UNIVERSITÉ DE NEUCHÂTEL FACULTÉ DES SCIENCES
Etude sur la réactivité du THP
(tris(hydroxyméthyl)phosphine)
et
Développement de composés
potentiellement efficaces en tant que
retardants de flamme
P
HO
OH
OH
Thèse soutenue à la Faculté des Sciences de l’Université de Neuchâtel le 19 février 2014 pour l’obtention du titre de Docteur ès Sciences
par
Christelle SCHENK
Chimiste diplômée de l’Université de Neuchâtel
Membres du jury :
Prof. Reinhard Neier, directeur de thèse Prof. Robert Deschenaux, rapporteur
Dr. Olivier Vallat, rapporteur Dr. Jean-Jacques Flat, rapporteur
III Ce travail a été effectué de septembre 2008 à octobre 2013 à l’Institut de Chimie de l’Université de Neuchâtel sous la direction du Prof. Reinhard Neier. J’aimerais lui adresser mes sincères remerciements pour m’avoir accueilli au sein de son laboratoire durant mes années de thèse. Il m’a non seulement permis d’approfondir mes connaissances scientifiques, mais m’a également apporté beaucoup sur le plan humain.
Mes remerciements s’adressent ensuite à Febex SA et au Dr. Olivier Vallat sans qui ce projet n’aurais jamais vu le jour et qui a été un précieux mentor pour mes débuts dans le monde industriel. Je remercie particulièrement Olivier pour son accueil, les connaissances scientifiques inestimables qu’il m’a transmises, sa générosité, son sens de l’humour et son goût pour la bonne cuisine qui a rendu ce projet particulièrement agréable !
Un grand merci au CERDATO (Arkema) et au Dr. Jean-Jacques Flat qui ont rendu possibles les tests de tenue au feu dans les polymères et pour leur accueil à Serquigny lors de notre séjour riche en informations.
Je tiens également à remercier le Dr. Olivier Vallat, le Dr. Jean-Jacques Flat et le Prof. Robert Deschenaux qui ont chaleureusement participé à mon jury de thèse et qui ont pris le temps de lire et de corriger mon manuscrit.
Ce travail a été soutenu financièrement par la Commission pour la Technologie et l’Innovation (CTI), à qui j’aimerais exprimer toute ma gratitude.
Je remercie les différents soutiens techniques sans qui une thèse ne serait pas possible : Les Dr. Claudio Dalvit et Julien Furrer pour les analyses RMN, la Dr. Armelle Vallat pour les analyses MS, le groupe du Prof. Deschenaux pour m’avoir permis de réaliser les ATG dans leur laboratoire, nos concierges fortement appréciés et notre irremplaçable Mme Tissot.
Un grand merci à mes apprenties : Elodie Allenbach (3ère année), Marion Gogniat (1ère année) que je remercie particulièrement pour sa perpétuelle bonne humeur et nos nombreux fous rires, et Aurélie Salgat (1ère année) et ses mains en or ! Je n’oublie pas de remercier également Lionel Burri pour ses bonnes idées et son humour.
Je remercie évidemment tous mes collègues qui ont rendu ces 5 ans fort agréables, lors des pauses, soirées foot, sorties à ski, séjour à Nottingham, semaines à Villars, … Un grand merci donc à mon groupe de recherche : Christian, Damien, Inga, Sara, Guillaume, William, Jihane (plein succès pour la suite du projet!), Anca, Christophe, Ana-Maria, Maria, Frédéric, Andrea, Björn, Csilla, Maude, Loïc, mais également à Julien, Luyen, Yo, Sébastiano, Sylvain, Anne-Flore, Michael, Thibaud, Thomas, David, Raja, Fifi, Jean-Luc, Lucienne, Marie-Eve, André et Mohamed.
Un remerciement tout spécial à Armelle, pour son amitié, sa générosité, son accueil des plus chaleureux, sa bonne humeur contagieuse, ses précieux conseils mais aussi ses bons petits plats ! Merci à mes amis de toujours : Joël, Carop, Cunhette et Jo ; ainsi qu’à tous les autres potes qui se reconnaîtront. Les moments de détente passés avec vous tous ont été des bienfaits inestimables pendant ma thèse.
IV
Un grand merci à toute ma famille, grands-parents, oncles, Thierry, ma marraine Huguette et Eric, Line, Daniel, et particulièrement à mon frère Marc et ma belle-sœur Christel qui ont illuminé la famille avec leurs deux rayons de soleil Malo et Lucas. C’est grâce à vous tous que je suis devenue celle que je suis !
Finalement, j’aimerais remercier du fond du cœur mes parents qui ont toujours été un exemple de courage et de détermination. Merci de m’avoir suggéré ce beau parcours universitaire, sans jamais m’obliger à faire des choix que je ne voulais pas. Cela n’a pas toujours été évident, mais je suis fière de pouvoir vous montrer le résultat. Ce travail est le vôtre…
Je termine par mon compagnon Vincent : aucun mot ne peut exprimer le bien que tu m’apportes au quotidien. J’aimerais donc simplement te remercier du fond du cœur pour ta patience, ton soutien et ton amour.
V
Abréviations
ABS Acrylonitrile butadiène styrène
ACES Acide N-(2-Acétamido)-2-aminoéthanesulfonique ADN Acide désoxyribonucléique
AG Aktien Gesellschaft (société par action) APO Oxyde de tris(aziridinyl)phosphine APP Polyphosphates d’ammonium
aq. Aqueux
ATG Analyse thermogravimétrique ATP Adénosine triphosphate AUT Autriche BDP Bisphénol-A-bis(diphénylphosphate) BEL Belgique Ca Calcium CD3OD Méthanol deutéré CH Suisse Co. Company conc. Concentré DDT Dichlorodiphényltrichloroéthane DE Allemagne DMMP Diméthylméthylphosphonate DMPP Diméthylpropylphosphonate DMSO Diméthylsulfoxyde DOPO 9,10-dihydro-9-oxa-10-phosphaphenantrene-10-oxide eau Eau désionisée
éq. Equivalent
ESI Ionisation par électro-spray EtOAc Acétate d’éthyle
EtOH Ethanol Fig. Figure
FR France
g, mg, μg, kg Gramme, milligramme, microgramme, kilogramme
h heure HCl Acide chlorhydrique HMP (hydroxyméthyl)phosphines Hz Hertz IR Infrarouge ISR Israël IT Italie J.-C. Jésus-Christ JPN Japon K Kelvin Ka Constante d’acidité Kb Constante de basicité kJ Kilojoule
l, ml, μl Litre, millilitre, microlitre LOI Limited Oxygen Index
Ltd Limited
MDPA N-méthyloldiméthylphosphonopropionamide MeOD Méthanol deutéré
MeOH Méthanol
Mg Magnésium
MHz Mégahertz
min Minute
mm, cm Millimètre, centimètre mol, mmol Mole, millimole MP Phosphate de mélamine MPP Polyphosphates de mélamine MS Spectroscopie de masse
n° Numéro
NaOH Hydroxyde de sodium N.B. Nota bene
VI
NL Pays-Bas
nm Nanomètre
31P-RMN Analyse du phosphore par résonance magnétique nucléaire P4 Phosphore blanc
PA Polyamide
p.a. pour analyse
PBT Polytéréphtalate de butylène pEb Point d’ébullition
PET Polytéréphtalate d'éthylène pF Point de fusion
pH Potentiel hydrogène pKa = - log Ka
ppm Partie par million PPO Polyoxyde de phénylène PVC Polychlorure de vinyle
RDP Résorcinol-bis(diphénylphosphate) RF Retardant de flamme
RMN Résonance magnétique nucléaire
s Seconde
SA Société anonyme t1/2 Temps de demie-vie
T Température
t.a. Température ambiante t/a Tonnes par année
taurine acide 2-aminoethanesulfonique TCP Tricrésylphosphate
TCPP Tris(2-chloroisopropyl)phosphate THP Tris(hydroxyméthyl)phosphine
THPC Chlorure de tétrakis(hydroxyméthyl)phosphonium THPO Oxyde de tris(hydroxyméthyl)phosphine
THPOH Hydroxyde de tétrakis(hydroxyméthyl)phosphonium THPS Sulfate de tétrakis(hydroxyméthyl)phosphonium THPX Sels de tétrakis(hydroxyméthyl)phosphonium TIPP Tris(isopropylphényl)phosphate TMM Triméthylolmélamine TMS Tétraméthylsilane TPP Triphénylphosphate UK Royaume-Uni
UL-94 Underwriters Laboratories-94 USA Etats-Unis d’Amérique UV Ultraviolet
δ Déplacement chimique °C Degré Celsius
° Degré d’un angle
® Marque déposée H Enthalpie de réaction € Euro % Pourcent < Inférieur à > Supérieur à
VII
Résumé
Depuis les années 70, les matériaux traditionnels, tels que le bois et les métaux, ont été progressivement remplacés par des matériaux polymères. Malgré tous les avantages que possèdent ces plastiques, ces derniers peuvent être particulièrement inflammables et peuvent dégager des composés hautement toxiques sous l’effet de la combustion. Ainsi, des additifs appelés « retardants de flamme » ont été développés pour améliorer la sécurité de notre quotidien face aux incendies. Un retardant de flamme est donc une substance que l’on ajoute aux matériaux au cours de leur processus de fabrication afin d’abaisser l’inflammabilité de ces derniers ou de ralentir leur combustion. Les premiers développements d’additifs retardants de flamme ont clairement démontré l’efficacité ignifugeante des composés halogénés, cependant leur toxicité avérée a accru l’intérêt pour des composés contenant du phosphore, de l’azote et/ou des charges minérales.
