UNIVERSITE TOULOUSE III-PAUL SABATIER
ECOLE DOCTORALE BIOLOGIE-SANTE-BIOTECHNOLOGIE
THESE
Pour obtenir le grade de
DOCTEUR DE L’UNIVERSITE TOULOUSE III
Discipline : Chimie Organiqueprésentée et soutenue par
Marie-France LAROCHE
Le 12 décembre 2007 à 14 hTitre :
HEMISYNTHESE de MEROTERPENES ISSUS de
DICHROSTACHYS CINEREA
________ Directeur de thèse :Dr Georges MASSIOT
_________________JURY
Mlle Janine COSSY Professeur, ESPCI, Paris Présidente M. Ange PONCRAZI Directeur de Recherche CNRS, Rapporteur
Cergy Pontoise
M. Ahcene BOUMENDJEL Maître de conférence, Rapporteur Université Joseph Fourier, Grenoble
M. Georges MASSIOT Directeur de Recherche CNRS, Examinateur Toulouse
M. Arnaud MARCHAND Directeur de la Chimie Médicinale, Examinateur CISTIM, Leuven
M. Rémi CHAUVIN Professeur, Université Paul Sabatier, Examinateur Toulouse
à mon Kater,
à Alain,
Avant d’exposer les travaux qui ont conduit à la réalisation de ce mémoire, je tiens à remercier tous ceux qui m’ont permise d’accomplir ce travail.
Monsieur Georges MASSIOT, Directeur du Centre de Recherche des Substances Naturelles (CRSN) à l’Institut des Sciences et Techniques du Médicament de Toulouse (ISTMT), qui a supervisé et suivi l’ensemble de mes travaux.
Monsieur Jacques FAHY, Directeur du Département de Chimie Médicinale 1 au Centre de Recherche en Oncologie Expérimentale (CROE) de Pierre Fabre Médicament à l’ISTMT, qui m’a accueillie dans son équipe pendant ces trois années. Je tiens également à le remercier pour sa gentillesse et sa sympathie.
Monsieur Arnaud MARCHAND, ancien chercheur en Chimie Médicinale 1 au CROE, désormais Directeur de la Chimie Médicinale chez CISTIM à Leuven (Belgique), pour m’avoir suivie, conseillée et encouragée.
Je tiens à remercier aussi les membres du jury :
Mademoiselle Janine COSSY, Professeur à l’ESPCI, qui a su au travers de ses cours me communiquer le goût pour la chimie organique, qui m’a permis de poursuivre dans cette matière jusqu’au doctorat et qui m’a fait l’honneur de présider ce jury de thèse.
Messieurs Ange PANCRAZI, Directeur de recherche du CNRS à Cergy Pontoise, et Ahcene BOUMENDJEL, Maître de conférences à l’Université Joseph Fourier, Grenoble, qui ont accepté de juger et commenter ce travail.
Monsieur Rémi CHAUVIN, Professeur à l’Université Paul Sabatier, qui a accepté de participer à ce jury de thèse.
Mes remerciements s’adressent également à :
Messieurs Christian BAILLY, Directeur de la Recherche en Oncologie chez Pierre Fabre Médicament, et Nicolas GUILBAUD, Directeur du CROE, pour les marques d’encouragement qu’ils m’ont souvent témoignées.
Mademoiselle Laurence MARCOURT pour sa gentillesse, sa grande disponibilité et ses conseils.
Mademoiselle Isabelle CARLETTI, Madame Isabelle POUNY et Monsieur Christophe LONG pour leur disponibilité.
Madame Isabelle SARTORI, Mademoiselle Céline MORDANT, Monsieur Frédéric MARION et Monsieur Patrice MAYER pour leurs nombreux conseils, leur gentillesse, leur aide et leur disponibilité
Florence (Flo), Nathalie T. (p’tite Nat), Violaine, François, Nathalie de S.J. (NJU), Jésahelle (Jésa), Nathalie B. (Bobo), Mélanie, Vincent et Christine pour leur amitié et pour tous les moments de complicités passés en leur compagnie.
Sans oublier toutes les personnes que j’ai pu côtoyer au sein de l’ISTMT au cours de ces trois années.
Je tiens également à remercier le CNRS et les Laboratoires Pierre Fabre pour l’aide financière dont j’ai bénéficié.
Merci enfin à ma famille, à Arnaud et à sa famille pour m’avoir soutenue, supportée et encouragée tout au long de ma thèse.
Remerciements………3
Introduction Générale………....9
P
P
a
a
r
r
t
t
i
i
e
e
A
A
-
-
E
ET
TU
U
D
D
ES
E
S
B
BI
I
BL
B
LI
IO
OG
GR
R
AP
A
PH
HI
I
QU
Q
U
ES
E
S
……….13I. ETUDES BOTANIQUES sur Dichrostachys cinerea
………....14I.1. Classification botanique 14
I.2. Description 14
I.3. Habitat et Répartition 14
I.4. Utilisation 16
I.4.a. Utilisation traditionnelle 16
I.4.b. Utilisation industrielle 16
I.4.c. Autres utilisations 16
II. ETUDES BIBLIOGRAPHIQUES
………17II.1. Les Labdanes 17
II.1.a. Généralités 17
II.1.b. Un labdane particulier : l’acide communique 18
II.2. Les flavonoïdes 19
II.2.a. Généralités 19
II.2.b. Biosynthèse des flavonoïdes 21
P
P
a
a
r
r
t
t
i
i
e
e
B
B
-
-
M
MO
OD
DE
E
LE
L
ES
S
d
de
e
R
RE
EA
A
CT
C
TI
IO
ON
N
d
d
e
e
D
D
I
I
EL
E
LS
S-
-A
A
LD
L
DE
ER
R
………………......……2244I. LES MODELES : CHOIX et SYNTHESE
………..………..26I.1. Modèle pour la quinoflavone 26
I.1.a. Choix du modèle 26
I.1.b. Synthèse du modèle 26
I.2. Modèle pour le diterpène 27
I.2.a. Choix du modèle 27
I.2.a.i. Choix initial 28
I.2.a.ii. Choix final 30
II. LA QUINOFLAVONE et le COMMUNATE de METHYLE :
SYNTHESE
……….32II.1. La quinoflavone 32
II.1.a. Rappels sur les méthodes de synthèse des flavones 32
II.1.a.i. Méthode de Robinson-Venkataraman 32
II.1.a.ii. Méthode de Cushman et Nagarathnam 33
II.1.a.iii. Méthode utilisant les micro-ondes 34
II.1.b. Synthèse de la flavone 34
II.1.b.i. Choix de la méthode 34
II.1.b.ii. Synthèse 34
II.1.c. Synthèse de la quinone 35
II.2. Le communate de méthyle 37
II.2.a. Choix de la plante pour l’extraction de l’acide communique 37
II.2.b. Synthèse du communate de méthyle 38
III.1.a. Généralités 41
III.1.b. La réaction de Diels-Alder avec les quinones 42
III.2. Réaction de Diels-Alder entre le modèle de la quinoflavone et le modèle du communate de méthyle 43
III.2.a. Aspect expérimental 44
III.2.b. Détermination de la structure 76 44
III.2.b.i. Régiosélectivité 45
III.2.b.ii. Stéréochimie 48
III.2.c. Conclusion 49
III.3. Réaction de Diels-Alder entre le modèle de la quinoflavone et le communate de méthyle 49
III.3.a. Aspect expérimental 49
III.3.b. Détermination de la structure 77 50
III.3.b.i. Régiosélectivité 51
III.3.b.ii. Stéréochimie 53
III.3.c. Conclusion 54
III.4. Réaction de Diels-Alder entre la quinoflavone et le modèle du communate de méthyle 54
III.4.a. Aspect expérimental 54
III.4.b. Détermination de la structure 78 55
III.4.b.i. Régiosélectivité 56 III.4.b.ii. Stéréochimie 57 III.5. Conclusion 57
P
P
a
a
r
r
t
t
i
i
e
e
C
C
-
-
S
S
YN
Y
NT
TH
HE
ES
SE
E
de
d
e
la
l
a
MO
M
OL
LE
EC
C
UL
U
LE
E-
-C
CI
I
BL
B
LE
E
//
/
/
A
A
PP
P
PL
LI
IC
C
A
A
TI
T
IO
ON
N
d
d
e
e
l
la
a
R
RE
EA
A
CT
C
TI
IO
ON
N
D
D
E
E
D
D
IE
I
EL
LS
S
-A
-
A
LD
L
DE
ER
R
………………....…………..5588I. SYNTHESE de la MOLECULE CIBLE via une REACTION de
DIELS-ALDER sans CATALYSEUR
………59I.1. Aspect Expérimental 59
I.2. Détermination de la structure 32 59
I.2.a. Régiosélectivité 60
I.2.b. Stéréochimie 62
I.2.b.i. Analyse du spectre ROESY 62
I.2.b.ii. Modélisation moléculaire 63
I.2.b.iii. Comparaison de données 67
I.3. Conclusion 67
II. SYNTHESE de la MOLECULE CIBLE via une REACTION de
DIELS-ALDER avec un CATALYSEUR CHIRAL
………...69II.1. Rappels bibliographiques 69
II.1.a. Intérêt du catalyseur chiral dans la réaction de Diels-Alder 69
II.1.b. Les oxazaborolidines chirales 69
II.1.b.i. Généralités 69
II.1.b.ii. Oxazaborolidines chirales et réaction de Diels-Alder 70
II.1.b.iii. Oxazaborolidines chirales et réaction de Diels-Alder avec des quinones asymétriques 71
II.2. Synthèse de la molécule cible 32 avec le catalyseur de Corey 73
II.2.a. Synthèse du catalyseur 90 73
II.2.b.i. Aspect expérimental 74
II.2.b.ii. Détermination de la structure 32 75
II.2.b.iii. Conclusion 83
III.
