HAL Id: pastel-00727391
https://pastel.archives-ouvertes.fr/pastel-00727391
Submitted on 3 Sep 2012HAL is a multi-disciplinary open access
archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
peptide sur surfaces modèles d’acier inoxydable
Garrain Pierre-Alain
To cite this version:
Garrain Pierre-Alain. Etude ab initio de l’adsorption d’acides aminés et peptide sur surfaces modèles d’acier inoxydable. Chimie analytique. Université Pierre et Marie Curie - Paris VI, 2011. Français. �pastel-00727391�
THESE DE DOCTORAT DE L’UNIVERSITE PIERRE ET MARIE CURIE
Spécialité
Chimie Physique et Chimie Analytique Présentée par
Pierre-Alain GARRAIN
Pour obtenir le grade de
DOCTEUR de l’UNIVERSITÉ PIERRE ET MARIE CURIE
Etude ab initio de l’adsorption d’acides aminés et peptide sur surfaces modèles d’acier inoxydable
soutenue le 07 novembre 2011 devant le jury composé de
M. Jean François LAMBERT Professeur à l’UPMC Examinateur Mme. Marie Pierre GAIGEOT Professeur à l’Université d’Evry Rapporteur M. Gianfranco PACCHIONI Professeur à l’Université de Milan Rapporteur Mme. Chantal COMPERE Ingénieur IFREMER Examinateur M. Marc BAADEN Chargé de Recherche à l’IBPC Examinateur Mme. Dominique COSTA Chargé de Recherche Directeur de thèse
à Chimie ParisTech
M. Philippe MARCUS Directeur de Recherche Co-directeur de thèse à Chimie ParisTech
Université Pierre & Marie Curie - Paris 6
Bureau d’accueil, inscription des doctorants et base de données
Esc G, 2ème étage
15 rue de l’école de médecine 75270-PARIS CEDEX 06
Tél. Secrétariat : 01 42 34 68 35 Fax : 01 42 34 68 40 Tél. pour les étudiants de A à EL : 01 42 34 69 54 Tél. pour les étudiants de EM à MON : 01 42 34 68 41 Tél. pour les étudiants de MOO à Z : 01 42 34 68 51 E-mail : [email protected]
Introduction 7
I De la théorie à la pratique 19
I.1 Notions de chimie quantique . . . 21
I.1.1 Méthode Hartree-Fock . . . 22
I.1.2 Théorie de la fonctionnelle de la densité . . . 25
I.1.2.1 Théorèmes de Hohenberg et Kohn . . . 25
I.1.2.2 Approche Kohn-Sham . . . 26
I.1.2.3 Les différentes classes de fonctionnelles . . . 27
I.1.3 La méthode de DFT+U . . . 29
I.2 Application à la modélisation des systèmes périodiques . . . 30
I.2.1 Modèles périodiques . . . 30
I.2.2 Base à onde plane . . . 32
I.2.3 Pseudopotentiels . . . 33
I.3 Principe d’un calcul VASP . . . 34
I.4 Prise en compte des forces de dispersion . . . 35
I.5 Dynamiques moléculaires . . . 37
I.5.1 Dynamique moléculaire classique . . . 37
I.5.1.1 Forme générale du champ de force . . . 38
I.5.1.2 Principe de la dynamique moléculaire . . . 39
II Vers un modèle d’acier inoxydable 41
II.1 L’oxyde de chrome . . . 43
II.1.1 Détails calculatoires . . . 45
II.1.1.1 Mise au point des conditions de calcul . . . 45
II.1.1.2 Energie d’adsorption . . . 46
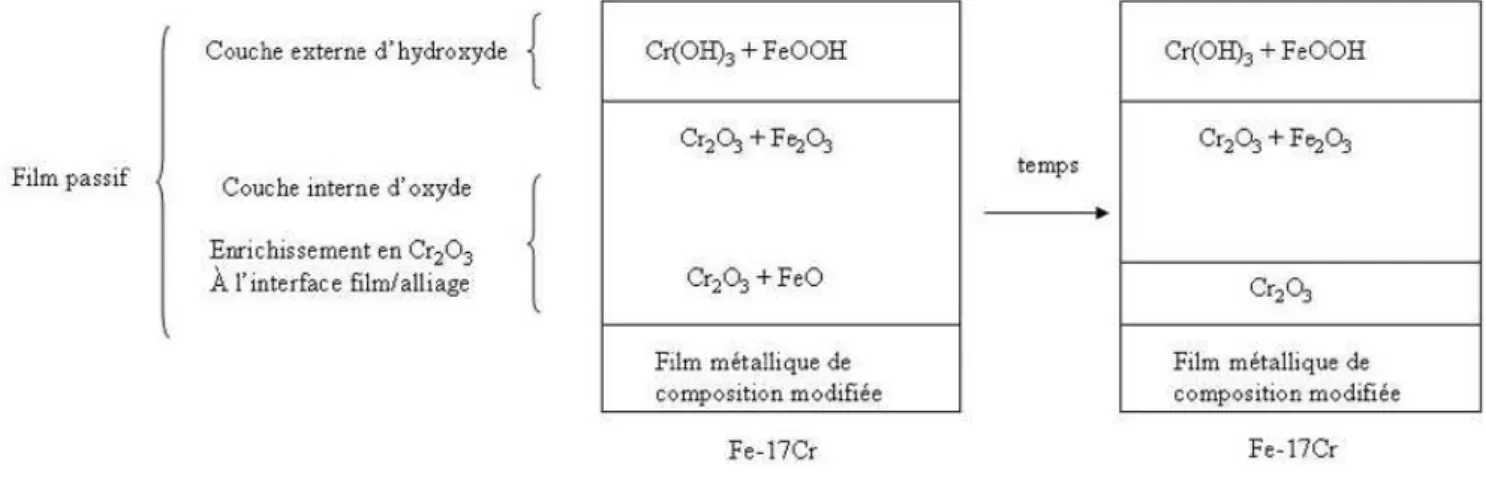
II.1.2 Propriétés structurales de ↵-Cr2O3 . . . 47
II.1.3 Propriétés électroniques de ↵-Cr2O3 . . . 48
II.1.4 Etude de la surface ↵-Cr2O3 (0001) . . . 49
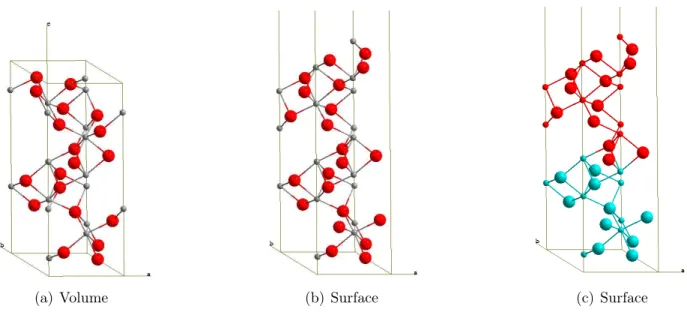
II.1.5 Hydroxylation de la surface ↵-Cr2O3 (0001) . . . 50
II.2 L’oxyde de fer . . . 52
II.2.1 Etude du volume de ↵-Fe2O3 . . . 53
II.2.2 Etude de la surface ↵-Fe2O3 . . . 54
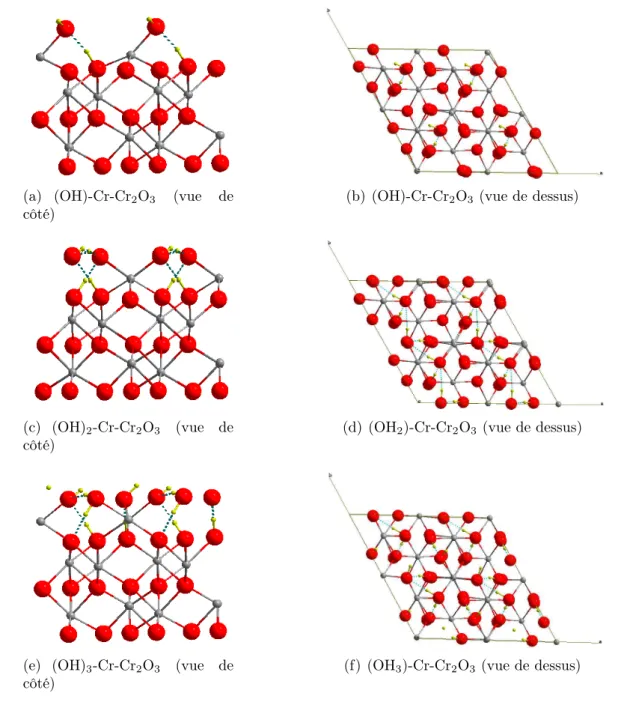
II.2.3 Hydroxylation de la surface de Fe2O3 . . . 55
II.3 Comparaison de la réactivité de l’eau sur les surfaces ↵-Cr2O3 et ↵-Fe2O3 . . . 56
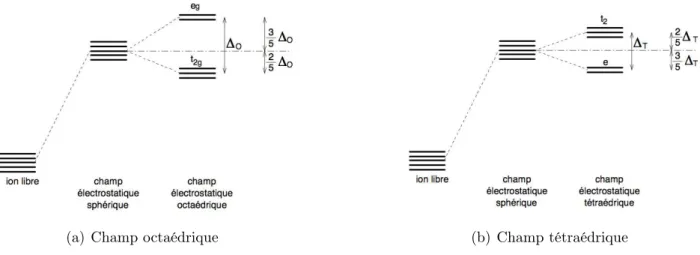
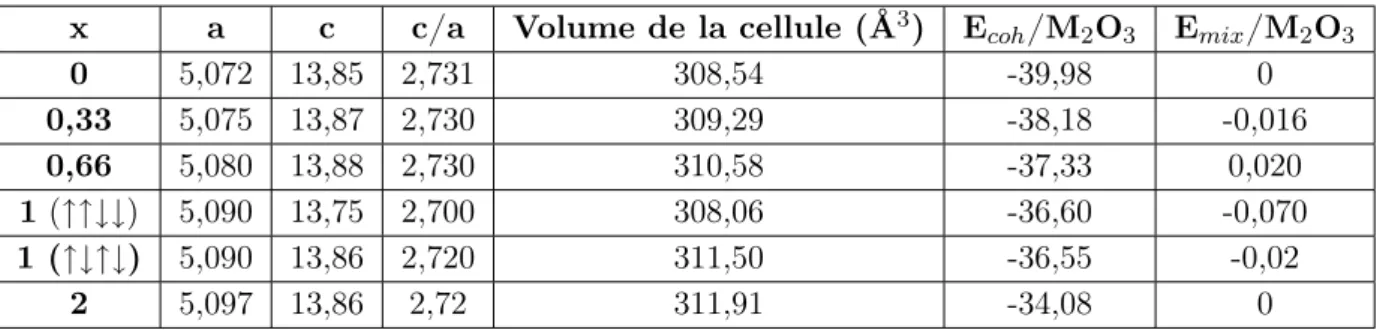
II.3.1 Aperçu de la théorie du champ cristallin . . . 57
II.3.1.1 Levée de dégénérescence des orbitales d dans un champ cristallin 58 II.3.1.2 Configuration électronique et champ cristallin . . . 58
II.3.2 Discussion . . . 59
II.4 Films minces FexCr2−xO3 . . . 62
II.4.1 Mélange FexCr2−xO3 dans le volume . . . 63
II.4.2 Localisation du fer dans un "slab" de Cr2O3 . . . 65
II.4.3 Adsorption de l’eau sur les films de Fe-O-Fe-Cr2O3 . . . 68
II.4.4 Bilan de la réactivité des surfaces de Cr2O3, Fe2O3 et Fe-O-Fe-Cr2O3 : une approche thermodynamique . . . 70
II.4.4.1 Approche thermodynamique . . . 70
II.4.4.2 Résultats . . . 72
III Etude de l’adsorption des acides aminés sur les surfaces de Cr2O3, Fe2O3 et Fe-O-Fe-Cr2O3 anhydres 77 III.1 Détails calculatoires . . . 81
III.1.1 Modèles utilisés . . . 82
III.1.3 Calcul énergétique . . . 84
III.1.3.1 Energie d’adsorption . . . 84
III.1.3.2 Energie de dispersion . . . 85
III.1.3.3 Energie de liaison . . . 85
III.2 Etude de l’adsorption de la glycine sur la surface ↵-Cr2O3 (0001) . . . 86
III.2.1 Adsorption de la glycine sur l’oxyde de chrome à faible taux de recouvrement (✓ = 0,25 ML) . . . 86
III.2.1.1 Contribution des forces de dispersion . . . 88
III.2.1.2 Déformation de la glycine et de la surface . . . 89
III.2.1.3 Analyse de DOS . . . 91
III.2.2 Adsorption de la glycine sur l’oxyde de chrome à fort taux de recouvrement (✓ = 0,5 à 1 ML) . . . 93
III.2.3 Discussion . . . 97
III.3 Etude de l’adsorption de l’acide glutamique sur la surface ↵-Cr2O3 (0001) . . . 97
