HAL Id: dumas-01863495
https://dumas.ccsd.cnrs.fr/dumas-01863495
Submitted on 28 Aug 2018
HAL is a multi-disciplinary open access
archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
La sclérose en plaques : histoire, physiopathologie et
thérapeutiques actuelles
Nadime Hoballah
To cite this version:
Nadime Hoballah. La sclérose en plaques : histoire, physiopathologie et thérapeutiques actuelles. Sciences pharmaceutiques. 2018. �dumas-01863495�
AVERTISSEMENT
Ce document est le fruit d'un long travail approuvé par le
jury de soutenance et mis à disposition de l'ensemble de la
communauté universitaire élargie.
Il n’a pas été réévalué depuis la date de soutenance.
Il est soumis à la propriété intellectuelle de l'auteur. Ceci
implique une obligation de citation et de référencement
lors de l’utilisation de ce document.
D’autre part, toute contrefaçon, plagiat, reproduction illicite
encourt une poursuite pénale.
Contact au SID de Grenoble :
bump-theses@univ-grenoble-alpes.fr
LIENS
LIENS
Code de la Propriété Intellectuelle. articles L 122. 4
Code de la Propriété Intellectuelle. articles L 335.2- L 335.10
http://www.cfcopies.com/juridique/droit-auteur
1
UNIVERSITÉ GRENOBLE ALPES
UFR DE PHARMACIE DE GRENOBLE
Année : 2018
LA SCLEROSE EN PLAQUES : HISTOIRE, PHYSIOPATHOLOGIE ET
THERAPEUTIQUES ACTUELLES
THÈSE
PRÉSENTÉE POUR L’OBTENTION DU TITRE DE DOCTEUR EN PHARMACIE
DIPLÔME D’ÉTAT
Nadime HOBALLAH
.
THÈSE SOUTENUE PUBLIQUEMENT À LA FACULTÉ DE PHARMACIE DE
GRENOBLE
Le : 27/08/2018
DEVANT LE JURY COMPOSÉ DE
Président du jury :
M. Christophe Ribuot, Professeur des universités (PU)
Membres :
M. Christian Drouet (directeur de thèse), Professeur des universités et Praticien
Hospitalier (PU-PH)
Mme. Delphine Morin Aldebert, Maître de conférences des universités (MCU)
M. Marc Hommel, Professeur des universités et Praticien Hospitalier (PU-PH)
L’UFR de Pharmacie de Grenoble n’entend donner aucune approbation ni improbation aux opinions émises dans les thèses ; ces opinions sont considérées comme propres à leurs auteurs.
2
5
Remerciements
Au Professeur Christian Drouet, un grand merci pour votre aide durant la rédaction de cette thèse
et l’intérêt que vous avez porté à l’élaboration de ce manuscrit.
Au Professeur Christophe Ribuot merci d’avoir accepté de présider mon jury, j’en suis très
honoré.
Au Professeur Delphine Aldebert, je vous remercie d’avoir accepté de faire partie de mon jury et
de m’avoir conseillé durant mes études.
Au Professeur Marc Hommel, je vous remercie d’avoir accepté de faire partie de mon jury et
d’avoir ajouté votre expertise médicale.
Je voudrais remercier également mon père et ma mère qui m’ont toujours conseillé et soutenu dans mes choix de parcours et dans les moments difficiles.
Merci à mon frère Rayane qui m’a supporté et guidé durant mes P1 et à ma sœur Mayla qui durant deux ans m’a montré le chemin du travail et de la persévérance.
Tous mes amis de Grenoble toutes filières confondues, qui m’ont suivi durant ces belles années, avec qui j’ai partagé des moments inoubliables. Spéciale dédicace à Dousson, Agathe, Pierre et Yann.
Un grand merci à tous ceux qui ont croisé mon chemin et qui ont apporté leur graine d’une manière ou d’une autre aux choix que j’ai pu entreprendre au cours de ces dernières années.
