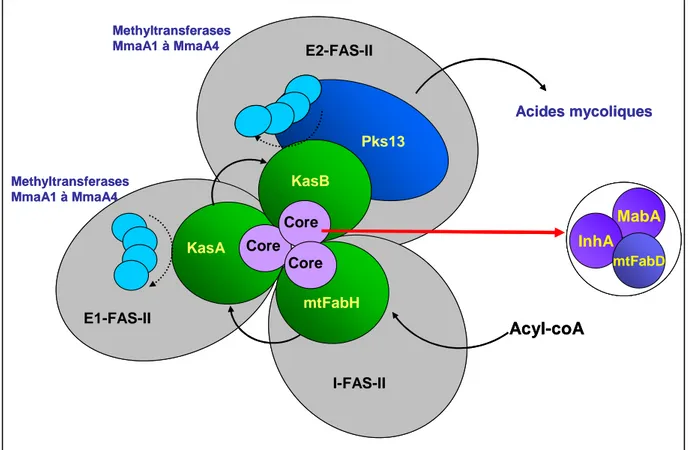
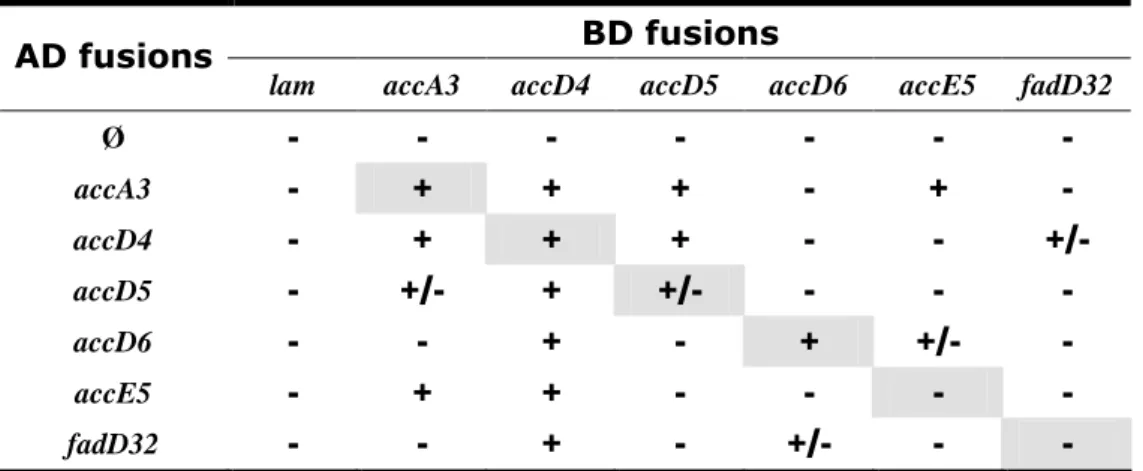
Documents relatifs
té allélique mais tant que le gène ne sera pas isolé, il serait prématuré de conclure car, chez la souris, il n'y a pas moins de treize gènes dont les mutations
A l'aide des techniques de biologie moléculaire permettant l'expression de protéines fusionnées à la GFP (et ses variants spectraux) en cellules et des méthodes utilisant les déclins
Ainsi, certains gènes sont encodés dans tous les génomes des bactéries d’un niveau taxonomique donné (et forment le génome de base) alors que d’autres ne sont encodés que
Le présent projet avait pour but de documenter les patrons de méthylation et d'expression de ce gène et de deux autres gènes, IL1RL1 et CCL26, qui sont directement
Les maladies génétiques monogéniques les plus connues et les plus anciennement observées sont les maladies récessives, comme la drépanocytose (gène HBB[Aki+11]),
2.3 Analyse de l’expression des gènes ALDH10A8 et ALDH10A9 chez les mutants Dans la mesure où les deux protéines ALDH10A8 et ALDH10A9 sont identiques à 80 % et similaires à 91 %
Les gènes qui spécifient les protéines de la membrane des globules gras du lait (« Milk Fat Globule Membrane », MFGM) sont en revanche plus conservés, depuis les monotrèmes
Les autres protéines identifiées dans ce groupe correspon- dent à des protéines impliquées dans le métabolisme lipidique, des protéines cytoplasmiques, d’autres protéines du