III.3.1 Adsorption de l’acide glutamique sur l’oxyde de chrome à faible taux de recouvrement (✓ = 0,25 ML) . . . 98
III.3.1.1 Energie d’adsorption totale . . . 99
III.3.1.2 Energie de liaison Molécule - Surface . . . 102
III.3.2 Adsorption de l’acide glutamique sur l’oxyde de chrome à fort taux de re-couvrement (✓ = 0,5 à 1 ML) . . . 103
III.3.3 Discussion . . . 105
III.4 Influence de la nature de la chaine latérale . . . 106
III.4.1 Adsorption des acides aminés sur l’oxyde de chrome à faible taux de recou-vrement (✓ = 0,25 ML) . . . 107
III.4.2 Adsorption de la cystéine et de la thréonine par les groupements -SH et -OH sur l’oxyde de chrome à faible taux de recouvrement (✓ = 0,25 ML) . . . . 109
III.4.3 Adsorption de la cystéine par les groupements -COOH et -SH . . . 111
III.4.4 Adsorption "tri-ancrée" : cas de l’arginine . . . 112
III.4.5 Adsorption de la cystéine et de la thréonine sur l’oxyde de chrome à fort taux de recouvrement (✓ = 1 ML) . . . 114
III.5.1 Adsorption de la glycine et de l’acide glutamique sur l’oxyde de fer à bas
taux de recouvrement (✓ = 0,25 ML) . . . 119
III.5.2 Influence de l’augmentation du taux de recouvrement pour la glycine . . . 121
III.5.3 Discussion . . . 123
IV Acidité et adsorption 127 IV.1 Propriétés acide-base . . . 128
IV.2 Concept acide-base pour les surfaces . . . 129
IV.3 Concept acide-base pour les acides aminés . . . 130
IV.4 Force des acides et adsorption . . . 130
IV.4.1 Acido-Basicité de Brønsted . . . 131
IV.4.2 Acido-Basicité de Lewis . . . 133
V Etude de l’adsorption des acides aminés sur des surfaces de Cr2O3 hydroxylées135 V.1 Rôle de l’acido-basicité de Brønsted de la surface . . . 139
V.1.1 Détails calculatoires . . . 139
V.1.2 Topologie des surfaces hydroxylées et acidité de Brønsted . . . 140
V.1.3 Adsorption de sphère externe . . . 143
V.1.3.1 Adsorption à faible taux de recouvrement (✓ = 0,25 ML) . . . 143
V.1.3.2 Co-adsorption de la glycine et de l’eau . . . 147
V.1.3.3 Autoassemblage de la glycine (✓ = 1 ML) . . . 148
V.1.4 Adsorption de sphère interne . . . 150
V.1.4.1 Cas de la glycine . . . 150
V.1.4.2 Autres acides aminés . . . 151
V.1.5 Discussion . . . 153
V.2 Extrapolations à l’interface solide/liquide . . . 154
VI Etude de l’adsorption du peptide Glu2 sur la surface ↵-Cr2O3 (0001) 161 VI.1 Analyse statistique du Glu2 en phase gaz et en solution aqueuse . . . 164
VI.1.1 Analyse de la conformation du Glu2 en phase gaz à 300K . . . 164
VI.1.2 Analyse de la conformation du Glu2 en solution aqueuse . . . 166
VI.1.3 Analyse des sites d’adsorption de la surface . . . 170
VI.2 Etude ab initio de l’adsorption du dipeptide de l’acide glutamique sur la surface
↵-Cr2O3 (0001) . . . 172
VI.2.1 Energie d’adsorption . . . 172
VI.2.2 Energie de liaison . . . 175
VI.2.3 Discussion . . . 176
Conclusion générale et perspectives 181
A Conformères des acides aminés en phase gaz 187
B Dynamique en phase gaz du dipeptide Glu2 191
Bibliographie 193
Les interactions entre une biomolécule et une surface inorganique sont vraisemblablement an-térieures à la vie sur terre (il y a 3,5-3,8 milliards d’années), puisque la synthèse de la liaison peptidique s’est probablement effectuée en phase adsorbée d’acides aminés sur les constituants de la croûte terrestre (voir la référence [1] pour une revue sur le sujet). La biogenèse et la biomi-néralisation (de silices, perles, coquille d’œuf ou squelette osseux) requièrent la participation des molécules biologiques en contact avec une surface inorganique. Une relation complexe existe donc entre la matière dure et la matière molle.
En surface d’un matériau inorganique immergé en milieu contenant des biomolécules, un bio-film se développe rapidement. Un biobio-film est une communauté multicellulaire de micro-organismes (bactéries, champignons, algues ou protozoaires), adhérant entre eux et à une surface, et marquée par la sécrétion d’une matrice adhésive et protectrice (MEC, matrice extra cellulaire). La figure 1 montre des exemples de biofilms en milieu naturel ou industriel. C’est un biofilm qui nous fait dé-raper sur les cailloux, ou souffrir de caries dentaires. . . Les conséquences de la présence non désirée de biofilms sont importantes, générant par exemple la biocorrosion des structures marines (Fig. 1(c)), l’érosion de bâtiments, la corrosion de structures immergées, ou des problèmes d’hygiène dans l’industrie agroalimentaire, dans les circuits d’eau ou en milieu hospitalier. Par exemple, un circuit d’eau chaude sur lequel adhère un biofilm peut être infecté par des Légionelles (Fig. 1(d)). L’implantation d’un biomatériau (greffe osseuse) contaminé peut générer des infections létales. L’adsorption de bactéries sur des surfaces est en effet un vecteur important de transmission de maladies. Dans les hôpitaux, à l’exception des blocs opératoires lavés entièrement quotidienne-ment, si les sols sont lavés quotidiennequotidienne-ment, ni les murs ni les conduits d’aération ne le sont,
des outils médicaux doit être la plus contrôlée possible.