6
Table des Matières
Liste des enseignants ... 2
Remerciements ... 5
Liste des abréviations ... 10
Liste des figures ... 14
Introduction ... 16
Première Partie : La maladie de la Sclérose en Plaques ... 17
I. Histoire... 18
I.1 Les premières descriptions ... 18
I.2 JM Charcot et la méthode anatomoclinique ... 20
I.3 Le XXème siècle ... 21
I.4 The Decade of the brain (1990-2000) ... 22
II. Epidémiologie et histoire naturelle ... 23
II.1 La prévalence en France ... 24
II.2 L’incidence en France ... 25
III. Étiologie ... 26
III.1 Facteurs génétiques ... 26
III.1.1. Rôle de l’immunogénétique HLA ... 27
III.1.2. Les régions non HLA ... 28
III.1.3. Les facteurs infectieux ... 29
III.2 Facteurs environnementaux ... 31
III.2.1. Hygiène ... 31
III.2.2. Tabac ... 32
III.2.3. Vitamine D ... 33
III.2.4. Migration ... 35
III.2.5. Alimentation ... 35
III.2.6. Obésité et régime ... 36
III.2.7. Vaccin HBV ... 38
IV. Physiopathologie ... 39
IV.1. Anatomopathologie : les plaques de démyélinisation ... 40
IV.2. Aspects cliniques... 42
IV.2.1. Phénotypes cliniques de la SEP ... 42
7
IV.2.3. Le syndrome radiologiquement isolé ... 43
IV.2.4. L'échelle Expanded Disability Status Scale (EDSS) ... 45
IV.3 Désordres auto-immuns ... 46
IV.3.1 Les lymphocytes T (LT) ... 46
IV.3.2 Implication des LT CD4+ helper Th1 et Th17 ... 47
IV.3.3 Les lymphocytes CD8+ ... 48
IV.3.4 Les Lymphocytes T régulateurs ... 48
IV.3.5 Les lymphocytes B ... 49
IV.3.6 Dérégulation périphérique ... 51
IV.3.7 La Barriere Hémato-Encéphalique (BHE) ... 52
IV.3.8 Mécanisme lésionnels au sein du système nerveux central ... 54
V. Diagnostic ... 55
V.1 Critères cliniques ... 55
V.2 L’Imagerie par résonnance magnétique ... 55
V.3 Etude du liquide céphalo-rachidien (LCR) ... 59
V.4 Potentiels évoqués ... 59
VI. Suivi clinique ... 60
VI.1 Phase de Début ... 61
VI.1.1 Les troubles visuels : La neuropathie optique ... 61
VI.1.2 Troubles moteurs ... 61
VI.1.3Troubles sensitifs ... 62
VI.1.4 Atteinte du tronc cérébral ... 62
VI.2 Phase d’état ... 62
VI.2.1 La fatigue ... 62
VI.2.2 Trouble vésicosphinctériens et sexuels ... 62
VI.2.3 Troubles neuropsychologiques et psychiatriques ... 63
VI.2.4 Troubles de l'humeur ... 63
VI.2.5 Troubles de la déglutition ... 63
VI.2.6 Troubles de la communication ... 63
VI.3 Cas particulier de la grossesse ... 64
VI.3.1 Effets de la grossesse sur la maladie ... 64
VI.3.2 Les options thérapeutiques ... 65
8
I. Thérapeutiques actuelles ... 68
I.1 Traitement de la poussée ... 68
I.1.1 Corticoïdes ... 68
I.1.2 Plasmaphérèse ... 69
I.2 Traitement de fond ... 70
I.2.1 Les traitements immunomodulateurs ... 71
I.2.2 Les traitements immunosuppresseurs ... 76
I.2.3 Les anticorps monoclonaux ... 81
I.2.4 Les traitements par voie orale ... 95
II. Réparation de la myéline et neuroprotection ... 112
III. Stratégies Thérapeutiques ... 113
III.1. Le switch ... 113
III.2 Escalade et Induction ... 113
IV. Traitements symptomatiques ... 114
IV.1. Traitement de la spasticité ... 114
IV.2. Traitement de la fatigue ... 115
IV.3. Traitement de la douleur ... 115
IV.4. Traitement des troubles urinaires, digestifs et sexuels ... 116
V. Prise en charge multidisciplinaire et rôle du pharmacien ... 117
V.1. Première délivrance d’un traitement de fond ... 118
V.1.1. Biothérapies injectables ... 118
V.1.2. Formes orales ... 119
V.2. Renouvellement d’un traitement de fond ... 120
V.3. Switch thérapeutique ... 121
Conclusion... ¡Error! Marcador no definido. Annexes ... 123
Annexe 1 : Détail de la cotation des paramètres fonctionnels de l'échelle EDSS ... 123