APPLICATION
de
la
REACTION
de
DIELS-ALDER
DITERPENE/QUINOFLAVONE
………...84III.1. Application de la réaction de Diels-Alder avec d’autres diterpènes 84
III.1.a. L’acide communique 84
III.1.a.i. Aspect expérimental 84
III.1.a.ii. Détermination de la structure 96 84
III.1.b. Le communol 87
III.1.b.i. Synthèse du communol 87
III.1.b.ii. Réaction de Diels-Alder 87
III.1.b.iii. Conclusion 91
III.2. Réaction de Diels-Alder entre le communate de méthyle et une autre quinoflavone 91
III.2.a. Synthèse de la 5,7-dihydroxy-2-(2,4,5-triméthoxyphényl)chromen-4-one92 III.2.a.i. Voie 1 : via une réaction de Friedel-Crafts 92
III.2.a.ii. Voie 2 : via l’utilisation des micro-ondes 96
III.2.b. Oxydation de la flavone 99 en quinoflavone 2 98
III.2.c. Réaction de Diels-Alder entre la quinoflavone 2 et le communate de méthyle 12 99
III.2.c.i. Aspect expérimental 99
III.2.c.ii. Détermination de la structure 110 99
III.3. Conclusion 102
P
P
a
a
r
r
t
t
i
i
e
e
D
D
-
-
PR
P
R
OD
O
D
U
U
IT
I
TS
S
NA
N
AT
TU
U
RE
R
EL
LS
S
I
I
SO
S
OL
LE
ES
S
//
/
/
H
H
E
E
M
M
I
I
S
S
Y
Y
N
N
T
T
H
H
E
E
S
S
E
E
………………………………………………………………………………………………………………………………………………......110033I. PRODUITS NATURELS ISOLES par le CRSN
………..105I.1. Configuration 1 106
I.1. Configuration 2 106
I.1. Configuration 3 107
I.1. Configuration 4 108
II. HEMISYNTHESE
………...110II.1. Modulations sur le composé 111 110
II.1.a. Estérification/méthylation en 7’ 110
II.1.a.i. Aspect expérimental 110
II.1.a.ii. Détermination de structure 111
II.1.b. Estérification/méthylation en 7’ suivie d’époxydation en C8-C17 et C13-C14 111
II.1.b.i. Aspect expérimental 111
II.1.b.ii. Détermination de structure 112
II.2. Modulation sur le composé 112 112
II.2.a. Méthylation en 5’ et 7’ 112
II.2.a.i. Aspect expérimental 112
II.2.a.ii. Détermination de structure 113
II.2.b. Double époxydation en C8-C17 et C13-C14 113
Conclusion Générale………...115
P
P
a
a
r
r
t
t
i
i
e
e
e
e
x
x
p
p
é
é
r
r
i
i
m
m
e
e
n
n
t
t
a
a
l
l
e
e
………………………………………………………………………………………………………………………………………………..112200I. GENERALITES
……….121II. LES MODELES : CHOIX et SYNTHESE
………..………..123III. LA QUINOFLAVONE et le COMMUNATE de METHYLE :
SYNTHESE
………...129IV. REACTIONS de DIELS-ALDER entre MODELES
………..136V. REACTION de DIELS-ALDER sans CATALYSEUR : SYNTHESE de
la MOLECULE CIBLE 32
………139VI. REACTION de DIELS-ALDER avec CATALYSEUR
……….141VII.
APPLICATION
de
la
REACTION
de
DIELS-ALDER
DITERPENE/QUINOFLAVONE
……….145VIII. PRODUITS NATURELS ISOLES par le CRSN
………..156IX. HEMISYNTHESE
……….158I
Introduction Générale
La nature est une source inépuisable de molécules douées d’activité biologique que l’habile chimiste pourra modifier pour mettre au point de nouveaux médicaments. Pourquoi les plantes synthétisent ces molécules ? Certainement le plus souvent en réponse à un milieu extérieur hostile.
Les seules informations disponibles qui le guideront dans la sélection des molécules cibles relèvent de la botanique, de la médecine traditionnelle, et de l’observation du terrain : des interactions de voisinage entre les espèces vivantes.
A l'image des plantes qui ont fourni de nombreuses drogues majeures (morphine, quinine, acide salicylique…), les substances naturelles ont toujours été une source importante de molécules utiles en thérapeutique. Environ 70% des médicaments mis sur le marché à l'heure actuelle sont d'origine naturelle, ce qui représente un marché de quelques 30 milliards d'euros par an (Données Pierre Fabre - CNRS). La nature étant loin d'avoir révélé tous ses secrets avec des millions d'espèces (plantes, insectes, procaryotes, algues…) encore non décrites, ce vivier gigantesque de molécules d'origine naturelle est une bonne source d'alimentation pour la recherche en chimie médicinale.
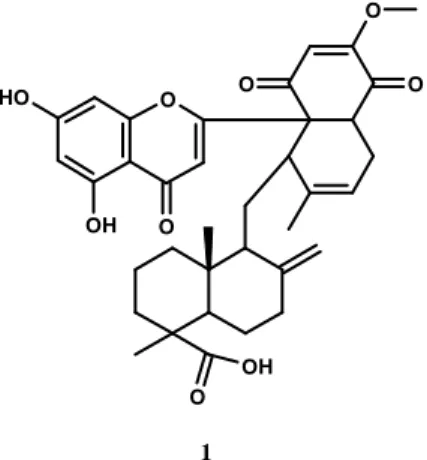
Le Centre de Recherche sur les Substances Naturelles (CRSN) s’appuie sur la biodiversité pour découvrir de nouvelles entités chimiques. Il a récemment isolé de Dichrostachys cinerea une nouvelle série de composés appartenant à la classe des méroterpènes dont la molécule 1 est un exemple (Figure 1). O OH O O O H OH O O O 1
Figure 1 : Nouvel archétype structural isolé de Dischrostachys Cinerea.
Ces composés sont probablement des adduits de Diels-Alder entre une quinone 2 issue d’un flavonoïde et un diène-labdane 3 (Schéma 1).
O OH O O O H OH O O O O O O H OH O O O O OH 1 2 3
+
Schéma 1 : Voie rétrosynthétique
L’originalité de cette structure et la rare littérature concernant les quinoflavones nous ont poussés à envisager la synthèse totale de composés de ce nouvel archétype structural. En partant de l’hypothèse que la réaction-clé de cette synthèse est la réaction de Diels-Alder, le travail de thèse a été fortement orienté autour de l’étude de cette réaction.
Le CRSN disposant d’une formidable échantillothèque botanique, le fragment diterpène n’a pas été synthétisé (bien que cela soit possible et soit décrit dans la littérature), mais simplement obtenu par extraction à partir de plantes. Cela a permis un gain de temps non négligeable et un gain écologique appréciable. Le fragment quinone a pour sa part été intégralement synthétisé à partir de produits disponibles commercialement.