(a) (b)
(c) (d)
(e) (f)
Figure 1 : Exemples de biofilms formés en milieu naturel (a) algues sur fond marin (b) carie dentaire (c) hublot de bateau (d) colonie de Légionelles dans une canalisation de chauffe eau (e-f) BIOGLYPHS, Images de biofilms, voir http ://www.microbialart.com/
Mais les biofilms ne sont pas uniquement néfastes. Outre le fait que ce sont peut être nos plus anciens parents, il faut leur concéder un certain caractère esthétique, comme illustré sur les figures 1(d) et 1(e). Dans certains cas, une bonne adhésion des cellules avec une surface peut être requise, par exemple dans le cas des exo-greffes osseuses ou des implants dentaires. La bonne intégration d’un matériau dans un organisme vivant est nommée la biocompatibilité. Elle se définit comme la capacité des matériaux à ne pas interférer, ne pas dégrader le milieu biologique dans lequel ces matériaux sont utilisés (un être vivant le plus souvent). Le terme biocompatibilité a trait princi-palement aux dispositifs médicaux en contact direct, bref ou prolongé, avec les tissus et fluides
internes du corps comme les sondes, les seringues ou les prothèses. Les matériaux biocompatibles sont appelés biomatériaux. Un biomatériau est donc un matériau non viable, d’origine naturelle ou artificielle, utilisé dans l’élaboration de dispositifs médicaux destinés à être mis en contact avec des tissus biologiques. Le titane, les alliages Cobalt-Cr et les aciers inoxydables sont des matériaux métalliques biocompatibles, utilisés notamment pour des prothèses et implants osseux, pour leurs propriétés mécaniques ainsi que pour leur absence de toxicité (Fig. 2). Il est donc nécessaire de
(a) (b) (c)
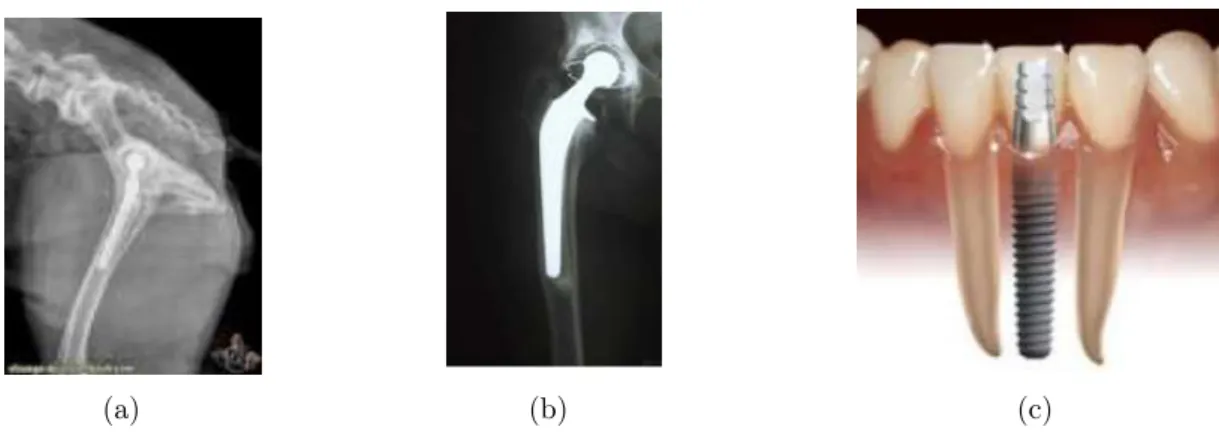
Figure 2 : Exemples de greffes de biomatériaux d’alliage de Ti ou d’acier inoxydable dans un tissu vivant (a) prothèse de hanche du lapin (b) prothèse de hanche de l’humain (c) implant dentaire
contrôler l’adhésion de cellules et bactéries sur les surfaces pour réduire la formation de biofilms lorsqu’ils sont indésirables, ou a contrario de favoriser la biocompatibilité d’un matériau. Cette dernière, qualifiée de "biocompatibilité passive" (absence de rejet et de toxicité), devient "biocom-patibilité active". Une très grande activité est vouée depuis quelques années à la compréhension et à l’optimisation de la biocompatibilité, notamment à travers la compréhension du rôle de la surface inorganique [2–4]. Cet effort est motivé notamment pour les greffes osseuses par le vieillissement de la population, qui induit l’exigence d’un temps de séjour de la greffe dans le corps humain croissant (il est actuellement d’une quinzaine d’années), le but étant d’éviter des opérations chirurgicales répétitives chez une population fragilisée par l’âge.
Les premières étapes de la formation d’un biofilm ou de l’immersion d’un matériau en milieu vi-vant consistent en l’adsorption de protéines et de sucres sur la surface [5,6]. De nombreuses études expérimentales ont porté sur le rôle de ce film primaire d’adhésion sur la croissance ultérieure de bactéries, dont des travaux du Laboratoire de Physico-Chimie des Surfaces (LPCS) [7, 8]. Par la fonctionnalisation des surfaces à l’aide de protéines ou de peptides, on peut favoriser l’intégration du biomatériau. Par exemple, la fonctionnalisation de surfaces de biomatériau par les protéines
Mais les études montrent aussi qu’il est nécessaire, pour maîtriser la croissance cellulaire, de comprendre profondément l’interaction des protéines avec les surfaces. De nombreux travaux ont été réalisés par la communauté des chimistes, en utilisant les techniques spectroscopiques de surface, pour mieux comprendre l’adsorption de protéines sur des surfaces et la conformation en résultant. Citons la spectroscopie de photoélectrons X (XPS), les techniques vibrationnelles (Fourier Transformed InfraRed spectroscopy (FTIR), Infrared Reflection Absorption Spectroscopy (IRAS), High Resolution Electron Energy Loss Spectroscopy (HREELS), Sum Frequency Gene-ration (SFG)), la microscopie à champ proche (AFM) et la microbalance à quartz (QCM). La combinaison de techniques complémentaires comme l’XPS (analyse chimique de la surface) et la QCM (évaluation in situ de la masse adsorbée) permet par exemple de caractériser la quantité et l’état chimique de la protéine adsorbée [10, 11]. Malgré tout, la question de l’adsorption des protéines sur les surfaces et de la conformation résultante est loin d’être élucidée [12,13]. La raison
Figure 3 : Approche "bottom up" de l’interaction biomolécule-surface inorganique
en est que les trois protagonistes de l’adsorption, surface (matière dure), biomolécule (matière molle) et solvant répondent à des descripteurs différents, qui seront en partie abordés au cours de
cette thèse.
Pour faire face à cet important défi conceptuel, quelques équipes ont entamé une approche systématique de complexification graduelle du système, ou approche de type "bottom up", allant du plus simple au plus compliqué. Cette approche peut être déclinée pour les trois partenaires de l’adsorption, surface, molécule et solvant (Fig. 3). Nous mentionnons ici quelques exemples, présentés selon chacun de ces axes :
Complexification de la biomolécule
L’équipe d’Imamura et al a réalisé un travail remarquable (tableau 1) sur l’adsorption des acides aminés, peptides, fragments protéiques et protéines sur surfaces de biomatériaux métalliques (Ti, acier inoxydable) [14–23]. Ces travaux, principalement de FTIR, ont mis en évidence le rôle des résidus acides (carboxylates) sur l’adsorption. Ces résidus acides en interaction avec la surface sont à l’état déprotonnés, même lorsque l’adsorption a lieu à un pH où ceux-ci sont protonnés en solution. Une étude de l’adsorption de peptides (une dizaine de résidus) sur surface d’acier a montré que le nombre et la répartition des résidus acides influaient sur l’adsorption, mettant en évidence les rôles conjoints de la structure primaire et secondaire du peptide. Cette série de travaux est réalisée sur des surfaces pulvérulentes, moins bien définies que des surfaces de matériaux massifs. L’interaction exacte entre les résidus et la surface n’a pas été totalement élucidée.
Année Surface Molécule adsorbée Références
1998 Acier inoxydable BSA [14]
1999 Acier inoxydable β-Lactoglobuline [15]
2000 Acier inoxydable Acides aminés [16]
2003 Acier inoxydable Peptides (résidus acides) [17] 2004 Acier inoxydable BSA et fragments de peptides [18]
2004 Acier inoxydable Acides carboxyliques [19]
2005 Ti, Ta et Zr Acides carboxyliques, amines et peptides [20]
2006 Acier inoxydable β-Lactoglobuline [21]
2007 Ti Oligopeptides (résidus acides et basiques) [22]
2008 Ti Protéines [23]
Table 1 : Liste des principales études réalisées par Imamura et al concernant l’adsorption d’acides aminés, de peptides et de protéines sur les aciers inoxydables et le titane
Il est plus courant dans la littérature de trouver des travaux ponctuels sur l’adsorption d’acides aminés et de petits peptides sur surfaces métalliques et oxydées, travaux qui ont fait l’objet de
à l’interface solide-gaz dans le domaine des surfaces chirales (voir les références [25, 26] pour des revues sur ce thème), l’adsorption des biomolécules sur surfaces oxydées relevant certainement d’autres mécanismes que sur surfaces métalliques.
La communauté des géochimistes a réalisé un grand nombre de travaux d’adsorption de molé-cules organiques naturelles (naturally occuring molemolé-cules, NOM), dont des acides aminés, sur des surfaces minérales (alumine, boehmite, goethite) et sur TiO2, pour la compréhension de
phéno-mènes interfaciaux en milieu naturel. Dans ce domaine aussi des revues sont disponibles [27, 28]. Sans être exhaustifs, mentionnons les travaux de Persson et al [29–31] et Hazen et al [32–36]. Très brièvement, le mode d’adsorption est exploré de la manière suivante. Si l’adsorption est dépen-dante du pH, et si, lors de l’adsorption, la surface et la biomolécule sont de charges opposées, il est conclu que l’adsorption est régie par des lois électrostatiques. De plus, si les spectres IR enregistrés pour la molécule adsorbée sont semblables à ceux de la molécule en solution, il est conclu que l’adsorption est de type sphère externe, c’est-à-dire impliquant des liaisons hydrogène (voir chapitre V). A contrario, si l’adsorption est indépendante du pH, elle n’est pas régie par l’électrostatique. Si de plus, les spectres IR de la molécule adsorbée sont différents de la molécule en solution, il est déduit que l’adsorption est de sphère interne, c’est-à-dire, avec formation d’une liaison iono-covalente avec la surface. Il ressort de ces études que les deux mécanismes d’adsorption coexistent, notamment pour l’ancrage des carboxylates sur les surfaces oxydées (Fig. 4). L’ana-lyse des réactions entre la fonction carboxylate et la surface (ou spéciation à l’interface) permet de prévoir les modes d’adsorption en fonction du pH et de la concentration de l’acide aminé en solution, comme montré sur la Figure 4 pour l’aspartate sur TiO2 [33].