Le présent projet est donc né de la collaboration avec Febex SA, compagnie spécialisée dans la production de dérivés du phosphore de haute technicité. L’idée originale est de recycler un produit secondaire phosphoré, la phosphine PH3, provenant des lignes de production de Febex SA afin de le valoriser en tant que matière première. Cependant, la toxicité et l’inflammabilité de cette phosphine rend la manipulation de ce gaz relativement difficile. C’est pourquoi, elle est d’abord transformée en THP (tris(hydroxyméthyl)phosphine), molécules nucléophile permettant de développer une chimie variée. Le principal défi de ce projet est donc de transformer un produit secondaire toxique pour obtenir un composé non-toxique ayant un intérêt commercial en tant qu’additif retardant de flamme.
Le potentiel réactif du THP a donc été exploré, essentiellement par les réactions d’additions de Michael avec des oléfines activées et de condensations de Mannich avec des sources d’azote, révélant une bonne réactivité du centre phosphoré. Une nouvelle méthode de formation de sels de calcium, de magnésium et d’aluminium a également été développée à partir de dérivés du THP. Une bibliothèque de composés contentant des phosphines, des oxydes de phosphines, des sulfures de phosphines, des phosphoniums et des sels métalliques de dérivés phosphorés a ainsi été synthétisée en respectant certaines contraintes liées à l’approche industrielle. Une sélection de composés a ensuite été effectuée selon des critères imposés par l’application en tant qu’additif retardant de flamme, tels que l’aspect du produit, la stabilité thermique et l’absence d’halogénures. Les composés sélectionnés ont finalement subi les tests de tenues au feu en les intégrant dans des matrices polymères à des taux de charge inférieur à 30%.
Mots-clés : Tris(hydroxyméthyl)phosphine, retardant de flamme sans halogénures, dérivés
IX
Abstract
Since the 70s, traditional materials such as wood and metals were gradually replaced by polymer materials. In spite of all advantages offered by these plastics, they can be particularly flammable and can release highly toxic compounds under the influence of combustion. It’s why additives called "flame retardants" were developed to improve safety of our everyday life in front of fires. A flame retardant is thus a substance we add to materials during their manufacturing process to lower their flammability or slow down their combustion. The first developments of fireproofing additives clearly demonstrated the efficiency of the halogenated compounds, however their well-known toxicity increased the interest for phosphorous, nitrogen or mineral derivatives.
This project arose from the collaboration with Febex SA, company specialized in production of high technicality phosphorous derivatives. The original idea is to recycle a phosphorous by-product (phosphine PH3) resulting from Febex production lines to value it as raw material. However, toxicity and flammability of this phosphine makes handling of this gas difficult. That’s why it is first transformed into THP (tris(hydroxymethyl)phosphine), nucleophilic molecule allowing to develop a varied chemistry. The main challenge of this project is to transform a toxic by-product to obtain a non-toxic compound having a commercial interest as fireproofing additive.
The reactivity of THP was first explored, essentially by Michael additions with activated olefins and Mannich condensations with nitrogen containing reagents, revealing a good reactivity of the phosphorous center. A new method to form calcium, magnesium and aluminum salts from THP derivatives was also developed. A library of compounds like phosphines, phosphine-oxides, phosphine-sulfides, phosphoniums and metallic salts of phosphorous derivatives was synthesized respecting some constraints related to the industrial approach. A selection of compounds was then made according to criteria such as aspect of products, thermal stability and halogen-free composition that are relevant for flame retardant application. Selected compounds were finally subjected to flammability standard tests by integrating them into polymer matrices at rates of load lower than 30%.
Key-words : Tris(hydroxymethyl)phosphine, halogen-free flame retardant, phosphorous derivatives,
XI
Table des matières
1. PARTIE THÉORIQUE ... 1
1.1 INTRODUCTION ... 1
1.1.1 Collaboration Febex SA – Université de Neuchâtel ... 1
1.1.2 Le phosphore : un élément crucial ... 2
1.1.2.1 Historique ... 3
1.1.2.2 Propriétés et réactivité du phosphore... 4
1.1.2.3 Provenance naturelle ... 6
1.1.3 Importance industrielle du phosphore ... 7
1.1.3.1 Phosphore élémentaire ... 7
1.1.3.2 Engrais ... 8
1.1.3.3 Détergents ... 9
1.1.3.4 Acide phosphorique et phosphates ... 9
1.1.3.5 Organophosphorés ... 10
1.1.4 Retardants de flamme ... 11
1.1.4.1 Définition et histoire ... 11
1.1.4.2 Mécanismes d’action ... 12
1.1.4.3 Les différentes classes de RF ... 15
1.1.4.4 L’Exolit OP® : RF de 3e génération ... 22
1.1.4.5 Coûts et quantités de RF incorporé dans une matrice ... 23
1.1.4.6 Critères de sélection d’un RF ... 23
1.1.4.7 Tests normés ... 24
1.1.4.8 Commercialisation des RF ... 26
1.2 BUT DU TRAVAIL ... 27
1.3 LITTÉRATURE EXISTANTE ... 29
1.3.1 Synthèse, réactivité et utilisation du THP ... 29
1.3.1.1 Synthèse ... 29
1.3.1.2 Réactivité ... 32
1.3.1.3 Utilisation ... 44
1.3.2 Retardants de flamme phosphorés ... 48
1.3.2.1 Phosphore à l’état d’oxydation -I ... 50
1.3.2.2 Phosphore à l’état d’oxydation 0 ... 53
1.3.2.3 Phosphore à l’état d’oxydation +I... 53
1.3.2.4 Phosphore à l’état d’oxydation +III... 55
1.3.2.5 Phosphore à l’état d’oxydation +V ... 57
1.3.2.6 Synergistes - Systèmes intumescents ... 61
1.4 RÉSULTATS ET DISCUSSION ... 63
1.4.1 Tris(hydroxyméthyl)phosphine (THP) ... 63
1.4.1.1 Considérations préliminaires ... 63
1.4.1.2 Caractéristiques du THP produit par Febex SA ... 63
1.4.1.3 Purification du THP ... 65
1.4.2 Synthèses ... 69
1.4.2.1 Considérations préliminaires ... 69
1.4.2.2 Additions de Michael ... 70
1.4.2.3 Condensations de Mannich ... 86
1.4.2.4 Synthèse à partir de dérivés du THP ... 88
1.4.2.5 Formation de sels métalliques des dérivés phosphorés ... 90
1.4.2.6 Résultats infructueux ... 97
1.4.2.7 Conclusion sur le potentiel réactif du THP ... 102
XII
1.4.3.1 Aspect des composés purs obtenus ... 103
1.4.3.2 Considérations toxicologiques et environnementales ... 105
1.4.3.3 Taux de phosphore et d’azote des composés ... 105
1.4.3.4 Stabilité thermique des composés ... 106
1.4.3.5 Miscibilité du composé avec la matrice ... 117
1.4.4 Tests de tenue au feu ... 117
1.4.4.1 Evaluation l’oxyde de tris(2-cyanoéthyl)phosphine [2] ... 118
1.4.4.2 Evaluation du sulfure de tris(2-cyanoéthyl)phosphine [3] ... 119
1.4.4.3 Evaluation de l’oxyde de 5-(hydroxyméthyl)-1,3-diphényl-1,3,5-diazaphosphinane [12] ... 120