Dans la partie A, nous rapporterons dans un premier temps les informations botaniques relatives à Dichrostachys cinerea. Comme nous n’avons trouvé aucune information chimique et structurale sur le nouvel archétype isolé, nous avons choisi de faire quelques rappels généraux sur les deux fragments qui permettent de l’obtenir, à savoir les diterpènes (et plus particulièrement l’acide communique) et les flavonoïdes.
Dans une deuxième partie, afin de savoir si le composé 1 peut être obtenu par une réaction de Diels-Alder entre une quinoflavone et un diterpène, nous avons décidé d’évaluer cette approche en utilisant des réactions modèles. Après avoir justifié le choix et montré la synthèse des modèles, et rappelé quelques notions sur la réaction de Diels-Alder, nous décrirons les résultats obtenus avec ces réactions modèles.
La partie C sera consacrée à la réaction de Diels-Alder entre le fragment diterpène et le fragment quinoflavone. Une étude de la stéréochimie à l’aide de la modélisation moléculaire sera proposée pour indiquer précisément quelle structure a été obtenue. Après avoir étudié les résultats de
cette réaction (sans et avec catalyseur chiral), nous l’exemplifierons à l’aide d’autres diterpènes et d’une autre quinoflavone.
Dans une dernière petite partie, nous décrirons deux des produits naturels isolés par le CRSN de Dichrostachys cinerea : nous étudierons à l’aide de la modélisation moléculaire leur stéréochimie et comparerons ces résultats à celles des adduits synthétisés par réaction de Diels-Alder. Par ailleurs, des modulations sur ces produits naturels seront effectuées, en vue d’éventuels tests pharmacologiques possibles au sein même de la structure de l’ISTMT.
P
P
A
A
R
R
T
T
I
I
E
E
A
A
E
I. ETUDES BOTANIQUES sur Dichrostachys Cinerea
I.1. Classification botanique1
Règne Plantae Sous-règne Tracheobionta Superdivision Spermatophyta Division Magnoliophyta Classe Magnoliopsida Sous-Classe Rosidae Ordre Fabale Famille Leguminosae Sous-famille Mimosacea
Genre Dichrostachys (DC.) Wight et Am.
Espèce Dichrostachys cinerea (L.) Wight et Am.
I.2. Description2
Dichrostachys cinerea est un arbuste, à écorce grise et épines isolées souvent munies d’une ou
deux feuilles, pouvant atteindre jusqu’à huit mètres de haut, aux rameaux terminés en épines. Les feuilles sont bi-pennées, chaque penne portant une glande. Les fleurs (nommées aussi « clochettes de mimosa ») pendantes de 2,5 cm de long sont composées d’une partie supérieure hermaphrodite jaune et d’une partie inférieure stérile allant du mauve au rose (Figure 2). Les fruits sont des gousses tordues, indéhiscentes, d’un effet décoratif original. Comme beaucoup de légumineuses, cette espèce est résistante à la sécheresse et supporte bien l’air marin.
I.3. Habitat et Répartition3
Les Dichrostachys sont des espèces sahélo-soudaniennes à guinéennes, ils poussent sur des sols lourds, envahissant les endroits perturbés (jachères, talus de route). Ils sont communs, localement abondants. Ils sont présents du Sénégal au Cameroun, jusqu’au Soudan ; Afrique tropicale et australe.
Branche de Dichrostachys cinerea
Fruits
Dichrostachys cinerea
I.4. Utilisation
I.4.a. Utilisation traditionnelle4-6
Plusieurs parties de Dichrostachys cinerea sont utilisées comme des « médicaments traditionnels » :
- les racines : ce sont des diurétiques, antivenimeux, et sont employées contre les rhumatismes, la blennorragie, les abcès, l’orchite, la lèpre, la toux infantile et la dysenterie ;
- les feuilles : elles sont utilisées pour le soin des abcès, de l’eczéma, de la blennorragie, des rhumatismes, de la gingivite et des caries dentaires, mais aussi de la rougeole ;
- les écorces : ce sont des ténifuges et antivenimeux, et sont employées contre la dysenterie ; - les fruits : ils sont utilisés contre l’otite, la hernie ombilicale, le paludisme chez les enfants.
I.4.b. Utilisation industrielle4,5,7
Le bois de Dichrostachys cinerea est dur, jaune à brun clair résistant aux termites et peut donc être utilisé en architecture ou comme manche d’outil ; il est aussi un excellent bois de feu et charbon.
I.4.c. Autres utilisations5,6
Dichrostachys cinerea est aussi utilisé comme vermifuge ; pour la fixation d’azote ; les fleurs
sont considérées comme mellifères ; elles sont localement consommées (Ghana), ou broutées par le bétail. De plus, cette espèce constitue une haie défensive très efficace grâce à la présence de nombreuses épines.
A ce jour, seules des études botaniques ont été réalisées sur Dichrostachys cinerea,8 aucune étude chimique ni pharmacologique n’a été entreprise.
En l’absence de données sur la composition chimique de Dichrostachys cinerea, l’étude bibliographique se porte plus particulièrement sur deux espèces de substances chimiques : les labdanes et les flavonoïdes, les deux précurseurs du nouvel archétype structural isolé (Figure 1).
II. ETUDES BIBLIOGRAPHIQUES
II.1. Les Labdanes II.1.a. Généralités
Les labdanes sont un vaste ensemble de composés diterpènes (C20) issus du métabolisme du
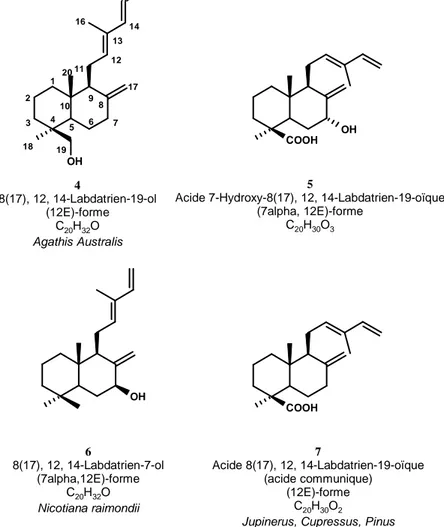
2E, 6E, 10E-géranylgéranylpyrophosphate (GGPP). Présents chez certains insectes et chez divers organismes marins, ils sont surtout répandus chez les végétaux.9 Certains d’entre eux sont représentés dans la Figure 3.10 OH COOH OH OH COOH 4 8(17), 12, 14-Labdatrien-19-ol (12E)-forme C20H32O Agathis Australis 5
Acide 7-Hydroxy-8(17), 12, 14-Labdatrien-19-oïque (7alpha, 12E)-forme C20H30O3 6 8(17), 12, 14-Labdatrien-7-ol (7alpha,12E)-forme C20H32O Nicotiana raimondii 7 Acide 8(17), 12, 14-Labdatrien-19-oïque (acide communique) (12E)-forme C20H30O2
Jupinerus, Cupressus, Pinus 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 20 19 18 17 16
Figure 3 : Exemples de Labdanes
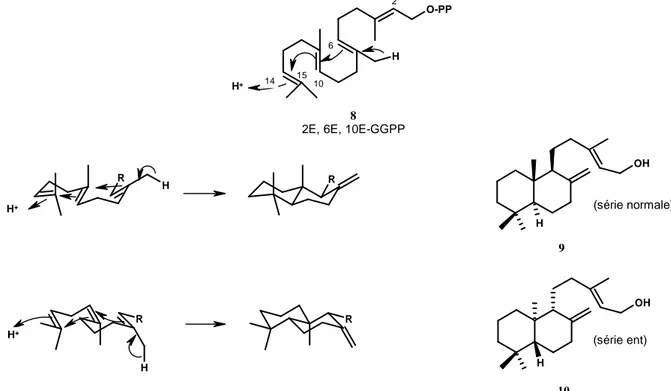
La cyclisation acido-catalysée du GGPP forme un décahydronaphthalène substitué. Ce type de cyclisation conduit à deux séries énantiomères, différant par les configurations opposées des carbones C5, C9 et C10. La série est dite « normale » lorsque la fusion des cycles A et B est identique à celle des
stéroïdes et « ent » (énantio) lorsque c’est l’antipode (Schéma 2). L’orientation vers l’une ou l’autre série est gouvernée par la conformation du précurseur linéaire (GGPP) sur la surface de l’enzyme
C14-C15 du précurseur et addition 1,2-antiparallèle des liaisons C6-C7 et C10-C11 pour former, dans les
deux cas, une transdécaline.