Figure4 :Modes d’adsorption de l’aspartate sur le rutile, calculés en considérant les pK des différentes réactions de surface. Le mode "bridging bidentate" fait référence à une adsorption de sphère interne
Le rôle des acides carboxyliques est confirmé par les études d’adsorption de peptides sur sur-faces oxydées. L’adsorption de peptides de la lysine (n = 2 à 5) et de la polylysine sur TiO2
est due à la formation de liaisons Ti-OCO avec le carboxylate alpha d’une part, d’autre part à des interactions électrostatiques entre les résidus basiques chargés positivement et la surface de TiO2 chargée négativement [37]. Les mêmes auteurs trouvent que sur un film de Cr hydroxylé,
ni la lysine ni ses peptides ne s’adsorbent à pH physiologique, à cause de l’absence d’interactions électrostatiques entre la surface et les résidus. L’adsorption de mutants du peptide EAK16 (H-AKAKAEAE-AKAKAEAE-NH2) sur TiO2 et Au a été caractérisée par NEXAFS, XPS et IR. Le
"design" du peptide permet de contrôler le degré d’organisation de la couche peptidique adsor-bée. Selon les auteurs, l’adsorption sur la surface a lieu via la formation de liaisons Ti-O avec les carboxylates, et l’organisation entre peptides via les liaisons inter-moléculaires [38]. Cette interpré-tation est connexe avec les études de couches auto-assemblées d’acides carboxyliques sur métaux ou métaux oxydés [39]. Le rôle des interactions électrostatiques a aussi été mis en évidence lors de l’adsorption de peptides enrichis en résidus basiques sur des surfaces de TiO2 et SiO2[40,41].
L’ad-sorption de fibrine sur Cr2O3 [42] et de mutants de l’Escherischia Coli sur Fe2O3 [43] a aussi été
attribuée aux résidus basiques. Au contraire, quelles que soient les conditions de pH, la polylysine ne s’adsorbe pas sur la surface d’Al2O3 [44]. L’adsorption de polyglutamate est, elle, dépendante
de la présence de cations en solution [44].
Notons ici que l’étude de l’adsorption d’acides aminés, peptides et oligopeptides comme briques élémentaires des protéines n’a pas seulement un intérêt académique. De possibles retombées pour-raient voir le jour dans la synthèse de nanoparticules de catalyseurs métalliques de taille et mor-phologie contrôlée, la formation de nano-alliages [45] ou la fonctionnalisation de surfaces pour des applications aussi différentes que la nano-électronique [46] ou la dépollution des eaux [47].
Complexification du matériau
L’étude de la complexification graduelle du matériau et de sa surface est réalisée historiquement au LPCS (voir dans le chapitre II la partie concernant la caractérisation des films passifs sur aciers inoxydables). L’adsorption de protéine albumine de sérum bovin (BSA), une protéine globulaire classiquement étudiée, sur des surfaces de complexité croissante, Cr, FeCr et acier inoxydable a été caractérisée au sein du LPCS, et dans le cadre d’un groupement de recherche (BASIS puis GDR 2614, coordonné par Ifremer) [7, 48, 49]. La BSA a une plus grande affinité avec le Cr pur
qu’avec la surface d’acier. Plus tard, l’adsorption de protéines de la MEC sur surfaces modèles des biomatériaux (Ti et Cr) a aussi été caractérisée [50]. Il se forme une couche continue de protéine sur la surface du biomatériau, dont une partie est adsorbée irréversiblement [51]. La quantité de BSA adsorbée sur du Cr pur est indépendante du pH [10], suggérant que les interactions électrostatiques ne sont pas les seules forces pilotant l’adsorption. L’adsorption ne modifie pas la composition cationique du film passif (Cr3+ dans un oxyde et un hydroxyde). De plus, l’adsorption
est irréversible, suggérant la formation de forces iono-covalentes entre la biomolécule et la surface. Dans un travail ultérieur, il a été montré que l’adsorption de BSA se fait au détriment d’un plan de groupements -OH de la surface suggérant une adsorption de sphère interne de la BSA sur Ti et Cr [52]. Notons cependant qu’une autre étude n’a pas permis de mettre en évidence un effet du
Figure 5 : Modèle issu des études de l’adsorption de BSA sur Cr pur, d’après Ithubide et al [10] et Payet et al [50]. La composition du film passif sur Cr et aciers est détaillée dans le chapitre II
degré d’hydroxylation de la surface de TiO2 sur l’adsorption de fibronectine [53].
Complexification de l’interface : activité de l’eau
Les études conjointes d’adsorption de la phase gaz et de la phase liquide sont rarissimes. Le système glycine/silice amorphe a été caractérisé à l’interface solide-gaz et solide-liquide par Lam-bert et al [54, 55]. Dans les deux cas, les résultats de l’étude IR suggèrent que l’adsorption a lieu par la formation d’un réseau de liaisons hydrogène. La glycine est adsorbée sous forme neutre de la phase gaz et sous forme zwitterionique de la phase liquide, avec une possible co-adsorption du solvant. La lysine a été adsorbée de la phase gaz et de la phase liquide sur une surface de Cu. L’adsorption à partir de la phase gaz a été réalisée sur surface métallique et préoxydée. Sur la surface métallique, il y a formation d’une couche ordonnée de lysine. La présence d’O préadsorbé
a pour effet la perte de l’ordre de cette couche. Il est aussi montré que le mode d’adsorption de la lysine à partir de la solution aqueuse dépend de la spéciation de la lysine en solution [56].
Même à partir de ces travaux sur des systèmes simplifiés, il est difficile de caractériser les liaisons entre la surface et la protéine à l’échelle atomique et moléculaire et un concept unifica-teur permettant de décrire l’interface protéine/surface inorganique est encore manquant. En effet, comme le montrent les précédents exemples, la théorie de Derjaguin and Landau, Verwey and Overbeek (DLVO), qui prend en compte les interactions électrostatiques, de van der Waals et hydrophiles-hydrophobes, actuellement majoritairement utilisée, est insuffisante pour décrire l’en-semble des interactions en jeu et prévoir les modes d’adsorption des biomolécules. Par exemple, les acides aminés acides, glutamique et aspartique s’adsorbent sur l’acier inoxydable à pH acide, alors que les acides aminés et la surface ont une charge positive [16]. La pepsine s’adsorbe dans la SBA-15 (silice microporeuse) dans des conditions où la surface et la protéine sont de même charge [57]. L’adsorption de la BSA sur le Cr est indépendante du pH appliqué et donc de la charge de la surface. Notamment, une monocouche de BSA est formée à un pH de 4 où les deux entités, surface et BSA, sont de charge positive. Ces exemples montrent que les interactions électrostatiques ne peuvent tout expliquer, du moins dans le cas de ces surfaces hydroxylées. En outre, le facteur entropique peut expliquer l’adsorption, un changement conformationnel pouvant compenser des interactions répulsives.
Dans ce contexte, un certain nombre d’études théoriques utilisant des méthodes atomistiques ont été engagées et sont en fort développement, compte tenu de l’importance des enjeux sociétaux sous jacents à cette question d’une part, et des fortes avancées des capacités de calcul d’autre part. Nous pouvons schématiquement distinguer deux stratégies, dépendant directement de la méthode de calcul :
– Les études ab initio traitent de l’adsorption d’acides aminés sur des surfaces modèles (mé-talliques, oxydes anhydres ou oxydes hydroxylés), en général en absence de solvant. Ici aussi, nous ne mentionnerons pas les études d’adsorption sur surfaces métalliques. Les études réa-lisées sur surfaces anhydres et hydroxylées sont détaillées dans les chapitres respectifs. De très rares études prennent en compte le solvant, soit sous la forme de micro-solvatation [58], soit explicitement [59–62]. Notons un travail récent utilisant une méthode hybride, DFT-B,
études ab initio permettent une exploration exhaustive du mode d’adsorption, incluant la formation de liaisons hydrogène, de liaisons covalentes et, plus récemment, de liaisons faibles (forces de dispersion). Ces études, peu nombreuses en ce qui concerne les surfaces d’oxydes, sont décrites en détail dans les chapitres III et V.
– Les études par champ de force classique traitent de l’adsorption de protéines ou de peptides sur des surfaces en présence de solvant. Les surfaces envisagées sont TiO2 [64], MgO [65],
calcite [66], hydroxyapathite [67], or [68, 69] et des surfaces de type graphite, graphène. Citons aussi les travaux de Robert Latour sur l’adsorption de protéines sur des couches organiques auto-assemblées [70]. Deux travaux ont retenu notre attention. Kang et al ont étudié l’adsorption de la protéine HSA (Human Serum Albumine) sur TiO2 rutile [71]. Les
auteurs ont montré que des liaisons covalentes sont formées sur la surface anhydre entre les fonctions Glu et les Ti de surface, et des liaisons hydrogène sur la surface hydroxylée. Il existe à notre connaissance une seule étude théorique par champ de force de l’adsorption de protéine sur surface d’or [72]. Dans cette étude (Fig. 6), la β-Lactoglobuline interagit avec une surface d’Au chargée positivement (l’or est utilisé dans cette étude comme "modèle" de surface d’acier). Les auteurs ont identifié que les résidus s’approchant le plus de la surface étaient localisés dans le fragment 102-135, et comporte des résidus Thr125 (rouge), Lys100 (orange), Pro50 (jaune), Gln13 (vert), et Thr18 (bleu). Cependant le modèle utilisé pour représenter la surface d’acier inoxydable n’est pas très réaliste. Dans les travaux par champ de force classique, la possibilité d’échange de ligands n’est pas encore envisagée. Sur la surface hydroxylée, seules les liaisons hydrogène et les interactions faibles sont prises en compte.
Il existe donc un vide méthodologique à combler ainsi qu’un grand nombre de résultats de référence ("benchmarks") à apporter à la communauté sur l’adsorption d’acides aminés et de petits peptides sur surfaces de biomatériaux. Cette thèse présente une approche "bottom up" de l’interaction d’acides aminés et d’un peptide sur surface d’acier inoxydable. Dans ce cadre, nous présenterons tout d’abord les méthodes théoriques utilisées lors de ce travail. Puis nous l’appliquerons à l’étude de l’adsorption de l’eau sur les surfaces de Cr2O3, Fe2O3 et Fe-O-Fe-Cr2O3
permettant ainsi de modéliser un film passif en surface des aciers inoxydables. Nous étudierons alors l’adsorption des acides aminés sur ces modèles anhydres, puis dans un deuxième temps sur ces modèles hydroxylés. Enfin, nous présenterons une nouvelle méthodologie pour l’étude de l’adsorption de peptides sur des surfaces bien définies. Cette méthodologie est appliquée à l’exemple de l’adsorption du dipeptide de l’acide glutamique (Glu2) sur Cr2O3.