1.5 CONCLUSION ET PERSPECTIVES ... 121
1.5.1 Conclusions ... 121
1.5.1.1 Conclusion sur les synthèses effectuées ... 121
1.5.1.2 Conclusion sur la stabilité thermique des composés... 122
1.5.1.3 Conclusion sur les tests au feu ... 122
1.5.2 Perspectives ... 123
2. PARTIE EXPÉRIMENTALE ... 125
2.1 MÉTHODES ANALYTIQUES ET APPAREILS ... 125
2.1.1 Spectroscopie infrarouge (FT-IR) ... 125
2.1.2 Spectroscopie par résonance magnétique nucléaire (RMN) ... 125
2.1.3 Spectroscopie de masse (MS) ... 125
2.1.4 Analyse thermogravimétrique (ATG) ... 125
2.1.5 Point de fusion (pF)... 126
2.2 RÉACTIFS, GAZ ET SOLVANTS ... 126
2.2.2 Gaz ... 127
2.2.3 Solvants ... 127
2.3 DÉTERMINATION DU TITRE DU THP ... 128
2.4 MÉTHODES POUR LA PURIFICATION DU THP ... 128
2.4.1 Préparation des colonnes de résine ... 128
2.4.2 Régénération des colonnes de résine ... 128
2.4.3 Préparation du charbon actif ... 128
2.4.4 Purifications... 128
2.5 SYNTHÈSES ... 129
2.5.1 Considérations préliminaires ... 129
2.5.2 Additions de Michael ... 129
2.5.2.1 Synthèse du tris(2-cyanoéthyl)phosphine [1]... 129
2.5.2.2 Synthèse de l’oxyde de tris(2-cyanoéthyl)phosphine [2] ... 130
2.5.2.3 Synthèse du sulfure de tris(2-cyanoéthyl)phosphine [3]... 131
2.5.2.4 Synthèse de l’hydroxyde de tétrakis(3-(tbu)-3-propylamide)phosphonium [4] ... 132
2.5.2.5 Synthèse du chlorure de (3-(tbu)-3-propylamide)tris(hydroxyméthyl)phosphonium [5] ... 133
2.5.2.6 Synthèse du méthanesulfonate de (3-(tbu)-3-propylamide)tris(hydroxyméthyl)-phosphonium [6] ... 134
2.5.2.7 Synthèse de l’oxyde de N-(tbu)-3-propylamide)-3-bis(hydroxyméthyl)phosphine [7] ... 135
2.5.2.8 Synthèse du propanoate de 3-(tris(hydroxyméthyl)phosphonium) [8] ... 136
2.5.2.9 Synthèse de l’acide 3-(bis(hydroxyméthyl)phosphoryl)propanoique [9] ... 137
2.5.3 Condensations de Mannich ... 138
2.5.3.1 Synthèse du 1,3,5-triphényl-1,3,5-triazinane [10] ... 138
2.5.3.2 Synthèse du 1,3-diphényl-1,3,5-diazaphosphinan-5-yl)méthanol [11] ... 139
2.5.3.3 Synthèse de l’oxyde de 5-(hydroxyméthyl)-1,3-diphényl-1,3,5-diazaphosphinane [12] ... 140
2.5.3.4 Synthèse du sulfure de 5-(hydroxyméthyl)-1,3-diphényl-1,3,5-diazaphosphinane [13] ... 142
2.5.4 Synthèses à partir de dérivés du THP ... 143
2.5.4.1 Synthèse du chlorure de tris(2-carboxyéthyl)phosphonium [14] ... 143
XIII
2.5.4.3 Synthèse de l’oxyde de tris(2-amidoéthyl)phosphine [16] ... 145
2.5.5 Formations de sels métalliques de dérivés phosphorés... 146
2.5.5.1 Synthèse du sel de calcium de 3-(bis(hydroxyméthyl)phosphoryl)propanoate [17] ... 146
2.5.5.2 Synthèse du sel de calcium de dichlorure de bis(3,3’,3’’-phosphoniotriyltripropionate) [18] ... 147
2.5.5.3 Synthèse du sel de calcium de 3,3’,3’’-(oxo-phosphanetriyl)tripropionate [19] ... 148
2.5.5.4 Synthèse du sel de magnésium de 3-(bis(hydroxyméthyl)phosphoryl)propanoate [20] ... 148
2.5.5.5 Synthèse du sel de magnésium de 3,3’,3’’-(oxo-phosphanetriyl)tripropionate [21] ... 149
2.5.5.6 Synthèse du sel d’aluminium de 3-(bis(hydroxyméthyl)phosphoryl)propanoate [22] ... 150
1
1. Partie théorique
1.1 Introduction
1.1.1 Collaboration Febex SA – Université de Neuchâtel
Le présent projet est né en septembre 2008, grâce à la collaboration de Febex SA avec l’Université de Neuchâtel. Febex SA est une compagnie spécialisée dans la production de dérivés phosphorés tournée essentiellement sur la production de produits de haute qualité. L’entreprise, située à Bex en Suisse, a été fondée en 1917. A cette époque, Febex SA était une Fonte Electrique spécialisée dans la métallurgie, puis elle s’est tournée vers la chimie du phosphore dans les années 70 pour devenir rapidement un des leaders dans le domaine des dérivés phosphorés de haute technicité. La société emploie actuellement une cinquantaine de collaborateurs, et elle est depuis 1976 une filiale du groupe français CECA, lui-même filiale du groupe Arkema (chimie de spécialités et des matériaux de performance).
Sa production se concentre sur 5 types de produits : l’acide phosphorique (différentes qualités, 10'000 t/a), l’acide pyro/polyphosphorique (différentes concentrations), l’hypophosphite de sodium (cristaux ou solutions, 3'600 t/a à Bex et 4'000 t/a à Shanghai), l’acide hypophosphoreux et des sels de phosphonium (sulfate de tétrakis(hydroxyméthyl)phosphonium (THPS), chlorure de tétrakis-(hydroxyméthyl)phosphonium (THPC)). La gamme des produits proposés par Febex SA est vendue principalement à l’export, en Europe (leader pour l’acide phosphorique électronique et l’hypophosphite de sodium), en Asie et aux Amériques. Le phosphore blanc (P4) est la matière première commune à tous ces produits. La voie de synthèse thermique, par combustion du P4, permet d’obtenir un acide phosphorique de très haute pureté, utilisé dans l’industrie électronique et l’industrie pharmaceutique.
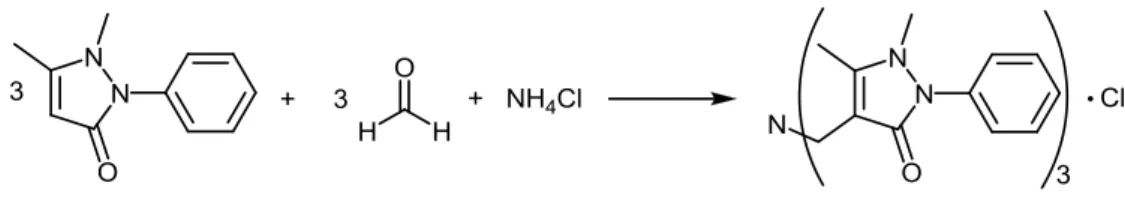
L’hypophosphite de sodium est obtenu par la réaction du phosphore blanc en milieu alcalin (soude et chaux) et est utilisé principalement pour le nickelage chimique de pièces diverses (disques durs, horlogerie, voitures, électronique, …). Lors de sa synthèse, de la phosphine PH3 gazeuse et du phosphite de calcium sont obtenus comme produits secondaires (environ 40% du P4 engagé). Actuellement, la phosphine est valorisée à travers sa combustion pour fabriquer de l’acide phosphorique de haute pureté et à travers la synthèse de deux sels de phosphonium, le THPC (utilisé comme retardant de flamme pour le coton) et le THPS (utilisé comme biocide pour les champs pétroliers). Febex SA cherche cependant à mieux valoriser ce gaz, étant conscient que c’est une matière première unique tant par sa rareté que par sa chimie.
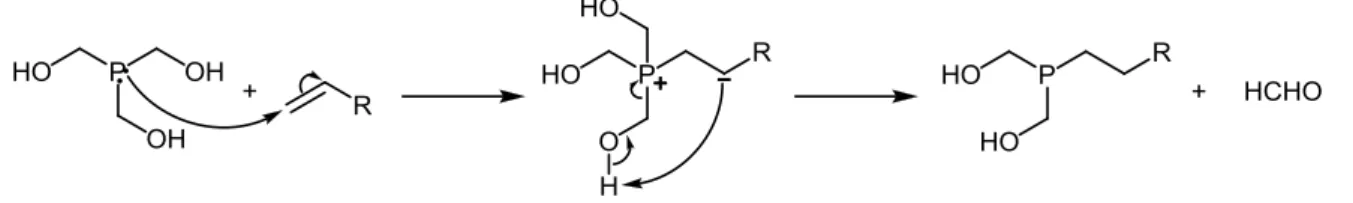
Le gaz brut obtenu est un mélange de phosphine (environ 35%) et d’hydrogène, produits de la réaction, ainsi que d’azote provenant de l’inertisation de l’installation. La dilution de la phosphine ainsi que sa toxicité et son inflammabilité en font un gaz relativement difficile à manipuler. Ces différentes contraintes ont amené Febex SA à réfléchir aux divers dérivés de phosphine permettant ensuite de développer une chimie variée. Le choix s’est porté sur le THP (tris(hydroxyméthyl)-phosphine) de par son large potentiel synthétique. En effet, le THP est une molécule nucléophile apportant une bonne réactivité grâce à son centre phosphoré portant un doublet libre et à la possibilité d’élimination d’une molécule de formaldéhyde sur le phosphonium correspondant. Ces
2
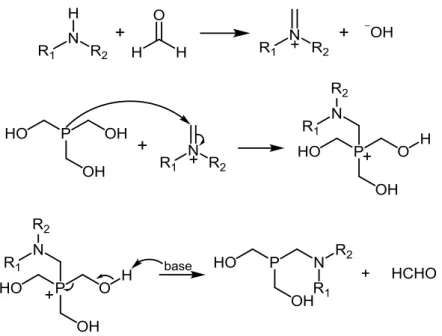
deux caractéristiques permettent par exemple de faire réagir le THP avec des sources d’azote type amine, amide ou urée par condensation de Mannich (Fig. 1a), mais aussi avec des oléfines activées type cétone, ester, nitrile, nitro ou sulfonyle par addition de Michael (Fig. 1b).