O-PP H R R H OH H R R H OH H H+ 2 6 14 15 10 8
2E, 6E, 10E-GGPP
H+ H+ (série normale) (série ent) 9 10
Schéma 2 : Cyclisation du géranyl-PP : formation de labdane et ent-labdane
Certains labdanes ont des propriétés pharmacologiques intéressantes telle qu’une activité cytotoxique,11 anti-inflammatoire et analgésique.12
II.1.b. Un labdane particulier : l’Acide Communique
L’acide communique 11 (Figure 4) est aussi connu sous le nom de acide 8(17),12,14-labdatrien-19-oïque (forme E).
COOH 11
Figure 4 : Acide communique
L’acide communique a été isolé de l’écorce de cinq espèces de Jupinerus,13 à 16 et de l’écorce du Cupressus arizonica.17 Il est également présent dans les baies de genevrier, mais aussi, entre autres, dans les cônes de cyprès de Provence (Cupressus sempervirens),18 la résine d’Agathis robusta,19 les
feuilles de cyprès de l’Himalaya (Cupressus torulosa)20 ou encore les cônes de pin de Luchu au Japon (Pinus luchuensis).21
L’isolement de l’acide communique nécessite les précautions suivantes : les manipulations doivent être effectuées très rapidement afin de laisser le composé le moins longtemps possible à l’état non estérifié.18 En effet, si l’acide communique pur semble stable,16 les fractions acides extraites des cônes ne se conservent guère plus d’une journée et l’acide se polymérise facilement.13 L’ester méthylique dérivé (communate de méthyle 12) lui est donc préféré car il est plus stable. La réaction d’estérification s’effectue toujours par utilisation du diazométhane.15-19
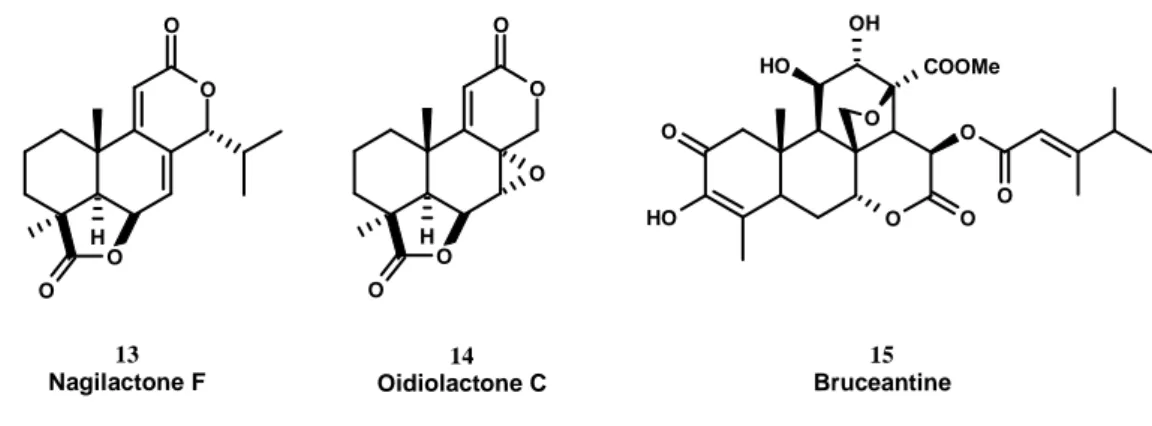
L’acide communique est aussi une molécule de choix comme point de départ de synthèse de composés biologiquement actifs comme l’oidiolactone C 13 (antifongique),22 la nagilactone F 14 (potentiel allelopathique)23 ou la brucéantine 15 (quassinoïde pentacyclique, anti-tumoral)24,25 (Figure 5). O O O O H O O O O O H HO O O O H OH COOMe O O O O 13 Nagilactone F 14 Oidiolactone C 15 Bruceantine
Figure 5 : Exemples de composés biologiquement actifs synthétisés à partir de l’acide communique
II.2. Les Flavonoïdes26
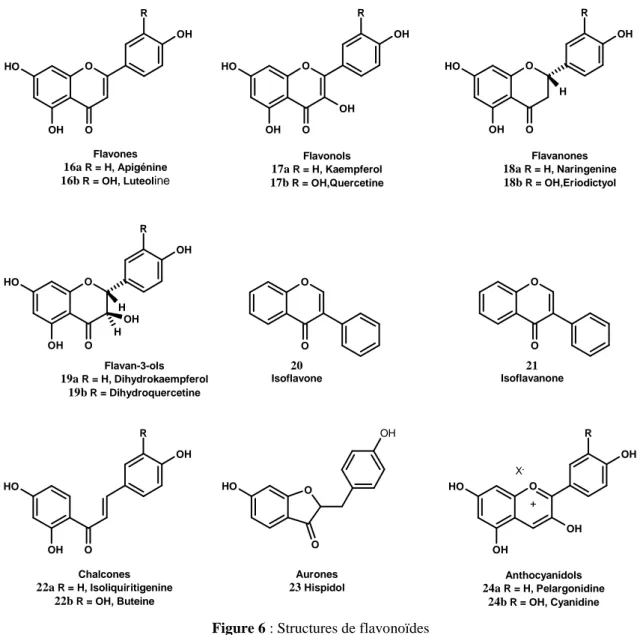
II.2.a. Généralités
Les flavonoïdes sont des pigments quasiment universels des végétaux. Ils sont responsables de la coloration des fleurs, des fruits et parfois des feuilles. Structuralement, les flavonoïdes se répartissent en quinze familles de composés, dont les plus importantes sont les suivantes : flavones, flavonols, flavanones, flavononols, isoflavones, isoflavanones, chalcones, aurones et anthocyanes (Figure 6). Ces diverses substances se rencontrent à la fois sous la forme libre ou sous la forme de glycosides, et plus de 6500 d’entre elles ont été décrites récemment.27 D’une manière très générale, les flavonoïdes se trouvent parmi toutes les plantes vasculaires, où ils peuvent être localisés dans les divers organes : racines, tiges, bois, feuilles, fleurs et fruits, bien qu’un type particulier de flavonoïdes ait en général une compartimentation définie (anthocyanes dans les fleurs).
O O O H OH R OH O O O H OH R OH OH O O O H OH R OH H O O O H OH R OH H OH H O O O O O O H OH R OH O H O O OH O O H OH R OH OH Flavones 16a R = H, Apigénine
16b R = OH, Luteoline
Flavonols 17a R = H, Kaempferol 17b R = OH,Quercetine Flavanones 18a R = H, Naringenine 18b R = OH,Eriodictyol Flavan-3-ols 19a R = H, Dihydrokaempferol 19b R = Dihydroquercetine 20 Isoflavone 21 Isoflavanone Chalcones 22a R = H, Isoliquiritigenine 22b R = OH, Buteine Aurones 23 Hispidol X -+ Anthocyanidols 24a R = H, Pelargonidine 24b R = OH, Cyanidine
Figure 6 : Structures de flavonoïdes
L’une des nombreuses fonctions des flavonoïdes est de contribuer à la couleur des plantes, et notamment à celle des fleurs. Par ailleurs, les flavonoïdes présentent des propriétés intéressantes dans le contrôle de la croissance et du développement des plantes en interagissant de manière complexe avec les diverses hormones végétales de croissance. Certains d’entre eux jouent également un rôle de phytoalexines, c’est-à-dire de métabolites que la plante synthétise en grande quantité pour lutter contre une infection causée par des champignons ou par des bactéries, ou pour provoquer une symbiose.
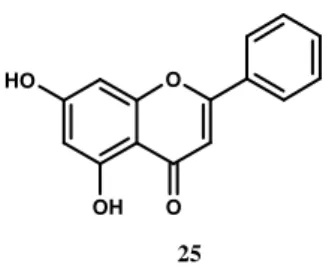
En complément du rôle joué par les flavonoïdes dans la vie des plantes (rôle dont tous les aspects sont loin d’être élucidés), il faut également signaler certaines autres de leurs propriétés. Par exemple, la chrysine 25 (extraite de la passiflore ou fleur de la passion, Passiflora coerulea, Figure 7) possède à elle seule les activités biologiques suivantes : anti-oxydant,28 anti-allergique,29 anti-inflammatoire30 (disponible commercialement, médecine des plantes), anti-cancéreux,31
antioestrogénique32 (disponible commercialement : empêche l’aromatisation de la testostérone en oestrogène, utilisée essentiellement par les culturistes pour augmenter la masse musculaire) et anxiolytique33 (les Indiens du Pérou et du Brésil avaient montré les propriétés sédatives de la fleur de la passion aux conquistadors).