Introduction
Depuis les années 1960, l’intérêt du calcul quantique n’a cessé d’augmenter. En effet, les cal-culs quantiques permettent une étude approfondie (à une échelle atomique) de la structure locale et des propriétés électroniques d’une surface, permettant ainsi la confrontation avec des données expérimentales.
La méthode Hartree-Fock est la première méthode de calcul à avoir vu le jour. Bien que cette méthode donne satisfaction dans de nombreux cas, ses limites ainsi que l’absence de prise en compte de la corrélation électronique ont conduit aux développements de nouvelles méthodes. Ces dernières peuvent être classées en deux catégories, les méthodes post Hartree-Fock basées sur la méthode Hartree-Fock mais avec une prise en compte de la corrélation électronique et les méthodes de la théorie de la fonctionnelle de la densité basées sur une observable, la densité électronique. La théorie de la fonctionnelle de la densité s’est distinguée ces dernières années par la bonne qualité des résultats obtenus et les temps de calcul associés. En effet, l’énergie exacte de l’état fondamental pour un système à n électrons est exprimée comme une fonctionnelle de la densité, permettant ainsi d’éviter les calculs de corrélation qui sont très couteux. Même si les études de type DFT sont utiles pour prédire les propriétés structurales et électroniques des systèmes carac-térisés par de fortes liaisons chimiques, la DFT pure ne tient pas compte des forces de dispersion. Ainsi, nous avons choisi d’utiliser la méthode proposée par Grimme et al [73] afin de prendre en compte ces forces de dispersion. Dans le cas d’une minimisation de l’énergie, nous nous déplaçons presque exclusivement vers le bas de la surface d’énergie potentielle (SEP), et le minimum atteint est en général le minimum local le plus proche de la configuration initiale. Dans ce cas il n’est pas possible de surmonter des barrières énergétiques et la dynamique moléculaire est un moyen plus efficace de recherche de configurations de plus basses énergies.
Dans le cadre de ce chapitre, nous effectuerons un bref rappel des notions théoriques dans le but de présenter la théorie de la fonctionnelle de la densité. Puis nous essayerons d’appliquer cette méthode à l’étude d’un système périodique. Nous présenterons le logiciel VASP qui nous a permis de réaliser l’ensemble des calculs basés sur la théorie de la fonctionnelle de la densité et nous introduirons la méthode proposé par Grimme et al [73] afin de prendre en compte les forces de dispersion. Pour finir, nous aborderons la dynamique moléculaire, une méthode de choix pour
l’exploration de la SEP et l’étude de milieux condensés en phase liquide.
I.1
Notions de chimie quantique
La quête de tout chimiste théoricien est de trouver les solutions de l’équation de Schrödinger dont la forme non-dépendante du temps est donnée par l’équation I.1 [74] :
ˆ
H(r, R) (r, R) = E (r, R) (I.1)
Cette équation décrit la dualité de l’électron possédant un caractère corpusculaire et ondulatoire. Les solutions sont données par les fonctions propres (fonctions d’onde ) et les valeurs propres E qui sont les valeurs numériques de l’énergie des états décrits par les fonctions qui dépendent à la fois de la position des noyaux R, ainsi que de la position (et du spin) des électrons r. Dans l’équation I.1, ˆH est l’opérateur Hamiltonien non relativiste défini par la somme des opérateurs associés à l’énergie cinétique ( ˆT ) et l’énergie potentielle ( ˆV ) :
ˆ
H = ˆT + ˆV (I.2)
Pour un système moléculaire comportant N noyaux de charge Zαe et de masse Mα et n électrons
de masse me, ce dernier peut également être décrit par la relation I.3 :
ˆ H = ˆTe+ ˆTN + ˆVeN + ˆVee+ ˆVN N (I.3) avec : ˆ Te= − n X i=1 ~2 2mer 2 i ˆ TN = − N X α=1 ~2 2Mαr 2 α ˆ VeN = − n X i=1 N X α=1 Zαe2 (4⇡✏0)|ri− Rα| ˆ Vee= n X i=1 n X j>i e2 (4⇡✏0)|ri− rj| ˆ V = N X N X ZαZβe2
où Rα et ri sont respectivement l’ensemble des coordonnées des N noyaux et n électrons. Etant
donné que les noyaux ont une masse beaucoup plus importante que celle des électrons, ils se dé-placent beaucoup plus lentement. Ainsi, dans la recherche d’une solution approchée, nous pouvons découpler le mouvement des électrons à celui des noyaux qui sont supposés fixes. Dans le cadre de cette approximation de Born-Oppenheimer [75], l’énergie cinétique des noyaux peut être négligée et le terme de répulsion entre noyaux supposé constant. Ainsi, nous pouvons écrire l’Hamiltonien électronique ˆHe selon la relation I.4 :
ˆ
He = ˆTe+ ˆVee+ ˆVeN + ˆVN N (I.4)
Etant donné que ˆVeN dépend de Rα, ˆHedépend évidemment des coordonnées nucléaires. Cet
opé-rateur ne commute donc pas avec l’opéopé-rateur ˆTN. Comme ces deux opérateurs apparaissent dans
l’Hamiltonien moléculaire ˆH, les mouvements électroniques et nucléaires ne sont pas séparables. Il n’est donc pas possible d’écrire la fonction d’onde totale (ri, Rα) sous la forme de deux fonctions
d’onde séparées, l’une pour les noyaux, l’autre pour les électrons. Ainsi, les fonctions d’onde propres
ei(ri, Rα) dépendent des coordonnées nucléaires et sont solutions de l’équation de Schrödinger
électronique. La fonction d’onde totale s’écrit de façon générale sous la forme d’une combinaison linéaire de produits de fonctions d’onde électroniques et de fonctions d’onde nucléaires :
(ri, Rα) =
X
i
Ni(Rα) ei(ri, Rα) (I.5)
I.1.1
Méthode Hartree-Fock
La résolution exacte de l’équation de Schrödinger (équation I.1) pour des systèmes avec plus d’un électron n’est pas possible car dans ce cas là, le mouvement des électrons est couplé à celui des autres électrons. Ainsi, afin de résoudre l’équation de Schrödinger pour des atomes ou des molécules polyélectroniques, il est nécessaire de faire certaines approximations comme dans le cas de la méthode Hartree-Fock. Le but est de résoudre l’équation de Schrödinger électronique
ˆ
He e = Ee e où l’Hamiltonien électronique ˆHe s’exprime en unités atomiques par la relation I.6 :
ˆ He = − n X i=1 1 2r 2 i − n X i=1 N X α=1 Zα |ri− Rα| + n X i=1 n X j>i 1 |ri− rj| + N X α=1 N X β>α ZαZβ |Rα− Rβ| (I.6)
Dans le cas de la méthode Hartree-Fock, chaque électron se déplace à l’intérieur d’un potentiel créé par les noyaux, mais aussi dans le potentiel moyen de tous les électrons. La fonction d’onde
e est décrite par un unique déterminant de Slater Φe [76] (équation I.7), lui même produit
antisymétrique de n fonctions d’onde monoélectroniques.
Φe = " " " " " " " " " φ1(1) · · · φn(1) · · · · φ1(n) · · · φn(n) " " " " " " " " " (I.7)
L’Hamiltonien s’exprime de la manière suivante : ˆ He = n X i=1 ˆh(i) + n X i=1 n X j>i ˆ g(i, j) + ˆVN N (I.8)
avec ˆh(i) un opérateur monoélectronique et ˆg(i, j) un opérateur biélectronique. Par analogie avec l’équation I.6, nous pouvons déduire que :
ˆh(i) = − 1 2r 2 i − N X α=1 Zα |ri− Rα| ˆ g(i, j) = 1 |ri− rj| (I.9)
L’énergie Hartree-Fock est alors calculée par :
E = hΦe| ˆHe|Φei (I.10)
Cette équation permet d’exprimer l’énergie du calcul Hartree-Fock en fonction d’intégrales mono et biélectroniques : E = n X i=1 hi+ n X i=1 n X j>i (Jij − Kij) + VN N (I.11) avec : hi =hφi(1)|ˆh(i)|φi(1)i Jij =hφi(1)φj(2)|ˆg(1, 2)|φi(1)φj(2)i K =hφ (1)φ (2)|ˆg(1, 2)|φ(2)φ (1)i
Jij est l’intégrale de Coulomb (répulsions entre deux électrons chacun dans une orbitale) et Kij
est l’intégrale d’échange. Les équations de Hartree-Fock sont données par la relation suivante : ˆ
F φi = ✏ijφi (I.12)
Il est possible, à l’aide d’une transformation unitaire, de diagonaliser la matrice des multiplica-teurs de Lagrange ✏ij, permettant ainsi d’obtenir un nouvel ensemble d’orbitales moléculaires φ0i
(appelées orbitales canoniques) associées aux valeurs propres telles que : ˆ F φ0 i = ✏ 0 iφ 0 i (I.13)
Roothaan [77] a proposé de construire les orbitales moléculaires φi comme une combinaison linéaire
de fonctions de bases centrées sur des atomes χµ(LCAO - Linear Combination of Atomic Orbitals)
[76]. φ0 i = M X µ=1 cµiχµ (I.14)
Nous pouvons donc écrire les équations Hartree-Fock (équation I.13) de la façon suivante : ˆ F M X µ=1 cµiχµ= ✏ 0 i M X µ=1 cµiχµ (I.15)
Par intégration à un système matriciel nous obtenons les équation de Roothaan-Hall [77, 78].