Figure 1 : condensation de Mannich (a) et addition de Michael (b) sur le THP.
L’avantage de cette démarche serait de produire des substances d’intérêt industriel avec une matière première plus stable et plus facile à manipuler que PH3, dont l’utilisation serait notamment axée sur l’application retardant de flamme. En effet, les composés riches en éléments phosphore et azote sont reconnus pour leur efficacité ignifugeante, principalement en tant qu’additifs dans la composition de polymères ou dans la formulation de traitement de textiles[1].
1.1.2 Le phosphore : un élément crucial
[2]"Sans phosphore, pas de vie sur Terre !"
Cette phrase résume parfaitement le rôle déterminant joué par le phosphore sur notre planète. Pour s’en convaincre, il suffit par exemple de se rappeler que cet élément est présent dans la structure même de l’ADN (molécule qui contient l’information génétique) et dans l’ATP (molécule qui stocke et transporte l’énergie créée pendant la photosynthèse et/ou la respiration cellulaire). L’organisme humain en contient environ 800 mg, dont environ 85% sont situés dans les os et les dents (associé au calcium), le reste entrant dans la composition des phospholipides, constituants importants des membranes cellulaires.
Le phosphore est un élément chimique (symbole : 31P, n° atomique 15, masse atomique = 30.973762) de la famille des non-métaux et des pnictogènes, c’est-à-dire appartenant à la quinzième colonne du classement périodique des éléments, tout comme l’azote (N), l’arsenic (As), l’antimoine (Sb), le bismuth (Bi) et l’ununpentium (Uup). Il possède 23 isotopes (1 stable et 22 radioactifs) et peut se présenter sous trois formes allotropiques selon la température et la pression à laquelle il est formé[3] :
phosphore blanc (P4 tétraédrique, spontanément inflammable au contact de l’air)
phosphore rouge (réseau amorphe, oligomères de phosphore blanc)
3 Son nom provient du grec phos (lumière) et phoros (porteur), le phosphore blanc émettant de la lumière visible dans l’obscurité quand il est exposé à l’air.
1.1.2.1 Historique
La découverte du phosphore est singulière, car il est le seul élément à avoir été extrait d’abord des êtres vivants, ensuite des plantes, puis il a été détecté seulement un siècle plus tard dans une matière minérale. En effet, sa découverte est attribuée à H. Brandt, alchimiste allemand à la recherche de la pierre philosophale, qui a isolé du phosphore blanc en 1669 à partir de l’urine[4]. En 1680, R. Boyle améliore le procédé, puis il réussit à obtenir l’oxyde et l’acide phosphorique, et démontre que l’exposition à l’air est nécessaire pour la luminescence (premier exemple de chimiluminescence)[5]. En 1688, le phosphore est observé pour la première fois dans le monde végétal par B. Albino. La méthode de distillation de l’urine ne permettant de se procurer que peu de phosphore, J. G. Gahn (chimiste suédois ayant découvert le manganèse) et C. W. Scheele (chimiste suédois ayant découvert le molybdène, le tungstène, l’acide citrique, le cyanure d’hydrogène, …) publient en 1769 un procédé à partir de poudre d’os calcinée puis décomposée par l’acide sulfurique permettant d’obtenir des quantités considérables de phosphore. Cette méthode, légèrement modifiée, est encore utilisée actuellement. C’est seulement en 1779 que J. G. Gahn découvre du phosphore dans la pyromorphite (Pb5(PO4)3Cl), puis T. Bergman et J. L. Proust dans l’apatite (Ca5(PO4)3(OH,Cl,F)).
Le phosphore étant devenu un élément plus commun, les chimistes ont ensuite pu étudier convenablement ses propriétés et ses dérivés. En 1783, P. Gengembre chauffe du phosphore blanc en milieu alcalin (KOH) et obtient un gaz qui s’enflamme spontanément à l’air qu’il identifie comme étant de la phosphine (PH3). Pour être précise, il a certainement obtenu un mélange PH3-P2H4, car la présence d’impuretés dans la phosphine (notamment de traces de diphosphine) est assez habituelle et rend le mélange gazeux particulièrement inflammable. De plus, son expérience lui a probablement permis d’être le premier à synthétiser de l’hypophosphite, ce qui est passé complètement inaperçu à cette époque !
En 1808, J. L. Gay-Lussac et L. J. Thenard, forment du trichlorure de phosphore (PCl3) et du pentachlorure de phosphore (PCl5) qui sont deux composés de grande importance en chimie de synthèse. Le premier composé organique est isolé en 1811 par N. L. Vauquelin : la lécithine, extraite de tissus du cerveau, que T. N. Gobley identifiera 40 ans plus tard comme étant un phospholipide. En 1820, J. L. Lassaigne synthétise, lui, les premiers composés organophosphorés en faisant réagir l’acide phosphorique avec des alcools pour obtenir des phosphates alkylés. La première manufacture de phosphore élémentaire voit le jour en Angleterre en 1844, à l’initiative de A. Albright qui en commercialise 0.75 tonne en 1844 et 26.5 tonnes en 1851, pour la fabrication d’allumettes principalement[6]. En 1848, le phosphore rouge est découvert par A. Schrötter, puis 2 ans plus tard est créé la première production commerciale d’acide phosphorique par procédé par voie humide (attaque des roches phosphates par l’acide sulfurique). En 1868, E. F. Hoppe-Seyler découvre dans le noyau des cellules une substance riche en phosphore : la nucléine, premier acide nucléique.
A partir des années 30, le phosphore est à nouveau projeté au-devant de la scène, d’abord par C. H. Fiske et Y. SubbaRow et leur travail sur le métabolisme des tissus musculaires qui amena à la découverte de la phosphocréatine et de l’ATP[7-9], ceci grâce à la méthode de détermination colorimétrique du phosphore qu’ils développèrent[10] et qui reste une des références les plus citées
4
de l’histoire de la biochimie (citée 2379 fois) ! Ensuite, les divers travaux sur les processus de glycolyse et d’oxydation du glucose, puis sur les acides nucléiques en tant que constituants des chromosomes, mettent particulièrement en avant le rôle joué par le phosphore dans de nombreuses réactions biochimiques[11].
D’autres recherches plus physico-chimiques sur le phosphore élémentaire permettent en 1935 de créer le premier atome de 32P radioactif à partir de 31P, qui sera par la suite intensément utilisé comme traceur en médecine nucléaire, biochimie et science des plantes et, en 1951, de mesurer la première 31P-RMN (spin = ½) grâce aux essais effectués par W. C. Dickinson[12].
Finalement, 4 Prix Nobel vont être décernés pour des sujets en relation étroite avec le phosphore : en 1957 (chimie) à A. R. Todd pour ses travaux sur les nucléotides et les coenzymes nucléotidiques[13], en 1962 (médecine) à F. H. C. Crick, J. B. Watson et M. H. F. Wilkins pour l’élucidation de la structure en double hélice des acides nucléiques[14], en 1978 (chimie) à P. D. Mitchell pour la formulation de la théorie chimiosmotique qui permet de comprendre le transfert d’énergie biologique en utilisant un gradient de protons à travers une membrane pour créer l’ATP[15], et en 1979 (chimie) à G. Wittig pour les progrès apportés aux méthodes de synthèse organique grâce à ses travaux sur le phosphore (réaction de Wittig)[16].
1.1.2.2 Propriétés et réactivité du phosphore
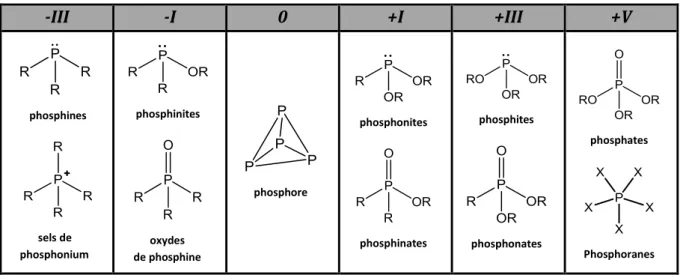
De nos jours, le phosphore présente une chimie très variée, car il forme des liaisons stables avec beaucoup d’éléments, particulièrement l’oxygène mais aussi l’hydrogène, le carbone, l’azote, le fluor ou encore le chlore. La valence des électrons du phosphore (3s2 3p3), mais également ses orbitales 3d vides, contribuent à beaucoup d’aspects étonnants de la chimie du phosphore, notamment à la stabilité particulière du système ylure (R3P+-C—R’2), à la force de la double liaison P=O et à l’existence de radicaux phosphoranyls (R4P∙) dans lesquels le phosphore est entouré par plus de 8 électrons de valence. Cette extension de la valence du phosphore permet aussi d’observer des composés pentavalents (PF5) et hexavalents (PCl6-).