O O O H OH 25
Figure 7 : Structure de la chrysine
D’autres flavonoïdes ont une activité anti-HIV-134 ou encore sont des anti-fongiques.35
Dans le spectre UV de flavones et de flavonols, il existe deux pics d’absorption majoritaires dans la région 240-400 nm. Ces deux pics correspondent à la bande I (généralement à 300-380 nm) et à la bande II (généralement à 240-280 nm). La bande I concerne l’absorption du cycle B, système cinnamoyle et la bande II concerne l’absorption du cycle A, système benzoyle (Figure 8).36
O
O
A
B
Figure 8 : Cycles A et B de flavone
II.2.b. Biosynthèse des Flavonoïdes29,37
Robinson38 remarqua que les différents systèmes d’hydroxylation des deux cycles aromatiques des flavonoïdes témoignent d’origines biogénétiques divergentes. Cependant, la principale voie de biosynthèse de cette classe de composés a été correctement identifiée par Birch.39
L’acide aminé phénylalanine 26 est la base du cycle B des flavonoïdes et est issu de l’acide shikimique (Schéma 3).40 Le cycle A est formé lors de la synthèse de la 4-2’,4’,6’-tétrahydoxychalcone 30 par la condensation graduelle de trois molécules de malonyl-CoA.41,42 Par l’action d’enzymes, cette chalcone de couleur jaune est métabolisée en différentes classes de flavonoïdes : flavanones 18a et 18b (qui conduisent à la biosynthèse des flavones), dihydroflavanols
Des étapes ultérieures, surtout de glycosylation et d’acylation, amènent les flavonoïdes à la forme définitive dans laquelle ils se trouvent in vivo.
O O O H OH OH OH OH O O O H OH OH H O O O H OH OH OH H OH H O O H OH OH OH O O O H OH OH H OH O O O H OH OH H OH H O O O H OH OH OH OH COCoA-S OH CO2H CO2H CO2H NH2 18a 18b 19b 19a 17b 17a 30 26 27 28 29 PAL C4H 4-CL CHS CHI F3'H F3H F3H F3'H F3'H FLS FLS FNS FNS DFR DFR Synthèse Flavones Synthèse Flavones Synthèse Anthocyanines Synthèse Anthocyanines Flavanones Dihydroflavonols Flavonols
PAL = Phenylalanine ammonia lyase C4H = Cinnamate-4-hydroxylase 4-CL = 4-coumarate-CoA ligase CHS = Chalcone synthase CHI = Chalcone isomérase F3'H = Flavonoïde 3'-hydroxylase FNS = Flavone synthase F3H = Flavanone 3-hydroxylase DFR = Dihydroflavonol 4-réductase FLS = Flavonol synthase
Schéma 3 : Biosynthèse des flavonoïdes
Ces idées ont été prouvées expérimentalement. Les équipes de Watkin et al.43 et Geissman et Swain44 ont montré que la quercétine 17b était formée dans le sarrasin à partir du [1-14C]- et [2-14 C]-acétate et [U-14C]phenylalanine 26, et que cette distribution des marqueurs isotopiques était compatible avec l’hypothèse de Birch. De plus, l’acide shikimique, l’acide cinnamique 27 et l’acide p-hydroxycinnamique 28 sont aussi incorporés. La même année, Grisebach45 prouva que le cycle A de la cyanidine 24b dérivait de l’acétate, et le cycle B de la phénylalanine.
Durant les quarante dernières années, beaucoup d’efforts ont été fournis pour prouver les interconnexions au sein même du vaste groupe des flavonoïdes, incluant les anthocyanines. Parallèlement, d’immenses progrès ont été faits dans la purification et la caractérisation des enzymes responsables des étapes biosynthétiques individuelles et leurs facteurs génétiques déterminants.42
P
P
A
A
R
R
T
T
I
I
E
E
B
B
M
M
O
O
D
D
E
E
L
L
E
E
S
S
d
d
e
e
R
R
E
E
A
A
C
C
T
T
I
I
O
O
N
N
d
d
e
e
D
D
I
I
E
E
L
L
S
S
-
-
A
A
L
L
D
D
E
E
R
R
La molécule cible 32 (de structure voisine à celle de 1) à synthétiser possède une structure dans laquelle le labdane se cyclise avec la quinoflavone au travers d’un cycle à six chaînons. La formation de ce nouvel archétype structural est proposée via la réaction de Diels-Alder (Schéma 4) entre la quinoflavone 31 et le communate de méthyle 12. Nous nous sommes donc intéressés à la faisabilité de cette réaction en étudiant des modèles.
O O O O O O O O O O O O O O O O O O O O H OH O O O O O OH Réaction de Diels-Alder 12 31 32 1 Schéma 4
Ainsi, nous allons choisir un modèle pour la quinoflavone et un modèle pour le diterpène. Les réactions « test » seront donc les suivantes :
+
+
+
Dans un premier temps, nous allons nous intéresser au choix et à la synthèse des modèles, puis nous décrirons l’obtention des deux fragments indispensables de la molécule cible, à savoir la quinoflavone et le communate de méthyle. Enfin, nous étudierons la réaction de Diels-Alder entre ces modèles. Modèle Quinoflavone Modèle Diterpène Modèle Quinoflavone Diterpène Modèle Diterpène Quinoflavone Modèle Quinoflavone Modèle Diterpène Modèle Quinoflavone Diterpène Modèle Diterpène Quinoflavone
I. LES MODELES : CHOIX et SYNTHESE
I.1. Modèle pour la quinoflavone
I.1.a. Choix du modèle
La réaction de Diels-Alder ayant lieu sur la partie quinone de la quinoflavone 31, notre attention s’est portée sur les substituants de la quinone. Le groupe méthoxy, de par son effet mésomère donneur, enrichit en électrons la double liaison carbone-carbone qui le porte. Par conséquent, cette double liaison est rendue moins réactive vis à vis d’un diène. Le groupe méthoxy oriente donc la régiosélectivité de la réaction et est, par conséquent, indispensable pour notre modèle.
La partie chromone de la quinoflavone peut être identifiée à une énone. Ce groupe-ci a un effet mésomère attracteur sur la double liaison carbone-carbone qui le porte. Celle-ci est donc appauvrie en électrons et donc rendue plus réactive vis à vis d’un diène. Nous pouvons donc remplacer le fragment chromone de la quinoflavone par un groupe électro-attracteur. Nous avons opté pour le groupe ester pour des raisons de facilité de synthèse.
Ainsi, le modèle pour la quinoflavone est la 2-méthoxy-5-carbométhoxy-1,4-benzoquinone 33, que nous appellerons par abus de langage « quinone simple » (Figure 9) :
O O O O O 33 Figure 9
I.1.b. Synthèse du modèle
La synthèse de la quinone simple est décrite dans la littérature.46 Elle est obtenue en deux étapes à partir de l’acide 2,4,5-triméthoxybenzoïque 34 disponible commercialement.
L’acide est estérifié par du sulfate de méthyle, en présence de carbonate de potassium, au reflux de l’acétone (Schéma 5). L’ester attendu 35 est obtenu avec un rendement de 80%.
O O O OH O O O O O O Me2SO4 K2CO3 Acétone, reflux 80% 34 35 Schéma 5
La déméthylation oxydante de l’ester 35 est effectuée en présence d’oxyde d’argent(II), d’acide nitrique 6N, dans l’acétone à température ambiante et conduit à la benzoquinone 33 désirée avec un rendement en produit brut de 97% (méthode de Snyder-Rapoport, Schéma 6).47
O O O O O O O O O O 35 33 AgO HNO3 6N Acétone 97% Schéma 6
Il est rapporté que la quinone 33 est instable46 : nous l’avons également observé expérimentalement ; elle n’est donc pas purifiée et est utilisée immédiatement.
Le rendement global pour la synthèse de cette benzoquinone est de 78%.