Annexe 2 : définition du score EDSS ... 126
Annexe 3 : Critères diagnostic de Mc Donald 2010... 128
Annexe 4 : Critères IRM 2010 de Dissémination spatiale et Dissémination temporelle ... 129
Annexe 5 : Historique de la maladie, les dates clés (5) ... 130
Annexe 6 : Tableau Récapitulatif des traitements actuels ... 131
9 Bibliographie ... 134 Serment de Galien ... 147 Résumé ... 148
10
Liste des abréviations
AFFSAPS : Agence Française de Sécurité Sanitaire des Produits de Santé ALAT : Alanine-Aminotransférase
ALD : Affection de Longue Durée
AMM : Autorisation de Mise sur le Marché
ANSM : Agence nationale de sécurité du médicament et des produits de santé ASAT : Aspartate aminotransférase
ATU : Autorisation Temporaire d’Utilisation BCR: B Cell Receptor
BHE: Barriere Hémato-Encephalique
CHMP: Committee for Medicinal Products for Human Use CMH: Complexe Majeur d’Histocompatibilité
CNAMTS : Caisse Nationale d’Assurance Maladie des Travailleurs Salariés CPA: Cellules présentatrices d’antigènes
CRP: C-reactive protein
11 DHO-DH: dihydro-orotate déshydrogénase
DMF : Dimethyl fumarate
EAE: Experimental autoimmune encephalomyelitis EBM : Evidence Based Medecine
EBV: Epstein - Barr virus
EDSS: Expanded Disability Status Scale EMA: European Medicines Agency FDA: Food and Drug Administration FLAIR: fluid attenuated inversion recovery GWAS: Genome Wide Association Studies HERV: Human Endogenous Retrovirus HHV-6: Human Herpes Virus 6
HLA : Human Leukocyte Antigen HSV : Herpès simplex virus
ICAM: InterCellular Adhesion Molecule IFN: Interferon
IgG Immunoglobulines de type G IM: Intra musculaire
IRM : imagerie par résonance magnétique JC: John Cunningham
LB: Lymphocytes B
LCR : Liquide Cephalo Rachidien
LEMP: Leucoencéphalopathie multifocale progressive LFA1: Lymphocyte function-associated antigen 1 LLCB: leucémie lymphoïde chronique à cellules B LT: Lymphocytes T
12 MAI : Maladies auto-immunes
MBP: Myelin basic protein MDC: Myeloid Dendritic cell MMP: Matrix metalloproteases
MOG: Myelin oligodendrocyte glycoprotein MSA : Mutualité Sociale Agricole
MTX: Methotrexate
NFS : Numérotation de formule sanguine NO: Nitric oxide
OR: Odd ratio
PLP: Proteolopid protein
PTI : Purpura thrombocytopénique immunologique RAP: Réactions liées à la perfusion
SC: Sous cutanée
SCI : Syndrome clinique isolé SEP : Sclérose en plaque
SEP-PP : Sclérose en plaques progressive primaire SEP-RR : Sclérose en plaques récurrente-rémittente SEP-SP : Sclérose en plaques secondairement progressive SNC : Système nerveux central
SNP: Single Nucleotide Polymorphisms SRI : Syndrome radiologiquement isolé TCR: T Cells Receptor
TNF: Tumor Necrosis Factor TYK: Tyrosine Kinase
13 VDR: Vitamin D Receptor
VIH : virus de l'immunodéficience humaine VLA4: very late activation antigen-4
14
Liste des figures
Figure 1 : Illustration des premières lésions mises en évidence au niveau de la moelle épinière par Cruveilhier et Carswell
Figure 2: Prévalence mondiale de SEP en 2013 Figure 3 : La prévalence de la SEP en France
Figure 4 : Risque de développer la sclérose en plaques suivant le degré de parenté avec un individu atteint
Figure 5 : Localisation de la région HLA sur le chromosome 6
Figure 6 : Incidence des maladies infectieuses et auto-immunes au cours du temps Figure 7 : Cibles de la Vitamine D dans le système immunitaire
Figure 8 : Obésité : principaux mécanismes pouvant engendrer un dysfonctionnement immunitaire Figure 9. Physiopathologie de la sclérose en plaques
Figure 10 : Les 4 différents profils de plaques