Le phosphore est un non-métal, possédant un potentiel de première ionisation relativement haut, et une affinité électronique basse. En général, il forme des liaisons covalentes (à noter tout de même l’existence de l’ion phosphure). Ses degrés d’oxydation sont nombreux (Tableau 1) :
-III
-I
0
+I
+III
+V
phosphines sels de phosphonium phosphinites oxydes de phosphine phosphore phosphonites phosphinates phosphites phosphonates phosphates Phosphoranes5 Il est à noter que les états d’oxydation –II (diphosphine P2H4), +II (acide hypodiphosphorique H4P2O4) et +IV (acide hypophosphorique H4P2O6) existent également.
En essayant de comparer la réactivité du phosphore avec celle de l’azote, des différences notables sont rapidement mises en évidence. Contrairement à l’azote, le phosphore a tendance à la caténation puisque 3 liaisons simples P-P sont plus stables qu’une liaison triple (Tableau 2). Ainsi, à l’état moléculaire, la molécule linéaire de diazote triplement liés N
≡
N est très différente de la molécule tétraédrique de phosphore P4.P-P 200
P-H 321
P-C 264
P
≡P 486
P-O 360
P-F 503
P-Cl 322
N-N 159
N-H 391
N-C 292
N
≡N 946
N-O 222
N-F 278
N-Cl 193
Tableau 2 : comparaison des enthalpies [kJmol-1] des différentes liaisons du phosphore et de l’azote.
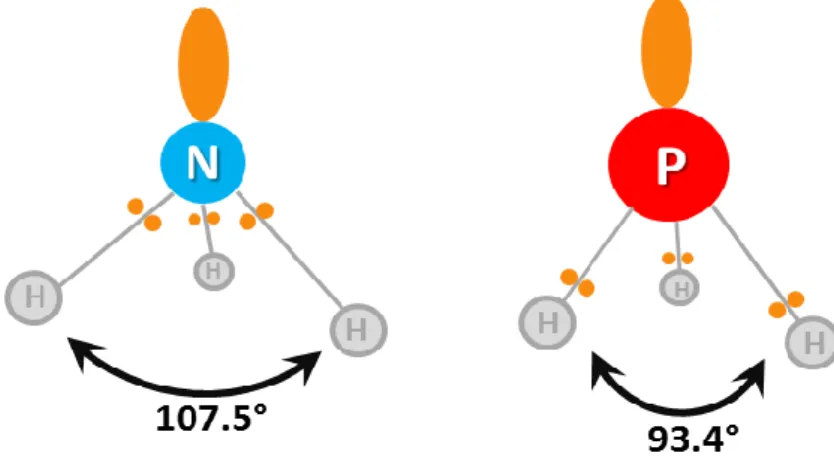
De plus, il est vrai que PH3 a une structure pyramidale analogue à NH3, pourtant l’angle HPH (93.4°) est inférieur à l’angle HNH (107.5°) (Fig. 2). Ceci s’explique d’une part par la taille et l’électro-négativité de l’atome central, mais également par l’hybridation de la molécule d’ammoniac. En effet, la structure de la molécule NH3, dont la valeur des angles est proche des angles tétraédriques, peut être expliquée par l’hybridation sp3 de l’atome d’azote avec les 4 paires d’électrons, formant 3 paires liantes et un doublet libre. La répulsion entre un doublet libre et un doublet liant est toujours plus forte qu’entre deux doublets liants. Ceci a pour effet que l’angle de liaison HNH est réduit à 107.5° au lieu des 109.3° attendus. Dans le cas de la phosphine, les électrons des doublets liants sont plus éloignés de l’atome central qui est plus gros, ainsi le doublet libre cause une répulsion plus forte ce qui fait décroître l’angle de liaison HPH à 93.4°.
Figure 2 : angles des liaisons HNH de l’ammoniac et HPH de la phosphine.
Ceci suggère que pour la phosphine, les orbitales liantes sont proches d’orbitales p pures, ce qui entraîne un caractère s très marqué du doublet libre. Ce dernier a donc moins d’énergie et est moins réactif que le doublet libre sp3 de l’ammoniac, ce qui explique pourquoi NH3 est plus basique (pKa (H2O) : NH4+ = 9.2, PH4+ = -14)[17] et moins réducteur.
Une autre propriété distingue les phosphines des amines : la stabilité conformationelle. Ayant des géométries de type tétraédrique, si trois groupes fonctionnels différents sont présents sur l’atome central et en comptant le doublet libre comme 4ème groupe fonctionnel (d’après les règles de
6
Cahn-Ingold-Prelog)[18], les 2 atomes pourraient être considérés comme potentiellement porteurs de chiralité. Pourtant, l’azote présente une inversion de configuration par changement d’état d’hybridation (sp3→sp2→sp3) appelé « effet parapluie » qui se produit naturellement à température ambiante. Cette inversion rapide transforme une forme énantiomère en l’autre forme (racémisation) et entrave donc l’observation de chiralité pour les amines, qui sont alors considérées comme centres asymétriques non chiraux. Le phosphore, lui, nécessite de l’énergie (chauffage) pour activer l’effet parapluie, il est donc conformationellement stable à température ambiante ce qui permet d’isoler et de séparer les deux formes énantiomères. Cette propriété présente un intérêt considérable dans le domaine de la catalyse asymétrique.
Les phosphines et organophosphines, de par leur versatilité, sont largement répandues en chimie organique et organométallique. Les voies de synthèse permettant d’obtenir des composés organophosphorés sont variées, mais la plupart utilisent le trichlorure de phosphore (PCl3) ou son dérivé oxydé, le chlorure de phosphoryle (POCl3). La triphénylphosphine, composé cristallin stable, est également couramment employée comme précurseur pour former d’autres organophosphines. De nombreuses réactions organiques font appel aux phosphines. La plus connue est certainement la réaction de Wittig qui permet de créer des doubles liaisons C=C par addition d’un ylure de phosphore sur un composé carbonylé. Les phosphines sont également utilisées pour la réaction de Staudinger[19] dans laquelle elles participent à la réduction d’azotures en amines. Elles sont aussi de très bons ligands utilisés pour produire des catalyseurs servant notamment aux synthèses asymétriques. Les exemples les plus connus sont le catalyseur de Wilkinson[20] permettant l’hydrogénation industrielle des alcènes en alcanes, ou pour les catalyseurs de Grubbs de première génération qui sont des complexes carbène-métal de transition utilisés pour la métathèse d’oléfines.
1.1.2.3 Provenance naturelle
Le phosphore est le douzième élément le plus abondant dans la croûte terrestre (0.09% en masse). On le trouve dans la nature essentiellement sous forme d’orthophosphates minéraux (phosphates et fluorophosphates d’aluminium, de fer, de manganèse, de terres rares, uranifères, de plomb) dont les plus répandus sont les phosphates calcaires, particulièrement les apatites. Selon l’origine du gisement, les apatites peuvent présenter une grande variation dans leur composition chimique, générant des propriétés physiques diverses.
Les gisements sédimentaires, provenant de matière organique, représentent environ 75% de la production mondiale. La plupart d’entre eux contiennent de la fluoroapatite carbonatée appelée francolite, dont la composition chimique est particulièrement adaptée à l’utilisation comme engrais en application directe. Les principaux gisements sédimentaires se trouvent au Maroc, aux USA et en Chine. Les gisements ignés (volcaniques) représentent 15-20% de la production mondiale. Ils fournissent des fluoroapatites moins efficaces en tant qu’engrais sous leur forme brute et sont en général purifiées et/ou transformées avant utilisation. Les principaux gisements ignés sont en Russie, au Canada, en Afrique du Sud, au Brésil et en Finlande.
Le phosphore blanc est extrait du minerai par réduction à haute température des phosphates par le carbone (charbon, coke) en présence de silice (sable). L’invention du four à arc électrique en 1890 et l’exploitation des gisements d’apatite ont conduit au fort développement de l’industrie du phosphore. La production annuelle de minerai phosphaté est de l’ordre de 190 millions de tonnes (environ 56 en Chine, 30 aux Etats-Unis, 27 au Maroc, …). La majorité du phosphore commercialisé
7 est utilisé en chimie inorganique (70% fertilisants, 15% détergents, 8% nourriture, 5% sous forme d’acide phosphorique pour le contrôle de la corrosion), le domaine des composés organophosphorés représentant 2% de l’utilisation totale du phosphore (industrie pharmaceutique, insecticides et plastiques) mais tend à augmenter.
1.1.3 Importance industrielle du phosphore
[21]1.1.3.1 Phosphore élémentaire
Historiquement, la première application connue du phosphore concerne la fabrication des allumettes. L’invention de l’allumette inflammable par friction est l’œuvre du chimiste anglais J. Walker en 1827 (d’après les travaux de R. Boyle en 1680 sur l’utilisation du phosphore et du soufre). Il mit au point un mélange de sulfure d’antimoine, de chlorate de potassium, de gomme et d’amidon qui s’enflammait en le frottant sur une surface rugueuse. En 1831, du phosphore blanc est ajouté au mélange afin d’atténuer l’odeur et d’améliorer l’efficacité, ce qui rendit les allumettes très populaires.