(F)(C) = (S)(C)(✏) (I.16)
où les éléments de matrice sont définis par : Fµl =hχµ|(F )|χli
Sµl =hχµ|χli
L’opérateur Hartree-Fock ˆF est monoélectronique et dépend de ses propres fonctions propres, non connues initialement. Ainsi ces équations doivent être résolues par une procédure itérative appelée champ auto-cohérent (SCF - Self Consistent Field) [74]. Pour cela, il est nécessaire de
choisir au départ un jeu d’essai, de construire l’opérateur de Fock correspondant puis de résoudre le système d’équations. Cette résolution apporte un nouveau jeu de fonctions, lui même réinjecté dans le système d’équations etc... Lorsque la valeur de l’énergie est stationnaire (ceci dépend du seuil fixé entre deux itérations), l’auto-cohérence est atteinte. Un calcul Hartree-Fock consiste à considérer le déplacement des électrons dans le champ des noyaux et dans le champ moyen dû aux autres électrons. Cette approximation ne tient pas compte de la corrélation de mouvement entre les électrons. En effet, à cause de la répulsion coulombienne, il est très improbable que deux électrons se retrouvent très proches l’un de l’autre. C’est la raison pour laquelle la limite Hartree-Fock de l’énergie obtenue variationnellement reste supérieure à la valeur exacte non relativiste de l’énergie. La différence entre l’énergie Hartree-Fock obtenue à l’aide d’une base infinie et la valeur exacte s’appelle l’énergie de corrélation.
I.1.2
Théorie de la fonctionnelle de la densité
La fonction d’onde électronique d’une molécule composée de n électrons dépend de 3n coor-données d’espace et de n coorcoor-données de spin. Cet aspect a motivé d’importantes recherches afin de trouver des fonctions dépendant de moins de variables que la fonction d’onde mais permettant de calculer l’énergie. L’idée élémentaire de la fonctionnelle de la densité (DFT - Density Functio-nal Theory) est que l’énergie de l’état fondamental est une fonctionnelle de la densité électronique ⇢ [79], en d’autres termes, E0= E[⇢]. Cette théorie est basée sur le postulat proposé par Thomas et
Fermi dans lequel les propriétés électroniques peuvent être décrites en termes de fonctionnelles de la densité. Thomas et Fermi ont utilisé leur théorie pour la description d’atomes, mais le manque de précision ainsi que l’impossibilité de traiter des systèmes moléculaires en ont fait un modèle trop simpliste lorsqu’il a été proposé.
I.1.2.1 Théorèmes de Hohenberg et Kohn
C’est Hohenberg et Kohn qui, en 1964, démontrèrent que la densité électronique pouvait être utilisée pour obtenir la solution exacte à l’équation de Schrödinger dans le cadre de l’approximation de Born-Oppenheimer. D’après le premier théorème de Hohenberg et Kohn, n’importe quelle propriété du système peut être obtenue à partir de la densité électronique ⇢(r) de la particule
dans son état fondamental. L’énergie de ce système est donnée par la relation suivante : E[⇢] = FHK[⇢] +
Z
⇢(r)⌫ext(r)dr (I.17)
avec FHK[⇢], appelée fonctionnelle universelle, qui dépend de l’énergie cinétique du système Te[⇢]
et de l’énergie potentielle de l’interaction électron-électron Vee[⇢] (équation I.18)
FHK[⇢] = Te[⇢] + Vee[⇢] (I.18)
et ⌫ext(r), le potentiel externe défini par la relation suivante :
⌫ext(r) = − n X i=1 N X α=1 Zα |ri− Rα| (I.19) Dans un second théorème, Hohenberg et Kohn ont démontré que la densité électronique à l’état fondamental pouvait être calculée à l’aide d’un principe variationnel.
E[⇢] ≥ E[⇢0] (I.20)
Cependant, le théorème de Hohenberg et Kohn ne dit pas la manière de calculer l’énergie à partir de la densité électronique ou la manière de calculer cette dernière sans connaître au préalable la fonction d’onde i.
I.1.2.2 Approche Kohn-Sham
Kohn et Sham introduisirent un Hamiltonien de référence décrivant un système de n particules sans interaction mais ayant la même densité que le système réel. Ce dernier point permet de réduire le problème à la résolution de n équations monoélectroniques couplées, analogues aux équations de Hartree-Fock. L’opérateur de Kohn-Sham est alors défini par la relation suivante :
ˆ HKS[⇢] = − 1 2r 2+ ⌫ ext[⇢] + ⌫H[⇢] + ⌫xc[⇢] (I.21)
avec ⌫H[⇢] potentiel de Hartree :
⌫H[⇢] =
Z ⇢(r’)
et ⌫xc[⇢] potentiel d’échange et de corrélation
⌫xc[⇢] =
@Exc[⇢(r)]
@⇢(r) (I.23)
Nous pouvons dès lors introduire un jeu d’orbitales i(r), appelées orbitales de Kohn-Sham afin
de résoudre l’équation aux valeurs propres : ˆ
HKS i = ✏i i (I.24)
L’utilisation des orbitales de Kohn-Sham permet de minimiser l’énergie du système en résolvant le jeu d’équations de manière auto-cohérente, comme dans le cas de la méthode Hartree-Fock. De plus, la densité exacte peut être déterminée à partie des i(r) :
⇢ =
n
X
i=1
| i|2 (I.25)
Pour finir, l’énergie du système s’exprime par la relation suivante : EKS[⇢] = TKS[⇢] +
Z
⇢(r)⌫ext(r)dr + J[⇢] + Exc[⇢] (I.26)
avec TKS[⇢] l’énergie cinétique des électrons sans interaction, J[⇢] l’énergie d’intéraction
coulom-bienne entre les différents électrons (équation I.27) et Exc[⇢] l’énergie d’échange et de corrélation.
J[⇢] = 1 2
Z Z ⇢(r)⇢(r’)
|r − r’| drdr’ (I.27)
D’après les théorèmes de Hohenberg et Kohn, EKS[⇢] doit être égale à l’énergie du système établi
par la relation I.17. Ainsi, l’énergie d’échange et de corrélation peut être exprimée par :
Exc[⇢] = Te[⇢] − TKS[⇢] + Vee[⇢] − J[⇢] (I.28)
I.1.2.3 Les différentes classes de fonctionnelles
Formellement, la DFT est donc une méthode exacte. Malheureusement, la forme exacte de l’énergie d’échange et de corrélation reste inconnue, nous obligeant ainsi à faire des
approxima-tions. L’énergie d’échange et de corrélation Exc[⇢] est alors calculée à l’aide de fonctionnelles et
est généralement séparée en deux termes distincts, l’un d’échange Ex[⇢] et l’autre de corrélation
Ec[⇢]. Plusieurs fonctionnelles ont donc été développées pour traiter chacune de ces contributions,
de façon simultanée ou indépendante.
Dans la plus simple des approximations (LDA - Local Density Approximation), l’énergie d’échange et de corrélation est uniquement fonction de la densité électronique :
ExcLDA[⇢] =
Z
⇢(r)✏xc[⇢(r)]dr (I.29)
où ✏xc[⇢(r)] correspond à l’énergie d’échange et de corrélation par particule pour un gaz d’électrons
uniforme de densité ⇢. Le potentiel d’échange et de corrélation correspondant est donc défini par :
⌫xcLDA(r) = ✏xc[⇢(r)] + ⇢(r)
@✏xc[⇢(r)]
@⇢ (I.30)
Cette approximation reste cependant une approximation locale dans laquelle l’inhomogénéité de la densité électronique n’est pas prise en compte.
Dans le but d’améliorer cet aspect, l’ajout d’une correction par gradient de densité a été utilisée (GGA - Generalized Gradient Approximation) donnant aux fonctionnelles de type GGA une relation de la forme :
ExcGGA[⇢] = Ax
Z
⇢(r)4/3FGGA(s)dr (I.31)
où s est le gradient de la densité réduite avec kF = (3⇡2⇢(r))1/3 :
s = |r⇢(r)| 2kF⇢(r)
(I.32) Dans ce cas, s fait apparaitre un terme quasi local dépendant non seulement de la densité élec-tronique mais également de son gradient au voisinage de r.
fonc-tionnelles hybrides. Une fraction d’échange calculée de manière exacte est alors introduite dans une fonctionnelle d’échange de type GGA. Dans ce cas, l’expression de Exc[⇢] devient :
ExcHybride[⇢] = (1 − ↵)ExcGGA[⇢] + ↵ExcHF[⇢] (I.33) avec le coefficient (↵) qui donne le rapport entre Hartree-Fock et la DFT.
I.1.3
La méthode de DFT+U
La DFT conventionnelle donne des résultats incorrects pour des systèmes à fortes corrélations électroniques. Le problème est dû à l’utilisation de fonctionnelles d’échange et de corrélation qui conduisent à une mauvaise estimation de la corrélation électronique, comme par exemple dans le cas des oxydes de métaux de transition dont les couches d ou f sont incomplètes. Pour résoudre ce problème, nous pouvons utiliser des méthodes qui représentent avec précision les énergies d’auto-interaction comme la méthode de DFT+U. Cette méthode de DFT+U permet d’obtenir des résultats corrects à la fois pour l’énergie de cohésion, la largeur de bande interdite (gap) et le moment magnétique. Dans cette méthode, les interactions entre électrons dans des états localisés sur le même centre atomique sont traités de la même façon que dans la méthode Hartree-Fock, le reste étant traité en DFT. L’énergie calculée en DFT+U est donnée par la relation suivante :
EDF T +U[⇢, nIlmσ] = EDF T[⇢] + Eon−site[nIlmσ] − Edc[NIlσ] (I.34)
avec EDF T +U l’énergie totale du système, EDF T l’énergie DFT du système, Eon−site l’énergie de
Hartree-Fock provenant des interactions sur site entre électrons localisés et Edc terme de double
comptage qui corrige les contributions à l’énergie totale incluses à la fois dans EDF T et Eon−site.
L’énergie d’interaction sur site Eon−site dépend du nombre d’électrons qui occupent les orbitales
localisées φIlmσ centrées sur un atome I et caractérisées par un moment angulaire l, un nombre
quantique magnétique m et un spin ↵. Les nombres d’occupation nIlmσsont obtenus par projection
des orbitales DFT de Kohn-Sham pour le système total en un ensemble de ces orbitales localisées. La valeur NIlσ correspond au nombre total d’électrons pour un spin et un moment angulaire
donnés qui sont localisés sur I. En d’autres termes,
Afin de calculer EDF T +U, il est nécessaire de connaitre au préalable les expressions de Eon−site
et Edc. Plusieurs expressions ont été proposées dans la littérature, mais nous ne retiendrons que
l’approche invariante d’un point de vue rotationnel proposée par Dudarev. Cette méthode conduit à l’énergie : EDF T +U[⇢, nIlmσ] = EDF T[⇢] + X I,l,m,σ UIl− JIl 2 (nIlmσ− n 2 Ilmσ) (I.36)
avec EDF T[⇢] l’énergie DFT obtenue en utilisant la densité électronique totale et incluant les
interactions sur site qui sont incorrectes. Le second terme quant à lui, permet de corriger cet aspect. En effet, ce terme permet une description améliorée de la structure électronique ayant pour effet de déplacer les énergies des états localisés de leur valeur DFT, de sorte que la bande interdite soit agrandie. Le paramètre UIl (équation I.37) décrit le surcoût énergétique pour placer
un électron supplémentaire sur le site I et le paramètre J correspond à une énergie d’échange.