I.2. Modèle pour le diterpène
I.2.a. Choix du modèle
Concernant la partie labdane, la conservation du système diénique avec le méthyle nous paraît indispensable (Figure 10) :
R
Figure 10
Il se trouve que l’équipe de Desai a rapporté une méthode de synthèse de diènes-1,3 substitués en 1,2.48 La réaction a lieu entre le 3-méthyl-3-sulfolène 36 disponible commercialement et un halogénure d’alkyle, en présence de bis(triméthylsilyl)amidure de lithium dans le tétrahydrofurane à – 90°C sous atmosphère inerte (Schéma 7).
S O O S O O R R X 36 LiHMDS THF, argon, -90°C
+
Schéma 7Le diène désiré s’obtient ensuite par désulfonylation du sulfolène au reflux de la pyridine (Schéma 8).
S O O R R Pyridine, reflux Schéma 8
I.2.a.i. Choix initial
•••• Discussion
Dans le cas du communate de méthyle 12, la partie diénique est reliée au système bicyclique de type décaline qui n’a pas d’effet électronique sur celle-ci. N’ayant pas trouvé de décaline substituée en position 2 disponible commercialement, notre choix s’est porté sur le fragment suivant (Figure 11) :
Figure 11
La molécule apportant ce motif est l’acide (1,2,3,4-tétrahydro-naphthalèn-2-yl)-2-carboxylique 37 (Figure 12) :
OH O
37
Figure 12
Afin d’effectuer l’alkylation du 3-méthyl-3-sulfolène, il nous faut transformer l’acide en composé portant un groupe nucléofuge. Le schéma réactionnel 9 est envisagé.
OH O O O OH O S O O 37 38 39 40 Schéma 9
L’acide 37 est estérifié en ester 38, puis réduit en alcool 39 qui est ensuite transformé en mésylate 40.
•••• Synthèse du diène modèle à partir de l’acide 37
L’acide (1,2,3,4-tétrahydro-naphthalen-2-yl)-2-carboxylique 37 est estérifié par du sulfate de méthyle en présence de carbonate de potassium, au reflux de l’acétone (Schéma 10).46
O O OH O 37 38 Me2SO4 K2CO3 Acétone, reflux Quantitatif Schéma 10
Le spectre RMN du produit brut étant propre, l’ester 38 est alors utilisé sans purification. Il est réduit en alcool primaire 39 par utilisation de l’hydrure d’aluminium et de lithium dans l’éther éthylique sous atmophère d’argon à température ambiante (Schéma 11).
O O OH 38 39 LiAlH4 Et2O, argon 84%
(sur deux étapes)
Schéma 11
Le dérivé attendu 39 est obtenu avec un rendement de 84% sur les deux étapes.
Cet alcool est ensuite transformé en mésylate en présence de chlorure de mésyle, de triéthylamine dans du tétrahydrofurane anhydre sous atmosphère d’argon à température ambiante (Schéma 12). OH O S O O 39 40 MsCl NEt3 THF anhydre argon Schéma 12
Après évaporation du solvant, le spectre RMN du produit brut indique que le mésylate s’est bien formé.
Ce mésylate 40 est ensuite directement utilisé dans la réaction d’alkylation du 3-méthyl-3-sulfolène 36 en présence de bis(triméthylsilyl)amidure de lithium 1M dans le tétrahydrofurane anhydre sous atmosphère d’argon à –90°C (Schéma 13).
S O O S O O O S O O 40 LiHMDS 1M THF THF anhydre argon,, -90°C
+
36 41X
Schéma 13Malheureusement, après traitement et purification, il s’avère que le produit récupéré n’est autre que le mésylate de départ. D’autres essais dans les mêmes conditions ont été tentés en vain. Ces échecs semblent être dus au manque de réactivité du fragment (1,2,3,4-tétrahydro-naphthalen-2-yl).
I.2.a.ii. Choix final
•••• Discussion
Nous nous sommes donc tournés vers le fragment benzyle plus réactif et dont la synthèse est décrite.48 Vu l’éloignement du cycle aromatique, il est légitime de penser qu’il n’aura que peu d’influence sur le système diénique et pourra constituer un modèle correct pour le communate de méthyle 12.
Le diène modèle est donc le 3-méthyl-5-phényl-1,3-butadiène 42 (Figure 13).
42
Figure 13
•••• Synthèse du diène 42
L’alkylation du sulfolène 36 avec le bromure de benzyle 43 est effectuée en présence de bis(triméthylsilyl)amidure de lithium à 1M dans le tétrahydrofurane anhydre à –90°C sous atmosphère d’argon. Le 2-benzyl-3-méthyl-3-sulfolène est obtenu avec un rendement de 65% (Schéma 14).
S O O Br S O O 43 36 44
+
LiHMDS 1M THFTHF, -90°C 65% Schéma 14La désulfonylation du composé 44 est conduite au refllux de la pyridine pour fournir le 3-méthyl-5-phényl-1,3-butadiène 42 avec un rendement de 56% (Schéma 15).
S O O 42 44 Pyridine, reflux 56% Schéma 15
Le rendement global de cette synthèse est donc de 36%.
Les modèles ayant été choisis et synthétisés, nous allons maintenant voir comment obtenir la quinoflavone 32 et le communate de méthyle 12.
II. LA QUINOFLAVONE et le COMMUNATE DE METHYLE :
SYNTHESE
II.1. La quinoflavone
II.1.a. Rappels sur les méthodes de synthèse des flavones
Bien qu’il existe de nombreuses méthodes de synthèse de flavones,49 la plupart d’entre elles souffre de conditions rudes, d’une pauvre tolérance aux substituants et de rendements faibles. L’importance croissante des nombreuses activités biologiques des flavones entraîne donc un réel intérêt à trouver de nouvelles approches de synthèse.50
Nous nous attarderons sur trois méthodes que nous estimons intéressantes : la méthode historique de synthèse des flavones, une méthode du type condensation de Claisen et enfin une méthode récente utilisant les micro-ondes.
II.1.a.i. Méthode de Robinson-Venkataraman
Historiquement la première méthode pour synthétiser une flavone fut mise au point par Robinson49a et Venkataraman.49b La 2’-hydroxyacétophénone 45 est estérifiée par un chlorure de benzoyle dans la pyridine (Schéma 16).
OH O R1 R2 R3 R4 Cl O R5 R6 R7 O O R1 R2 R3 R4 O R5 R6 R7
+
Pyridine 45 46 47 Schéma 16S’en suit un réarrangement de Baker51-Venkataraman52 de l’ester 47 résultant en une dicétone
O O R1 R2 R3 R4 O R5 R6 R7 OH O R1 R2 R3 R4 O R6 R7 R5 Pyridine, KOH 47 48 Schéma 17
Une cyclisation dans des conditions acides (acide sulfurique) de 48 conduit à la formation de la flavone 49 (Schéma 18). OH O R1 R2 R3 R4 O R6 R7 R5 O O R1 R2 R3 R4 R7 R6 R5 48 H2SO4 49 Schéma 18
II.1.a.ii. Méthode de Cushman et Nagarathnam49h
Un di-anion est formé à partir de la 2’-hydroxyacétophénone 45 en présence de bis(triméthylsilyl)amidure de lithium dans du tétrahydrofurane anhydre à –78°C sous atmosphère d’argon. Le di-anion réagit ensuite avec le chlorure de benzoyle 50 pour donner après traitement la dicétone 48 (Schéma 19). OH O R1 R2 R3 R4 Cl O R5 R6 R7 OH O R1 R2 R3 R4 O R6 R7 R5 48
+
LiHMDSTHF argon, -78°C 45 50 Schéma 19La flavone est ensuite obtenue par cyclisation de la dicétone en milieu acide (Schéma 18). Cette méthode permet de gagner une étape de synthèse par rapport à la précédente. Par ailleurs, le chlorure de benzoyle 50 peut être remplacé par l’ester méthylique correspondant.53
II.1.a.iii. Méthode utilisant les micro-ondes
Enfin, il existe une méthode intéressante, utilisant les micro-ondes et s’effectuant sans solvant, ce qui est économiquement et écologiquement favorable.49n Le phloroglucinol 51 et un β-cétoester 52 sont mis sous irradiation de micro-ondes à 240°C. Après traitement, une dihydroxyflavone 53 est obtenue (Schéma 20). O R1 R2 R3 R4 O O OH O H OH O O OH O H R4 R3 R2 R1 51
+
µW 52 53 Schéma 20II.1.b. Synthèse de la flavone
II.1.b.i. Choix de la méthode
Etant donné que pour la synthèse de la quinone simple nous sommes partis de l’acide 2,4,5-triméthoxybenzoïque (transformable en ester ou en chlorure d’acyle), nous avons donc décidé d’opter soit pour la Méthode de Robinson-Venkataraman, soit pour la méthode de Cushman et Nagarathnam. Cette dernière nous a séduits d’une part parce qu’elle ne nécessite que deux étapes et d’autre part, elle peut se faire directement à partir de l’ester 35 que nous avons synthétisé précédemment (gain de temps). Nous avons donc cherché une 2’-hydroxyacétophénone substituée si possible en 4’ et 6’ par des groupements hydroxy. Elle est disponible commercialement, mais pour des raisons de coût, nous avons préféré prendre la 2’-hydroxy-4’,6’-diméthoxyacétophénone 54.