Une autre application du phosphore blanc a été développée pendant la Première Guerre Mondiale : les bombes incendiaires au phosphore (Fig. 3). Les exemples les plus connus de leur utilisation massive sont les bombardements de Dresde et de Tokyo en 1945.
Figure 3 : l’USS Alabama (BB-8) touché lors du test d’une bombe incendiaire au phosphore en septembre 1921[22].
Officiellement, leur utilisation actuelle est restreinte à la signalisation, au marquage de cibles et à la création d’écrans de fumée. Leur utilisation contre des civils ou des cibles militaires situées à l’intérieur de concentrations civiles est strictement interdite par la Convention sur certaines Armes
Classiques (Protocole III pour les armes incendiaires)[23]. Malheureusement, sur le terrain la distinction entre ces deux utilisations n’est pas toujours évidente ce qui conduit à des situations ambigües, telles que l’utilisation de bombes au phosphore par l’armée américaine lors de la bataille de Fallujah en novembre 2004. Les Etats-Unis ont reconnu en avoir utilisé comme arme incendiaire contre des insurgés mais réfute avoir touché des civils, ce que contestent plusieurs médecins et témoins irakiens présents sur place.
8
Actuellement, le phosphore élémentaire est largement utilisé comme matière première dans certains domaines industriels, tels que la production d’engrais et de pesticides. Il est également très couramment employé en chimie de synthèse en tant que réactif pour obtenir des phosphates organiques via le pentoxyde de phosphore (P2O5), ou pour produire différents composés organophosphorés via les chlorures de phosphore (PCl3, PCl5).
1.1.3.2 Engrais
Le phosphore inorganique a également un usage notable directement liée à son importance dans les organismes vivants : le phosphore soluble (phosphates) est assimilé par les plantes et entre ainsi dans la chaîne alimentaire. Il est donc produit massivement pour la fabrication d’engrais, car il sert au bon développement des racines et à la résistance aux maladies. Par le passé, les écosystèmes agricoles s’adaptaient à la faible disponibilité du phosphore en le recyclant autant que possible (fumier, détritus, excréments des animaux pâturant dans les champs, …). Cependant, depuis la seconde moitié du XIXe siècle, l’exploitation minière des gisements de phosphates naturels a induit d’importants changements écologiques et agricoles. En 1843 déjà, J. Murray brevète sa production de fertilisants « superphosphate » qui est le produit de la réaction de l’acide sulfurique sur des roches de phosphates. Au fil du temps, le domaine des fertilisants est devenu un marché très lucratif[24], et la disponibilité du phosphore à travers les engrais artificiels ainsi que les rendements plus élevés qu’ils apportent ont réduit la nécessité de recycler les déchets organiques. En effet, le gain de productivité et la sécurité alimentaire plus élevée ont contribué à une augmentation de la population mondiale, ce qui accroît la demande alimentaire et donc demande plus de productivité. Entre 1972 et 2011, l’utilisation mondiale d’engrais est passée d’environ 74 à 198 millions de tonnes. En comparant la quantité de phosphore « consommée » dans les aliments par la population mondiale avec la quantité annuelle de phosphore dédiée à la production d’engrais, il est étonnant de constater que seul 20% du phosphore semé sur les cultures se retrouve dans les aliments. Bennet et
al.[25] estiment que 22 millions de tonnes de phosphore se retrouvent dans l’océan chaque année (contre 8 millions de tonnes à l’époque préindustrielle) par le lessivage des terres cultivées.
Dans les années 70, le phosphore présent dans les cours d’eau posait un véritable problème d’eutrophisation de l’eau (asphyxie des plans d’eau douce), c’est pourquoi des normes visant à réguler l’utilisation d’engrais phosphatés ont été imposées avec succès. En Suisse, l’Ordonnance sur la Protection des Eaux ne fixe aucune norme en matière de phosphore pour les plans d’eau, cependant des valeurs inférieures à 0.04mg/L de phosphore total définissent une qualité d’eau très bonne. A titre d’exemple, la valeur maximale de phosphore présent dans le lac de Neuchâtel ces trentes dernières années a été mesurée en 1983 (46µg/L), puis elle est descendue graduellement et s’est stabilisée en-dessous de 10µg/L depuis 2006[26].
Un second problème méconnu lié aux engrais phosphatés est que les minerais desquels ils sont extraits (gisements sédimentaires) contiennent une multitude d’éléments secondaires, dont de l’uranium (noyau-mère du polonium) et du cadmium qui se retrouvent en petite quantité dans les végétaux et donc dans la chaîne alimentaire. Les gisements d’apatites sont d’ailleurs souvent liés à des réserves exploitables d’uranium. Ces éléments sont non-seulement radioactifs mais également très toxiques et cancérigènes, et s’accumulent facilement dans les sols. Il est cependant difficile de dire précisément quelle quantité se retrouve dans notre organisme, car cela dépend de nombreux paramètres (type de sol, âge de la plante consommée, quantité d’engrais utilisé, …). Cependant, en
9 Europe on estime à 3 µg par jour d’uranium ingéré par personne (41% proviennent des boissons, 33% des légumes et 26% des viandes), dont 1-2% seulement sont absorbés par voie gastro-intestinale. Certains aliments en contiennent très peu (environ 1 µg d’uranium par kilo de matière sèche pour le miel et le lait) alors que d’autres l’assimilent bien (environ 100 µg pour les champignons). Pour le cadmium, l’estimation est de 3 à 35 µg de cadmium ingéré par jour et par personne dont 5% sont absorbés par voie gastro-intestinale. Ces quantités semblent négligeables, cependant le problème majeur de ces composés est leur capacité à s’accumuler dans l’organisme et leur élimination difficile[27].
1.1.3.3 Détergents
Traditionnellement, le savon a été le premier agent de nettoyage couramment utilisé dans les foyers. Déjà à l’époque romaine, il était connu que le chauffage d’un mélange de graisses animales et de cendres de bois produisait une substance soluble dans l’eau capable de dissoudre les graisses. Dès 1952, les détergents utilisant des polyphosphates dépassent le savon comme principal agent de nettoyage aux USA. En effet, des additifs polyphosphates appelés agents séquestrants, dont le plus connu est le triphosphate de sodium Na5P3O10, sont utilisés car ils chélatent les cations responsables de la dureté de l’eau (Mg2+, Ca2+) et empêchent les anions sulfonates (têtes hydrophiles des détergents) de précipiter et ainsi de perdre leur propriété nettoyante. De plus, le tripolyphosphate de sodium permet également de maintenir un pH de l’eau basique par effet tampon et a une action synergique avec les tensioactifs. En 2000, la consommation mondiale de tripolyphosphate de sodium était estimée à environ 2 millions de tonnes. En Suisse, l’emploi des phosphates dans les lessives est interdit depuis 1986 et des taux maximaux sont fixés pour la composition des autres détergents. De plus, un nombre croissant de détergents sans phosphates est mis sur le marché chaque année. Les zéolithes (minéraux microporeux dérivés des silicates) sont par exemple une bonne alternative pour l’usage domestique du lavage de linge. Ces mesures ont permis de diminuer l’eutrophisation du lac Léman qui était très atteint depuis les années 1950 (89 µg/L en 1979 contre 27.5 µg/L en 2008).
1.1.3.4 Acide phosphorique et phosphates
L’acide phosphorique et les phosphates sont également largement commercialisés dans le monde, puisqu’il est produit environ 25 millions de tonnes par année. Ils sont utilisés principalement sous forme d’engrais, mais également comme détartrant, traitement antirouille de surfaces métalliques, pour la phosphatation (protection contre la corrosion de pièces en acier par trempage), pour le décapage de pièces électroniques et de différentes surfaces (métalliques, tuiles, porcelaines), pour le traitement des eaux usées, le nettoyage des équipements de production de l’industrie alimentaire, mais il est aussi très répandu dans notre alimentation. En effet, particulièrement connu pour être un composant des boissons sodas (0.7 g/L), il fait partie des additifs alimentaires sous le code européen E338 et est utilisé comme agent de conservation, émulsifiant ou correcteur d’acidité. D’autres formes de phosphate se retrouvent dans l’alimentation : les orthophosphates de sodium (E339), de potassium (E340) et de calcium (E341), les polyphosphates (E450, E451, E452) qui servent de régulateurs de l’acidité, émulsifiants, agents de texture, stabilisants, conservateurs (fromages, viandes et produits de la mer) et absorbeurs d’humidité, le phosphate d’ammonium (E342) qui sert d’agent de traitement des farines, le phosphate de magnésium (E343) qui sert d’antiagglomérant (riz) et les phosphates d’amidon (E1410, E1412, E1413, E1414, E1442) qui servent d’épaississants et de liants (biscuits, cakes, gâteaux). Certains aliments contiennent naturellement des phosphates, tel que
10
le lait de vache (phosphate de calcium), les jaunes d’œuf ou les légumineuses. Un excès de phosphore alimentaire est rare, mais peut déclencher une hyperphosphatémie temporaire qui inhibe la synthèse de la vitamine D. De plus, un excédent de phosphates dans l’alimentation des enfants est suspecté de provoquer l’hyperactivité et certains troubles du comportement.