U = E(dn+1) + E(dn−1) − 2E(dn) (I.37)
La résolution de l’équation I.36 nécessite le choix des paramètres UIl et JIl. Ces valeurs ne sont pas
connues et peuvent être déduites à partir des calculs Hartree-Fock ou choisies de manière empirique de façon à obtenir la meilleure précision possible pour le calcul des différentes propriétés (paramètre de maille, gap, moment magnétique). Nous retiendrons cette méthode pour la suite car elle est plus simple à mettre en oeuvre et donne des résultats équivalents à la méthode ab initio.
I.2
Application à la modélisation des systèmes périodiques
I.2.1
Modèles périodiques
La structure de tout cristal peut être décrite par la répétition dans l’espace d’un motif ato-mique, appelé maille, cette dernière étant décrite par trois vecteurs fondamentaux a1, a2et a3. Les
coordonnées des atomes du motif par rapport à la maille sont indiquées en tant que coordonnées fractionnaires en fonction des vecteurs de base de la maille (module de base d’un solide).
avec 0 xi, yi, zi 1. Le choix de la maille est arbitraire, du moment qu’il puisse remplir l’espace
par translation. La maille avec le volume Va le plus petit est appelée maille primitive.
Va= |(a1⇥ a2) · a3| (I.39)
Une des formes spéciales de la maille primitive est la maille de Wigner-Seitz (WSZ - Wigner-Seitz Zelle), cette dernière correspondant à la région de l’espace localisée autour d’un noeud du réseau et à plus proche distance de ce dernier.
Dans le contexte de ce travail, les solides et les surfaces seront décrits à l’aide de modèles périodiques car ils conviennent particulièrement à la description des solides cristallins idéaux. Ces derniers sont des modèles infinis dont l’unité de base contient un nombre fini d’atomes ce qui implique que le potentiel extérieur agissant sur les électrons soit périodique. En d’autres termes,
V (r + T) = V (r) (I.40)
avec T un vecteur de translation du réseau. La fonction d’onde (r + T) est alors obtenue par application d’un opérateur de translation ˆT sur (r). Compte tenu de la périodicité du cristal, l’hamiltonnien ˆH est également périodique car l’opérateur d’énergie cinétique ˆT reste invariant par translation. Ainsi,
ˆ
T ˆH(r) (r) = ˆH(r + T) (r + T) = ˆH(r) ˆT (r) (I.41)
D’après le théorème de Bloch, les fonctions propres peuvent être choisies comme ayant la forme d’un produit d’une fonction d’onde par une fonction (r) ayant la périodicité du cristal.
(r + T) = ˆT (r) = exp(ik · T) (r) (I.42)
Dans cette expression, k = k1b1 + k2b2 + k3b3 est un vecteur du réseau réciproque, les ki sont
des entiers et les bi sont les vecteurs de base du réseau réciproque.
Dans le cas où le motif du modèle périodique est une supercellule, les conditions aux limites périodiques, appelées conditions de Born Von Karman, impliquent que les fonctions de Bloch
obéissent à la relation
(r + Njaj) = exp(iNjk · aj) (r) = (r) (I.43)
avec Ni le nombre de cellules répété dans la direction ai et kj = Nljj (lj, Nj 2 N et kj 2 R). A
cause des conditions aux limites, les vecteurs k sont réels. Le vecteur général κ à l’intérieur de la maille réciproque est défini avec ni 2 N < Ni comme :
κ= n1b1 N1 +n2b2 N2 +n3b3 N3 (I.44) Le nombre de points κ est le nombre de mailles dans le cristal soit N. Plus le cristal augmente en taille plus les points κ se rapprochent les uns des autres. Pour Ni ! 1, κ devient continu et
peut avoir toutes les valeurs possibles dans l’espace réciproque. Grâce aux conditions de Born Von Karman, nous pouvons préciser la forme des fonctions d’onde κ
n(r), développées en un ensemble
fini de fonctions de Bloch χκ µ(r) : κ n(r) = X µ cκµnχκµ(r) (I.45) avec cκ
µn des coefficients déterminés variationnellement en résolvant un ensemble d’équations
ma-tricielles couplées.
HκCκ = SκCκEκ (I.46)
avec Hκ la matrice hamiltonienne dans l’ensemble de base des fonctions κ
n(r), Cκ la matrice des
coefficients cκ
µn, Sκ la matrice de recouvrement et Eκ la matrice diagonale des valeurs propres
d’une seule particule ✏κ n.
I.2.2
Base à onde plane
Le choix d’une méthode pour traiter un système chimique s’accompagne également du choix de la base qui va être utilisée pour décrire celui-ci. Dans le cadre de ce travail, nous avons utilisé la base à onde plane pour le développement de κ
système périodique peuvent être exprimées par la relation suivante : χκKn(r) = 1 p N Va exp(ir(κ + Kn)) (I.47)
avec Kn un vecteur du réseau réciproque, N = N1N2N3 et Va le volume de la maille primitive.
Ainsi, d’après l’équation I.45, κ
n(r) est exprimée comme une combinaison linéaire d’ondes planes.
La taille de la base est en théorie infinie. En pratique, celle-ci est tronquée afin de rendre possible le calcul lors de la diagonalisation de l’hamiltonien. Un nombre raisonnable d’ondes planes est retenu selon le critère suivant :
(κ + Kn)2 Ecut (I.48)
avec Ecut le cutoff d’énergie cinétique. Plus ce dernier est grand, plus la représentation de la
fonction d’onde est précise. Cette précision est aussi recherchée pour la modélisation des éléments locaux au sein des fonctions d’onde. Pour cela un grand nombre d’ondes planes est utilisé et permet de définir les orbitales de coeur à basse énergie et celles subissant des oscillations rapides à cause de leur proximité du noyau. Mais, dans ce cas, il sera nécessaire d’avoir un ensemble de bases d’ondes planes plus grand que les ensembles de gaussiennes.
I.2.3
Pseudopotentiels
Le concept de pseudopotentiel (PP - PseudoPotential) fournit une solution élégante aux li-mites des ondes planes. En effet, l’intérêt des pseudopotentiels est de réduire la taille du calcul en remplaçant les orbitales de coeurs par un potentiel analytique. Dans un cristal, les électrons de coeur restent localisés près du noyau. La probabilité de trouver un électron de coeur loin du noyau est quasi-nulle. La méthode des pseudopotentiels considère que les électrons de coeur sont "gelés" près du noyau. En effet, l’essentiel des propriétés de la liaison chimique est contenu dans les orbitales de valence.
Le code de calcul VASP utilise trois types différents de pseudopotentiels, les pseudopotentiels à conservation de norme (NC NormConserving PP), les pseudopotentiels ultradoux (USPP UltraSofts PP ) et les combinaisons de pseudopotentiels et d’ondes augmentées linéarisées (PAW
-les pseudopotentiels PAW pour la représentation des électrons de valence.
I.3
Principe d’un calcul VASP
Les calculs ont majoritairement été réalisés à l’aide du logiciel VASP - Vienna Ab initio Simu-lation Package, ce programme étant tout particulièrement adapté aux systèmes périodiques. Ce dernier est basé sur la théorie de la fonctionnelle de la densité et les calculs sont réalisés sur la base d’ondes planes. Le logiciel VASP permet de résoudre les équations de Kohn-Sham de manière itérative et offre le choix de deux méthodes d’approximation de la fonctionnelle, l’approximation de la densité locale (LDA) et l’approximation du gradient généralisé (GGA). Parmi les fonction-nelles existantes, nous avons choisi pour l’étude des solides et des surfaces la fonctionnelle de Perdew-Wang (PW91) [80].
La résolution du système de Kohn-Sham s’effectue non pas dans l’espace réciproque tout en-tier mais sur une grille de point k appartenant à la zone de Brillouin. Il est possible de limiter le calcul de la fonction d’onde électronique à un nombre fini de points k de la zone de Brillouin, en s’assurant de la convergence de l’énergie totale. Cet ensemble de points k peut s’obtenir par diverses méthodes comme celle implémentée dans le code VASP, la méthode de Monkhorst et Pack. Pour les semi-conducteurs et isolants, un nombre réduit de point k suffit, par contre, pour les métaux, une description plus fine de la région proche de la surface de Fermi nécessite d’ac-croître le nombre de points k. Des améliorations ont été implémentées dans VASP pour traiter des systèmes métalliques avec un nombre réduit de points k, telles que l’occupation partielle des niveaux proches de Fermi, comme cela a été proposé dans la méthode de Methfessel et Paxton [81].
En résumé, la résolution du système Kohn-Sham se fait de manière itérative et peut être représentée par la figure I.1. La nouvelle densité ⇢0
inest calculée à chaque étape par une combinaison
linéaire des densités précédemment calculées : ⇢0
in=
X
i
Figure I.1 : Cycle autocohérent sur un calcul de convergence VASP
I.4
Prise en compte des forces de dispersion
Même si les études de type DFT sont utiles pour prédire les propriétés structurales et électro-niques des systèmes caractérisés par de fortes liaisons chimiques, la DFT pure ne tient pas compte des forces de dispersion. Dans ce travail, nous avons donc choisi d’utiliser la méthode proposée par Grimme et al [73] afin de prendre en compte ces forces de dispersion. Nous ne développons pas la méthode en détail dans cette partie, mais essayons de donner un aperçu des équations utilisées pour le calcul des forces de dispersion. L’énergie du système est alors définie par :
EDF T −D = EKS + EDisp (I.50)
avec EKS l’énergie de Kohn-Sham obtenue par un calcul de type DFT et EDisp la correction de la
dispersion.