II.1.b.ii. Synthèse
L’ester 35 réagit avec la cétone 54 lors d’une réaction du type condensation de Claisen en utilisant comme base le bis(triméthylsilyl)amidure de lithium à 1M dans le tétrahydrofurane anhydre à –78°C sous atmoshpère d’argon (Schéma 21).
O O O O O O O O OH O O O O O O H O O 54 35 55
+
LiHMDS 1M THFTHF, -78°C Schéma 21Le spectre RMN du produit obtenu indique la présence du composé 55 et de sa forme céto-énolique 55’ en proportion 85/15 en faveur de la forme di-cétone (Schéma 22).
O O O O O O H O O O O O O OH O H O O 55 55' Schéma 22
La dicétone 55 est immédiatement engagée dans une réaction de cyclisation en présence de 0,5% d’acide sulfurique au reflux de l’acide acétique (Schéma 23). La pentaméthoxyflavone 56 est obtenue avec un rendement de global de 83%.
O O O O O O H O O O O O O O O O 55 0.5% H2SO4 AcOH, reflux 83% (sur 2 étapes) 56 Schéma 23
II.1.c. Synthèse de la quinone
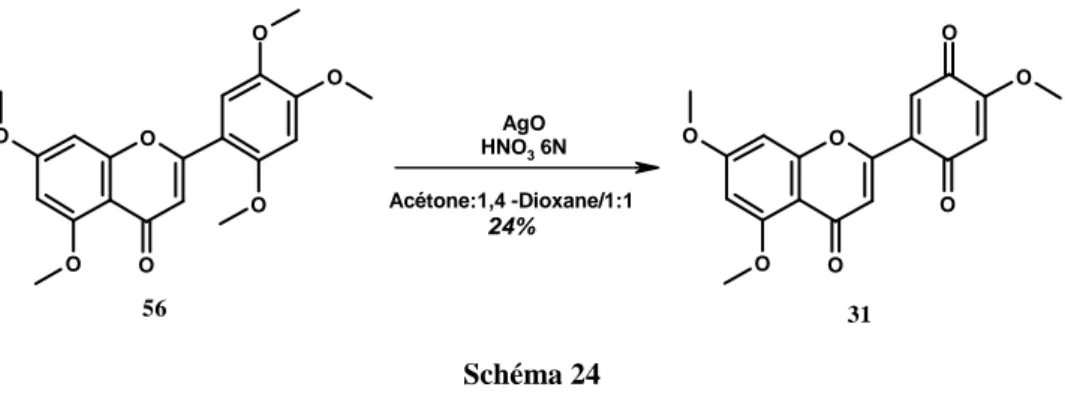
Pour transformer la pentaméthoxyflavone 56 en quinoflavone 31, nous nous sommes inspirés de la déméthylation oxydante utilisée lors de la synthèse de la benzoquinone 33.
Cette déméthylation oxydante sur le cycle B de la pentaméthoxyflavone 56 est effectuée en présence d’oxyde d’argent(II), d’acide nitrique 6N dans un mélange acétone:1,4-dioxane 1:1 (Schéma 24). La quinoflavone désirée est obtenue après purification par chromatographie sur gel de silice avec un rendement de 24%.
O O O O O O O O O O O O O O 31 56 AgO HNO3 6N Acétone:1,4 -Dioxane/1:1 24% Schéma 24
Le rendement global de cette synthèse (à partir de l’acide 2,4,5-triméthoxybenzoïque 34) est de 16%.
Dans cette dernière étape d’oxydation, un mélange acétone/dioxane est nécessaire pour une meilleure solubilité, ainsi que l’utilisation du bain à ultrasons lors de la mise en solution.
Comme nous pouvons le constater, le rendement de cette oxydation n’est pas très bon. L’essentiel de la quinoflavone est perdu lors de la phase de purification sur gel de silice. D’autres méthodes de purifation ont été tentées en vain : HPLC préparative, phase inverse (silice greffée C18),
alumine. Par ailleurs, les spectres HPLC et RMN révèlent que le produit brut n’est pas assez propre pour être utilisé tel quel.
Nous avons donc recherché d’autres méthodes de déméthylation oxydante dans le but d’augmenter le rendement de la dernière étape de la synthèse de la quinoflavone.
D’après un examen des données de la littérature, cette oxydation peut être réalisée par utilisation de cerium (IV) ammonium nitrate (CAN),54 de dioxyde de manganèse (MnO2)-acide
nitrique,55 de fluorure de cobalt(III) (CoF3), 56
de NBS-H2SO4, 57
et de bis(trifluoroacétate) de phényliodine(III) (PIFA) et de diacétate de phényliodine(III) (PIDA).58 Nous avons tenté les méthodes utilisant le CAN, le PIFA et le PIDA pour oxyder la pentaméthoxyflavone. Les résultats des essais sont rassemblés dans le Tableau 1.
Essai Réactif (equiv.) Solvant Temps de réaction Rendement
1 CAN (3) AcCN/H2O (1/1) 24 h Pas de réaction 2 PIFA (1.5) H2O + 2.5% MeOH 24 h Pas de réaction 3 PIDA (1.5) H2O +2.5% MeOH 24h Pas de réaction 459 PIFA (1.5) AcCN/H2O (7/3) 24 h Pas de réaction 5 PIDA (1.5) AcCN/H2O (7/3) 24 h Pas de réaction
Pour l’essai 1, la flavone se dissout bien dans le mélange AcCN/H2O, mais au bout de 24 h il
n’y a toujours pas de réaction et le produit de départ est récupéré. Concernant les essais 2 et 3, la flavone se dissout mal dans le mélange H2O/MeOH ; au bout de 24 h, le produit de départ est récupéré.
La quantité de méthanol ne peut être augmentée (pour améliorer la solubilité) car celui-ci risque de désactiver le PIFA/PIDA. C’est pourquoi les essais 4 et 5 ont été tentés ; le milieu est bien homogène et dans les deux cas, au bout de 24 h, la flavone de départ est récupérée.
Compte tenu de ces échecs, nous n’avons pas tenté d’autres méthodes et avons gardé la méthode de Snyder-Rapoport (utilisation d’oxyde d’argent(II) et d’acide nitrique 6N). En effet, bien que conduisant à un faible rendement, cette méthode de préparation de la quinoflavone 31 s’est avérée très reproductible.
II.2. Le communate de méthyle
Le communate de méthyle 12 est issu de l’estérification de l’acide communique 11. Celui-ci est présent dans de nombreuses plantes communes.