1.1.3.5 Organophosphorés
Les organophosphorés ont été synthétisés pour la première fois au début du XIXe siècle, puis leur développement s’est emballé dès le moment où leurs propriétés biochimiques ont été découvertes. L’activité biocide a été la première propriété mise en avant des organophosphorés, à la fin des années 30 en Allemagne par I.G. Farbenindustrie AG (aujourd’hui Bayer AG) qui voulait développer des insecticides plus performants. Des études étaient réalisées sur le parathion (aujourd’hui interdit), mais l’armée allemande à l’aube de la 2ème Guerre Mondiale, s’empare de ces développements pour en faire des armes chimiques neurotoxiques (sarins, somans, tabuns). Ces gaz ont encore été utilisés récemment pendant la guerre Iran-Irak dans les années 80 et en Syrie en août 2013, mais l’exemple de leur utilisation le plus connu est sans doute l’attentat au gaz sarin dans le métro de Tokyo en 1995. De nombreux organophosphorés sont donc de puissants neurotoxiques qui agissent par inhibition de l’acétylcholinestérase dans les cellules nerveuses[28], ils entrent d’ailleurs dans la composition de certains venins, dont le seul antidote est l’atropine.
Les composés organophosphorés sont très répandus dans notre société, principalement sous forme de pesticides. Leur principale attractivité en tant que produits phytosanitaires est qu’ils sont efficaces en très petites quantités et que leur stabilité dans la biosphère est relativement limitée, ayant des temps de ½ vie de 2 à 10 jours alors que leurs homologues chlorés (DDT et dérivés) persistent plusieurs années. Le développement des pesticides est un avantage économique considérable pour l’industrie agro-alimentaire. L’utilisation des composés organiques du phosphore s’étend également aux retardants de flamme (tissus, polymères, fluides hydrauliques dans l’aviation), aux composés médicinaux et pharmaceutiques, aux catalyseurs, aux agents stabilisants et antioxydants pour les plastiques (phosphites), aux additifs pour les produits pétroliers (contrôle de la préignition), …
Comme on l’a vu, les sources de phosphore proviennent de gisements naturels et sont donc non renouvelables. Cependant, contrairement à d’autres ressources telles que le pétrole, les phosphates sont seulement dispersés et non dégradés, ce qui permet d’envisager leur récupération. D’ailleurs, plusieurs projets étudient la possibilité de recycler efficacement le phosphore (récupération des sédiments des hauts-fonds océaniques, recyclage des déchets carnés, …). De plus, il est fort probable que quelques gisements restent encore à découvrir. Cependant, au vu de la consommation actuelle de phosphore et de l’épuisement des gisements de phosphates à travers le monde, la réserve calculée en 2011 se montant à environ 65 milliards de tonnes, les chercheurs estiment que l’épuisement du phosphore se produira dans environ 350 ans. Cette prévision, bien que contestée, soulève néanmoins quelques questions : le phosphore étant un facteur limitant du vivant qui n’a aucun substitut potentiel, pourra-t-on en extraire assez pour approvisionner les 10 milliards d’habitants prévus d’ici la fin du siècle ?
11
1.1.4 Retardants de flamme
1.1.4.1 Définition et histoire
"Nouvelles technologies et nouvelles applications apportent de nouveaux dangers"
Un retardant de flamme (RF) est une substance que l’on ajoute aux matériaux au cours de leur processus de fabrication afin d’abaisser leur inflammabilité ou de ralentir leur combustion. A noter que le terme « retardant de flamme » décrit une fonction et non une famille de substances chimiques. Cependant, il existe pratiquement autant de produits possédants des propriétés RF qu’il existe de matériaux de natures chimiques différentes. En effet, depuis les années 70, les matériaux traditionnels (bois, métaux) sont progressivement remplacés par des matériaux polymères (plastiques) qui certes, ont des avantages certains, mais peuvent être particulièrement inflammables et peuvent également dégager des composés hautement toxiques sous l’effet de la combustion. La majorité des matières plastiques qui nous entourent sont effectivement de très bons combustibles, ce qui conduit au développement rapide et violent des incendies (600°C en 3 minutes) et laisse peu de temps pour se mettre à l’abri. Les RF sont donc utilisés pour améliorer la sécurité de notre quotidien face aux incendies, essentiellement dans les domaines de la construction (isolation, équipements électriques), du transport (agencement automobile, sécurité aérienne) et des aménagements intérieurs des habitations (canapé, rideaux, fibres synthétiques).
Chaque jour en Europe, les incendies entraînent le décès de 12 personnes (environ 60 décès par année en Suisse), provoquent plusieurs milliards de dommages matériels, et 80% de ces incendies ont lieu dans des bâtiments résidentiels[29]. Avec l’utilisation croissante de polymères et la concentration toujours plus élevée de population dans les milieux urbains, le développement de RF efficaces a donc un réel intérêt. Ils contribuent de manière notable à la réduction des risques de départ de feu, à l’amélioration de la sécurité dans les habitations et lieux publiques, à l’accroissement du temps de fuite lors d’un incendie et à la diminution de gaz toxiques formés par la combustion. Il semblerait d’ailleurs que le nombre de décès liés aux incendies est en baisse depuis les années 70[29], ce qui coïncide parfaitement avec les débuts de l’utilisation répandue des RF.
L’histoire nous prouve que l’idée d’empêcher la destruction par le feu est bien plus vieille que la compréhension même des mécanismes de combustion. En effet, les Egyptiens utilisaient de l’alun pour réduire l’inflammabilité du bois vers 450 avant J.-C. déjà. Puis, dès 360 avant J.-C., les Romains préconisent le traitement des poutres en bois des bâtiments importants par un mélange de vinaigre et d’alun. Bien plus tard, en 1638, Nicolas Sabbatini suggère d’utiliser de l’argile et du gypse pour ignifuger les décors et les sièges de théâtres qui sont bien trop souvent la proie des flammes. Mais le premier brevet déposé dans ce domaine est celui de l’anglais O. Wyld en 1735 concernant l’utilisation d’un RF à base d’alun, de borax et de vitriol pour protéger les papiers et les textiles[30]. En 1783, les célèbres frères Montgolfier utilisaient également un enrobage d’alun afin de réduire l’inflammabilité de leurs ballons. Au début du XIXe siècle, L. J. Gay-Lussac va être mandaté suite aux incendies répétés de théâtres afin de trouver une solution efficace pour l’ignifugation des textiles. Il suggéra un mélange de polyphosphates d’ammonium, de chlorure d’ammonium et de borax qui se révéla très efficace et est encore utilisé de nos jours[31]. Mais c’est seulement une centaine d’années plus tard que W. H. Perkin entreprend une étude systématique sur les mécanismes d’action des RF,
12
dont les résultats sont encore d’actualité aujourd’hui[32,33]. Comme discuté précédemment, c’est à partir des années 70 que les RF sont véritablement devenus une science en soi, conséquence du développement rapide des polymères et des fibres synthétiques. Pour exemple, en 1971 aux USA, 78% des fibres utilisées pour produire les habits d’enfants provenaient du coton alors que 2 ans plus tard, le taux chutait à moins de 10%.
Les tout premiers développements ont clairement démontré l’efficacité ignifugeante des composés halogénés, cependant des quantités importantes de fumées opaques et toxiques se dégagent lors de leur combustion. La toxicité problématique observée pour ces composés, par analogie aux inconvénients observés pour les pesticides chlorés, a fortement encouragé le développement de composés moins toxiques. Les recherches actuelles s’orientent ainsi vers l’utilisation de RF contenant du phosphore, de l’azote et/ou des charges minérales.
1.1.4.2 Mécanismes d’action
Pour mieux appréhender les mécanismes mis en jeu dans l’efficacité d’un RF, il faut évidemment comprendre en détail ce qu’est la combustion. Tout d’abord, il est intéressant de savoir que les principales causes d’incendie sont la foudre (30%), les foyers déterminés (23%), incluant cigarettes, bougies, feux d’artifice, …), les installations électriques défectueuses ou utilisées de manière inappropriée (19%), les installations de chauffage (12.5%), puis viennent ensuite la malveillance et les causes inconnues[34]. Ces diverses causes représentent la source d’énergie nécessaire à l’initiation d’un feu, cependant deux autres éléments doivent obligatoirement être présents pour que le feu soit entretenu (Fig. 4) : un apport d’oxygène (comburant) et un produit inflammable (combustible).
Figure 4 : interaction entre les trois éléments composant le feu.
