Détection, caractérisation et comparaison des interactions protéine - ligand
205
0
0
Texte intégral
Figure
![Figure 1 Structure et classification des 20 acides aminés naturels présents dans l'organisme humain selon les propriétés chimiques [1]](https://thumb-eu.123doks.com/thumbv2/123doknet/2282222.22458/15.892.177.721.578.1075/figure-structure-classification-naturels-presents-organisme-proprietes-chimiques.webp)
![diagramme de Ramachandran, de l'ensemble des angles φ et ψ observées dans les structures protéiques [5, 6]](https://thumb-eu.123doks.com/thumbv2/123doknet/2282222.22458/17.892.111.782.115.593/diagramme-ramachandran-angles-f-ps-observees-structures-proteiques.webp)
+7

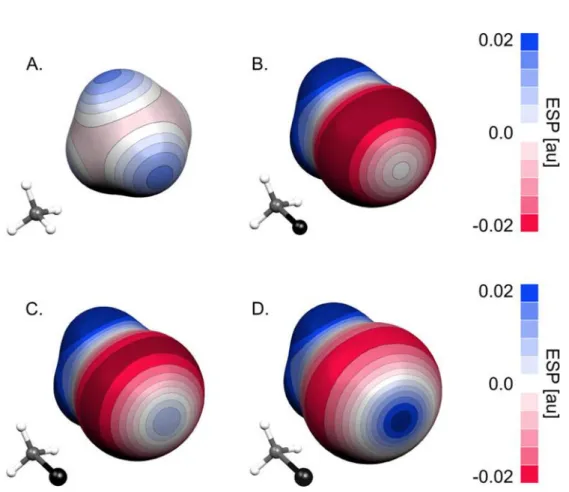
![Figure 14 Surface de potentielle électrostatique du benzene et de l'hexafluorobenzene mettant en avant la distribution distincte des charges sur l'aromatique [37]](https://thumb-eu.123doks.com/thumbv2/123doknet/2282222.22458/33.892.212.698.106.292/figure-surface-potentielle-electrostatique-hexafluorobenzene-distribution-distincte-aromatique.webp)
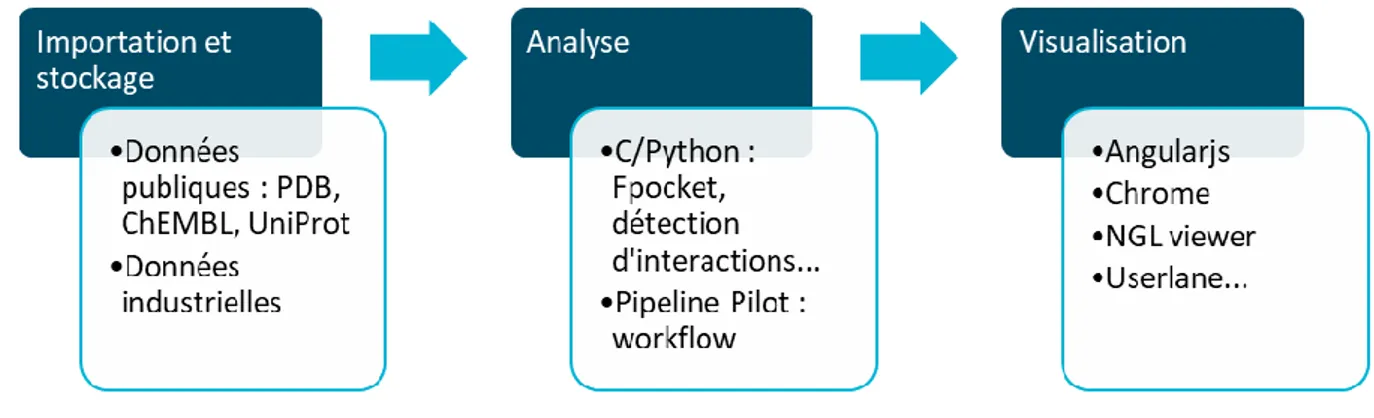
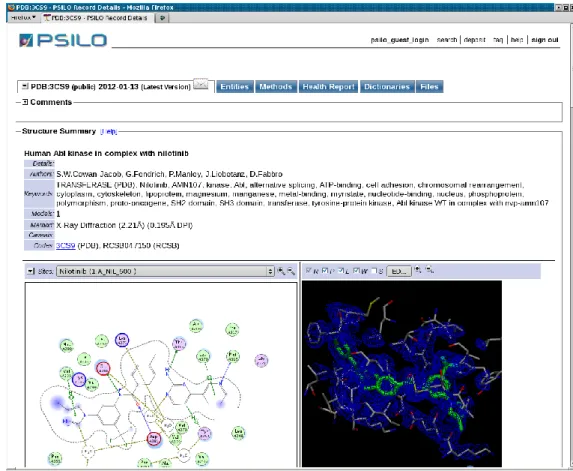
Outline
Méthodes de descriptions de l’interaction
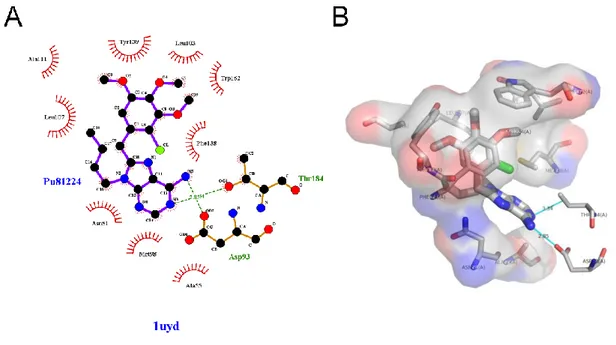
Retraitement et caractérisation des contacts
Interactions polaires a Liaisons hydrogènes
Description des interactions
Interactions autour des halogènes lourds
Regroupement des conformations d’un complexe
Génération d’un jeu de données non-redondant
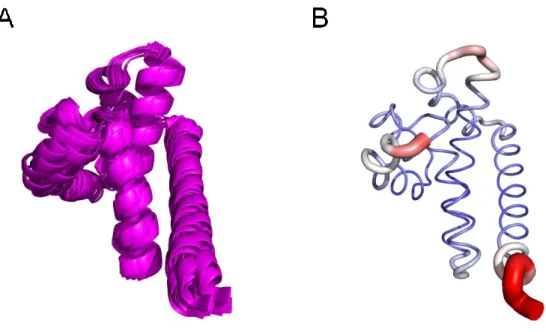
Analyse du comportement dynamique des hélices
Flexibilité protéique et alphabet structural
Documents relatifs