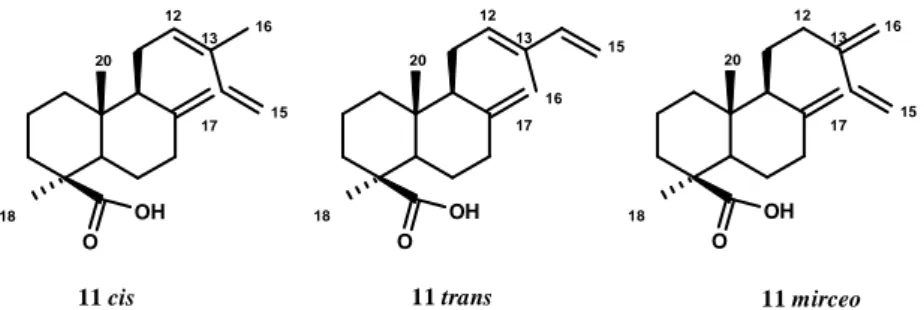
II.2.a. Choix de la plante pour l’extraction de l’acide communique
Une première méthode a consisté à extraire l’acide communique à partir de baies de genevrier (Jupinerus communis). Cependant, un mélange de trois isomères de l’acide est obtenu : l’isomère cis, l’isomère trans et l’isomère mirceo (Figure 14). Ces trois isomères sont difficilement séparables par chromatographie éclair sur gel de silice simple (même après estérification de l’acide communique). Nous nous sommes donc intéressés à d’autres plantes contenant l’acide communique et simples d’approvisionnement. O OH O OH O OH 11 cis 18 20 16 15 13 12 17 18 20 16 15 13 12 17 18 20 16 15 13 12 17 11 trans 11 mirceo
Figure 14 : Différents isomères de l’acide communique
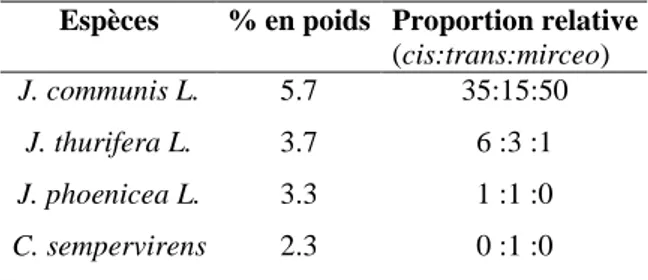
Une publication de l’équipe de A.F. Barrero a étudié la proportion de chacun des isomères de l’acide communique dans les fruits de Jupinerus communis, Juniperus thurifera, Jupinerus phoenicea
Espèces % en poids Proportion relative (cis:trans:mirceo) J. communis L. 5.7 35:15:50 J. thurifera L. 3.7 6 :3 :1 J. phoenicea L. 3.3 1 :1 :0 C. sempervirens 2.3 0 :1 :0
Tableau 2 : Distribution des acides communiques dans les fruits de différents conifères
Certes les baies de genevrier sont les plus riches en acide communique (5.7% en poids), mais elles contiennent les trois isomères, avec majoritairement l’isomère mirceo qui ne nous intéresse pas. Pour J. thurifera, nous retrouvons les trois isomères dont majoritairement l’isomère cis. J. phoenicea ne contient que les isomères cis et trans en proportions égales. Néanmoins, ce sont les cônes de cyprès qui sont les fruits les plus intéressants (malgré leur plus faible proportion en poids en acide communique) car seul l’isomère trans de l’acide communique y est présent.
Une petite quantité de cônes de cyprès broyés étant déjà disponible au CRSN, nous avons donc opté pour Cupressus sempervirens pour obtenir facilement et rapidement l’acide
trans_communique. Par ailleurs, l’approvisionnement de cette plante est aisé pour le laboratoire.
II.2.b. Synthèse du communate de méthyle
L’hexane de l’extrait hexanique des cônes de cyprès broyés est évaporé et une huile verte est obtenue. Après traitement et extraction, la RMN du produit brut révèle la présence de l’acide trans-communique 11 et de l’acide 13β-hydroxylabda-8(17)-14 diénoïque 57 en proportion 7:3 (Figure 15). Ce composé 57 est effectivement présent dans Cupressus sempervirens.61
O OH OH O OH 57 11 Figure 15
Ce mélange est purifié par chromatographie sur gel de silice ; l’acide communique 11 est obtenu avec un rendement massique d’extraction de 2%.
L’acide 11 est ensuite estérifié en présence de (diazométhyl)triméthylsilane et de méthanol dans l’hexane à température ambiante avec un rendement de 53% (Schéma 25).
O OH O O 11 N2CHTMS 2M hexane MeOH Hexane 53% 12 Schéma 25
La stéréochimie indiquée est celle de la littérature. Nous l’avons contrôlée en effectuant un spectre RMN ROESY sur ce composé.
Le Tableau 3 indique les données ROESY de 12 (Figure 16).
Position des carbones RMN 1H δ en ppm (Mulplicité, J en Hz) ROESY 1 H/1H 1 1a 1,96 à 1,83 (m) 1b 1,14 (m) 1a/1b, 2, 1b/3b, 1b/5, 1a/20, 1b/9 2 2a 1,96 à 1,83 (m) 2b 1,54 (m) 2a/2b, 1, 3, 2a/20 3 3a 2,23 à 2,09 (m) 3b 1,06 (m) 3a/3b, 3b/1b, 2, 3b/5, 18 4 / / 5 1,34 (dd, J = 12,3 ; 2,6) 7b, 6b, 9, 18, 1b, 3b 6 6a 1,99 (m) 6b 1,82 (m) 6a/6b, 7, 6b/5, 6b/18, 6a/20 7 7a 2,42 à 2,38 (m) 7b 1,96 à 1,83 (m) 7a/7b,6, 7b/5, 7b/9, 7a/17a 8 / / 9 1,74 (m) 5, 1b 10 / / 11 11a 2,42 à 2,38 (m) 11b 2,23 à 2,09 (m) 11a/11b, 20, 12, 11b/17b 12 5,42 (dd, J = 5,2) 14, 17b 13 / / 14 6,33 (dd, J = 17,4 ; 10,7) 12, 15 15 15a 5,05 (d, J = 17,4) 15b 5,89 (d, J = 10,7) 15a/15b, 14 16 3H 1,76 (s) 12, 15a 17 17a 4,85 (s) 17b 4,47 (s) 17a/17b, 17b/12, 17b/11b 18 3H 1,20 (s) 6b, 5, 3b 19 / / 20 0,56 (s) 11, 21 21 3H : 3,63 (s) 20
O O 1 2 3 4 5 6 7 8 9 10 11 12 12 13 14 15 16 17 18 19 20 21 Figure 16
D’après le Tableau 3, les protons 20 et 21 corrèlent, cela signifie donc qu’ils sont du même côté du bicycle décaline, tout comme les protons 20 et 11. Par conséquent, les carbones 20 et 19 sont en position axiale β et le carbone 11 est en position équatoriale β.
La corrélation H5/H9 indique que ces protons sont du même côté du bicycle décaline, et
l’absence de corrélation H20/H5 entraine une jonction trans entre les deux cycles. Les protons 5 et 9
sont donc situés en position axiale α. L’effet ROE entre H18 et H5, l’absence d’effet entre H20 et H18 et
le déplacement chimique du carbone 18 (28,8 ppm) vont dans ce sens et le carbone 18 est donc en position équatoriale α.
Ainsi le composé 12 a bien la stéréochimie suivante (Figure 17) :
O O 1 2 3 4 5 6 7 8 9 10 11 12 12 13 14 15 16 17 18 19 20 21 H CO2Me 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 Figure 17
Nous allons à présent passer aux réactions de Diels-Alder entre modèles et entre fragments indispensables de la molécule cible.
III. REACTIONS de DIELS-ALDER entre MODELES
III.1. Rappels sur la réaction de Diels-Alder
III.1.a. Généralités
Cette réaction de cycloaddition [4+2] révolutionnaire a été découverte en 1928 par le Professeur Otto Diels et son élève Kurt Alder en identifiant parfaitement les produits 60 et 61 issus de la réaction entre le cyclopentadiène 58 et la benzoquinone 59 (Schéma 26).62
O O O O O O H H O O HH O O H H H H
+
endo 60 monoadduit Diels-Alder endo Diels-Alder 61 diadduit 58 59Schéma 26 : Mécanisme de la réaction de Diels-Alder
L’importance et la puissance de la réaction de Diels-Alder en synthèse ont vraiment commencé dans les années 50-60 avec une élégante utilisation de celle-ci dans les synthèses totales de produits naturels complexes.63 Le développement de la réaction de Diels-Alder continua et s’étoffa. De nombreuses versions différentes de la réaction de Diels-Alder ont été élaborées, incluant les cycloadditions intramoléculaires [4+2], les réactions d’hétéro-Alder, les réactions de Diels-Alder sous pression et les réactions de Diels-Diels-Alder catalysées par les acides de Lewis.64 Son intérêt est principalement dû à sa versatilité et à sa haute régio et stéréosélectivité. Elle conduit à la formation d’une large variété de composés carbocyclique et hétérocyclique à six chainons.
La réaction de Diels-Alder (inter et intramoléculaire) est une étape importante dans la synthèse de nombreux produits naturels. Les réactions intermoléculaires sont généralement faites au début des synthèses et les stéréocentres alors formés lors de la cycloaddition sont utilisés pour contrôler