Ainsi, la combustion est une réaction chimique exothermique d’oxydoréduction qui nécessite une interaction entre ces 3 éléments (transfert d’énergie et contact entre combustible et comburant). Lorsque l’apport d’un de ces éléments s’amenuise, le feu décline et finalement s’éteint. C’est d’ailleurs de cette manière que travaillent les pompiers pour combattre un incendie, en refroidissant par exemple la source d’énergie avec de l’eau, ou en créant une couche isolant le combustible du
13 comburant à l’aide de poudres composées de sels inorganiques (phosphate ou sulfate d’ammonium et/ou bicarbonate de sodium en général).
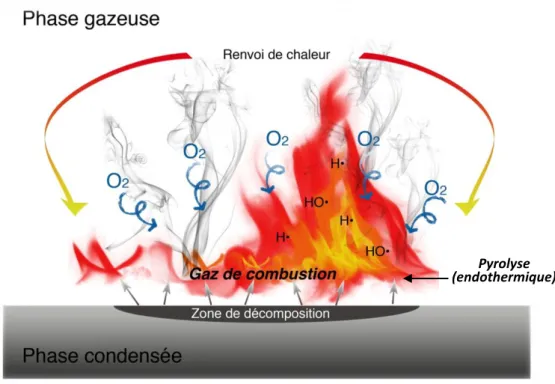
La combustion des polymères nous intéresse plus particulièrement dans le présent travail, c’est pourquoi ils sont pris comme modèles pour imager les mécanismes menant à un incendie (Fig. 5). L’initiation d’un feu passe d’abord par l’échauffement du matériau par la source de chaleur. Au-delà d’une température critique, un processus endothermique de dégradation se met en place : les liaisons les plus faibles du polymère se cassent et engendrent des radicaux qui se recombinent en molécules de bas poids moléculaires et inflammables. Il faut cependant que ces molécules soient relativement volatiles et que la concentration en oxygène du milieu soit suffisante pour que le mélange combustible/air soit adéquat. Cette étape dépend donc directement de la nature physique ou chimique du combustible (polymère) : composition, point d’inflammation, conductivité thermique, chaleur spécifique de combustion, mais également épaisseur du matériau, surface exposée à l’air ou encore dispersion sont autant de paramètres qui peuvent influencer l’ampleur de la combustion.
Figure 5 : phénomènes entourant le processus de combustion.
La formation de gaz inflammables (pyrolyse) est essentielle, car la combustion est uniquement une réaction en phase gazeuse (un solide ne brûle donc pas !). Une fois que l’inflammation a débuté, la décomposition du polymère génère assez de gaz combustibles pour que la flamme s’autoalimente. Le feu peut donc se propager exponentiellement, soit par conduction, par convection et/ou par rayonnement. Ce phénomène est souvent rapide et très violent, la température pouvant facilement atteindre les 1200°C.
Il est reconnu que les substances contenant les atomes azote, phosphore, chlore, brome, fluor sont particulièrement efficaces en tant que RF, et que le pourcentage de ces atomes contenu dans la substance RF est directement proportionnel à l’efficacité de l’ignifugation. Les RF peuvent intervenir chimiquement ou physiquement à plusieurs stades du processus de combustion, mais en général ils
Pyrolyse (endothermique)
14
agissent dans la phase décisive du départ de feu, c’est-à-dire qu’ils préviennent l’ignition ou la propagation de la flamme. Il est même assez fréquent d’utiliser plusieurs RF différents afin d’intervenir à divers stades de la combustion et ainsi augmenter l’efficacité de l’ignifugation. La description des mécanismes chimiques et physiques du comportement des RF dans la phase gazeuse et la phase condensée a été décrite précisément en 1990 par J. Troitzsch[35].
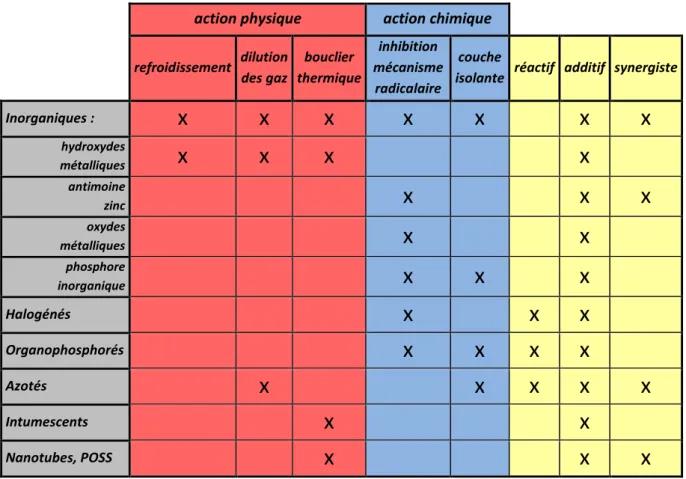
L’action physique d’un RF peut se faire par refroidissement, dilution des gaz ou formation d’une couche protectrice.
Refroidissement : l’additif se décompose de manière endothermique ce qui permet demaintenir la surface du polymère à une température empêchant l’auto-alimentation de la combustion (Fig. 6A).
Dilution des gaz : certains RF libèrent des gaz inertes (H2O, CO2), ce qui va diminuer la concentration en gaz combustibles, appauvrir le mélange combustible/air et donc provoquer un affaiblissement puis une extinction de la flamme. Les RF de ce type ne sont pas très courants dans l’application polymères (Fig. 6B).
Formation d’un bouclier thermique : le RF se transforme en couche protectrice (solide,liquide ou gazeuse) qui isole le polymère de l’oxygène ou du transfert de chaleur nécessaire au processus de combustion (Fig. 6C).
15
>210°C
L’action chimique d’un RF peut se faire soit en phase gazeuse ou alors en phase condensée et est en général plus efficace que l’action physique.
Phase gazeuse : le RF inhibe les réactions radicalaires, le processus exothermique est alors
arrêté provoquant ainsi un abaissement de l’énergie, un ralentissement de la réaction de combustion et une réduction de gaz combustibles formés (Fig. 6D).
Phase condensée : le RF forme une couche carbonée, éventuellement expansée, à la surface
du polymère, par déshydratation par exemple, générant des doubles liaisons qui peuvent former la couche carbonée par cyclisation ou réticulation (Fig. 6E).
De plus, une distinction est faite entre les RF réactifs et additifs. Les premiers sont des composants qui réagissent chimiquement et font partie intégrante du polymère (plus couteux et moins évidents à mettre en œuvre), alors que les seconds sont incorporés dans le polymère avant, pendant ou, le plus fréquemment, après la polymérisation[35].
1.1.4.3 Les différentes classes de RF
Les RF peuvent être divisés en trois principaux groupes : les produits inorganiques, les halogénés et les organophosphorés[35,37,38].
RF inorganiques : les principaux RF inorganiques sont le trihydroxyde d’aluminium, le
dihydroxyde de magnésium, le polyphosphate d’ammonium, le phosphore rouge, les argiles, les phyllosilicates. Ce groupe représente environ 50% en volume de la production mondiale. Ils agissent essentiellement par voie physique dans la phase condensée et dans la phase gazeuse. Pour être efficaces en tant que RF, les minéraux doivent se décomposer dans une gamme de température relativement basse (150 à 400°C), ce qui est peu répandu pour cette famille de substances. Ils se décomposent de manière endothermique, ce qui fait baisser la température du matériau et ainsi de diminuer sa vitesse de dégradation. De plus, leur décomposition libère des gaz inertes (eau et/ou CO2 le plus souvent) qui ont pour effet de diluer les mélanges inflammables et forment un écran isolant la surface du polymère de l’oxygène. Les minéraux sont donc typiquement utilisés de manière additive, et leur efficacité est en général proportionnelle à la quantité d’additif ajoutée au polymère ce qui peut poser certains problèmes au niveau des propriétés mécaniques des polymères. Le trihydroxyde d’aluminium, le RF minéral le plus largement répandu, est utilisé comme retardateur de flamme et suppresseur de fumée depuis les années 60[39]. Il est peu onéreux et très facile à incorporer dans les plastiques. Il se décompose vers 210°C selon la réaction suivante :
L’oxyde d’aluminium résultant de cette déshydratation forme une couche superficielle thermo-isolante. De plus, la proportion d’eau formée est de 30% en poids. Il est cependant très difficile de déshydrater complètement cette molécule. A 900°C, le solide contient encore une petite quantité d’eau qu’il ne perd que vers 1200°C. De plus, tant que la température n’est pas trop élevée (<1000°C), la réaction de déshydratation est réversible. Le problème majeur dans l’utilisation du trihydroxyde d’aluminium est la teneur requise (>60% en masse) pour atteindre le même niveau de protection que d’autres ignifugeants.
![Tableau 4 : critères de classification pour le test applicatif UL-94 vertical [54].](https://thumb-eu.123doks.com/thumbv2/123doknet/2192811.11658/41.892.181.771.104.333/tableau-criteres-classification-test-applicatif-ul-vertical.webp)
![Figure 12 : Mécanisme de la réaction entre la phosphine et le formaldéhyde [66] .](https://thumb-eu.123doks.com/thumbv2/123doknet/2192811.11658/47.892.106.790.953.1020/figure-mecanisme-reaction-phosphine-formaldehyde.webp)