EDisp = E(2)+ E(3) (I.51)
avec le terme à deux corps donné par : E(2) =X AB X n=6,8,10... sn CAB n rn AB fd,n(rAB) (I.52)
Dans cette équation la première somme porte sur toutes les paires d’atomes du système, CAB n
indique la moyenne au ne ordre du coefficient de dispersion pour la paire d’atome AB et r
AB leur
distance internucléaire. Le facteur de graduation sn (qui dépend du choix de la fonctionnelle) est
ajusté uniquement pour n > 6 afin de s’assurer de l’exactitude assymptotique. Afin d’éviter des étrangetés pour les courtes distances rAB et les effets de double comptage de la corrélation à des
distances intermédiaires, des fonctions d’amortissements fd,n(rAB) sont utilisées pour déterminer
la gamme de correction de la dispersion. fd,n(rAB) =
1
1 + 6(rAB/(sr,nRAB0 ))
−αn (I.53)
avec sr,n le facteur de graduation des rayons de cutoff RAB0 . L’interaction à longue portée entre
trois atomes n’est pas exactement égale à l’énergie d’interaction calculée entre paires. Pour cela, un terme à trois corps a été utilisé.
E(3) = X
ABC
fd,3(¯rABC)EABC (I.54)
avec une somme sur tous les triplets ABC du système, l’équation I.53 avec ↵ = 16, sr = 0,75 et
la moyenne des rayons ¯rABC est utilisée comme fonction de graduation.
EABC = C
ABC
9 (3cos✓acos✓bcos✓c+ 1)
(rABrBCrCA)3
(I.55) avec ✓a, ✓b et ✓c les angles internes du triangle formé par rAB, rBC, et rCA.
Dans le cadre de ce travail, deux stratégies ont été adoptées : un calcul a posteriori des forces de van der Waals, incluant les coefficients C6 et C8 (appelé D3), puis, lorsque cela a été disponible, un calcul incluant les forces de van der Waals pendant l’optimisation de géométrie, cette fois ci avec la correction C6 uniquement (D2). Nous ne mentionnons pas dans le texte quelle technique a été adoptée, car les résultats sont très proches. Dans tous les cas où les forces de van der Waals pouvaient jouer un rôle sur la géométrie pendant l’optimisation, les calculs ont été refaits en les incluant (forts taux de recouvrement).
I.5
Dynamiques moléculaires
La dynamique moléculaire consiste à étudier la trajectoire d’une molécule en appliquant les lois de la mécanique classique newtonienne c’est à dire à simuler les mouvements atomiques au cours du temps. Ces mouvements correspondent à des vibrations autour d’un minimum d’énergie, ainsi la dynamique moléculaire permet de s’extraire d’un minimum local. Nous pouvons utiliser la dynamique moléculaire pour étudier le comportement dynamique d’une molécule ou bien faire de la recherche conformationnelle. Dans une simulation de dynamique moléculaire standard, l’énergie totale E du système moléculaire est une constante. En général nous gardons également le nombre d’atomes N et le volume V de la boîte de simulation fixe. Ces simulations se déroulent alors dans l’ensemble microcanonique (N,V,E). Souvent il est préférable de maintenir la température constante plutôt que l’énergie, il s’agit alors de simulations dans l’ensemble canonique (N,V,T). Dans d’autres cas il peut être avantageux de simuler à pression constante plutôt qu’à volume constant, ce sont des simulations dans l’ensemble isobare-isotherme (N,P,T).
I.5.1
Dynamique moléculaire classique
Dans les simulations classiques, l’hamiltonien du système moléculaire prend la forme :
H(p, r, m, s) = T (p, m) + V (r, s) (I.56)
avec T le terme d’énergie cinétique qui dépend uniquement des masses atomiques m et définit par
T (p, m) = N X i=1 p2 i 2mi = N X i=1 1 2miv 2 i (I.57)
et V le terme d’énergie potentielle permettant de décrire l’énergie d’interaction en fonction des coordonnées r des particules et des paramètres du champ de force s.
Les simulations de dynamique moléculaire classique consistent à résoudre les équations du mouvement de Newton d’un ensemble d’atomes. Chaque atome de la molécule est considéré comme une masse ponctuelle dont le mouvement est déterminé par l’ensemble des forces exercées sur lui par les autres atomes en fonction du temps. La force fi qui agit sur une particule i est donnée par
l’équation : fi = −
@ @ri
V (r1, r2, ..., rN) (I.58)
Au temps t, chaque atome i, de coordonnée ri et de masse mi subira une accélération sous l’action
d’une force fi telle que :
fi(t) = mi dvi(t) dt (I.59) et vi(t) = dri(t) dt (I.60)
avec vi, les vitesses atomiques.
Dans l’hamiltonien du système moléculaire, l’énergie cinétique est entièrement décrite par l’équation I.57 et l’énergie potentielle par des paramètres du champ de force utilisé.
I.5.1.1 Forme générale du champ de force
De manière générale, l’énergie potentielle dans les champs de force prend la forme suivante :
V (r, s) = Vbon(r, s) + Vnonb(r, s) (I.61)
où Vbon(r, s) correspond aux interactions entre atomes liés par des liaisons covalentes et Vnonb(r,
s) aux interactions dites "non-liées" (van der Waals et électrostatique).
Vbon(r, s) = Vbond(r, s) + Vangle(r, s) + Vdihed(r, s) + Vimprop(r, s) (I.62) Vbon(r, s) est défini par quatre termes qui permettent de décrire l’élongation des liaisons, la
déformation des angles et les torsions pour les angles dièdres périodiques et "impropres".
Vbond(r, s) = Nb X n=1 Kbharmn (bn− b0n) 2 (I.63)
avec bn la longueur instantanée de la liaison n, b0n la valeur à l’équilibre et K harm bn la constante de force associée. Vangle(r, s) = Nθ X n=1 Kθharmn (✓n− ✓0n) 2 (I.64)
avec ✓n l’angle instantané et ✓0n la valeur à l’équilibre.
Vimprop/dihed(r, s) = Nϕ X n=1 pn X m=1 Kϕn,m[1 + cos(mn'n− γn)] (I.65)
avec l’angle dièdre 'n, les angles de phase des dièdres γn, les termes de la série de Fourier de 1 à
pn et les constantes de forces associées Kϕn,m.
Les interactions entre atomes non-liés sont décrites par deux termes, VLJ pour les interactions
de type Lennard-Jones et VC pour les interactions électrostatiques.
VLJ = ✏ij ✓R⇤ ij rij ◆12 − 2✏ij ✓R⇤ ij rij ◆6 (I.66) Le premier terme en r−12
ij correspond à la répulsion entre deux atomes due à l’exclusion de Pauli
et le deuxième terme en r−6
ij représente la dispersion attractive de London.
VC = qiqj 4⇡✏0✏1 ⇥
1 rij
(I.67) I.5.1.2 Principe de la dynamique moléculaire
La mécanique moléculaire nous permet de minimiser l’énergie calculée à partir de l’équation I.61 afin d’obtenir des configurations à basse énergie. Dans le cas d’une minimisation, nous nous déplaçons presque exclusivement vers le bas de la surface d’énergie potentielle (SEP), et le mi-nimum atteint est en général le mimi-nimum local le plus proche de la configuration initiale. Dans ce cas il n’est pas possible de surmonter des barrières énergétiques et la dynamique moléculaire est un moyen plus efficace de recherche de configurations de basses énergies. Les simulations de dynamique moléculaire sont en général utilisées pour étudier la structure, la dynamique et certains aspects thermodynamiques de systèmes moléculaires à l’équilibre et hors équilibre. Les équations du mouvement sont alors intégrées dans le temps ce qui permet d’obtenir la trajectoire de chaque
base est donnée par la relation I.68 : ri(tn+ ∆t) = 2ri(tn) − ri(tn− ∆t) +
fi(tn)
mi
∆t2 (I.68)
I.5.2
Dynamique moléculaire ab initio
En dynamique moléculaire ab initio, il faut prendre en compte le mouvement des noyaux en utilisant la mécanique classique et utiliser l’état électronique du système pour chaque configu-ration. Dans le code VASP, nous réalisons des dynamiques de type Born-Oppenheimer où l’état électronique fondamental est calculé d’une manière exacte à chaque fois que les positions ioniques R(t) sont déterminées. Les forces sont calculées après la détermination de l’état électronique fon-damental en utilisant le théorème de Hellmann-Feynman :
Fi(Ri) = −h'0 " " " " @H @Ri " " " " '0i (I.69)
A partir des forces interatomiques, les positions atomiques sont déterminées à l’instant t + ∆t par le biais de l’algorithme de Verlet qui exige une détermination précise des forces interatomiques à chaque pas de temps t.
Dans le cadre de ce travail, nous avons cherché à explorer la surface d’énergie potentielle (SEP) de différentes molécules afin de trouver la conformation de plus basse énergie et avons donc réalisé un recuit simulé. Ce recuit simulé consiste en une dynamique moléculaire qui se décompose en une montée lente en "température" et en un palier (NVT). Nous maintenons alors le système à cette "température" optimale (500K) pendant 10 ps. Compte tenu des petites tailles des systèmes étudiés (entre dix et vingt atomes), un temps de palier de 10 ps nous a paru suffisant pour explorer la surface de potentiel. Au cours de ce palier, le système balaie un grand nombre de configurations, et donc une large gamme d’énergie. Nous sélectionnons alors les configurations ayant les énergies les plus basses puis nous abaissons lentement l’énergie cinétique (c’est à dire la "température"). Une fois que le système a atteint une "température" de 10K, nous procédons à une optimisation de géométrie.
![Figure 6 : Etude de l’adsorption de la β-Lactoglobuline sur une surface d’Au chargée positivement [72]](https://thumb-eu.123doks.com/thumbv2/123doknet/2895219.74141/18.893.227.663.826.1143/figure-etude-adsorption-β-lactoglobuline-surface-chargée-positivement.webp)