Figure11 : Évolution du handicap pour les trois formes de sclérose en plaques au cours du temps Figure 12 : Evolution différentielle de la SEP
Figure 13 : L'échelle EDSS (Expanded Disability Status Scale)
Figure 14 : Différenciation des LT CD4+ naïfs en fonction des cytokines
Figure 15 : Développement et migration des lymphocytes B : représentation de leur implication dans la sclérose en plaques
Figure 16 : Dérégulation du système immunitaire en périphérie
Figure 17 : La Barrière Hémato-Encéphalique BHE en condition physiologique Figure 18 : Passage des lymphocytes activés à travers la BHE
Figure 19 :Imagerie cérébrale dans la sclérose en plaques
Figure 20 : Imagerie médullaire dans la sclérose en plaques. IRM en séquence T2 Figure 21 : Examen classique de potentiels évoqués magnétiques
Figure 22 : Tableau clinique varié de la SEP
Figure 23 : Taux annualisé de poussées pour chaque trimestre dans l’année avant la grossesse, pendant la grossesse, et dans les deux années suivant l’accouchement
15 Figure 24 : Grossesse et traitements de fond
Figure 25 : Prise en charge de la poussée, arbre décisionnel thérapeutique Figure 26 : Arbre décisionnel pour les traitements de fond
Figure 27 : Description des 4 immunomodulateurs disponibles dans le traitement de la SEP Figure 28 : Récapitulatif des différentes études de phase III dans la forme rémittente de SEP Figure 29 : Le processus normal de la migration leucocytaire à travers la BHE
Figure 30 : Estimation du risque de LEMP chez les patients traités par natalizumab
Figure 31 : Mécanisme d’action de l’alemtuzumab en 3 étapes : Sélection-Déplétion-Repopulation Figure 32 : Présentation générale du traitement de Lemtrada®
Figure 33: Comparaison du bras ocrelizumab 600 mg versus bras IFN- β1a dans OPERA I et II Figure 34 : La sortie des lymphocytes des ganglions lymphatiques dépend du gradient de concentration de S1P [S1P]
Figure 35: Action du fingolimod sur le système immunitaire
Figure 36 : Taux annualisé de poussée au cours de l’étude FREEDOMS comparant l’efficacité du fingolimod 0.5 mg et 1.25 mg au placebo
Figure 37 : Taux annualisé de poussées au cours de l’étude TRANSFORMS comparant l’efficacité du fingolimod à l’IFN-β1a IM
Figure 38 : Mécanisme d’action du tériflunomide
Figure 39 : Diminution de 49 % du risque de présenter une poussée sur 2 ans, critère principal Figure 40 : Taux annualisé de poussées : diminution de 44 % du taux annualisé de poussée à 2 ans Figure 41 : Représentation schématique des cibles des traitements actuels et futurs
Figure 42 : Algorithme thérapeutique proposé en cas d’escalade Figure 43 : Prise en Charge Multidisciplinaire du patient
16
Introduction
La sclérose en plaque (SEP), maladie inflammatoire chronique et autoimmune du système nerveux central est la première cause de handicap non traumatique chez le sujet jeune. Aujourd’hui, elle affecte plus de 2.3 millions de personnes dans le monde, en touchant deux fois plus de femmes. Plus de 100 ans après les observations de Jean-Martin Charcot, d’autres investigateurs, tels Carswell, Cruveilhier, ont contribué à décrire les phénotypes cliniques et caractéristiques pathologiques de la sclérose en plaque, cette énigmatique maladie qui touche le système nerveux central, continue aujourd’hui à intriguer les chercheurs et acteurs de santé du monde entier.
Les chercheurs poursuivent les efforts de recherche pour apporter des explications à l’immunopathologie, tels les facteurs de déclenchement, et ouvrir à l’innovation thérapeutique des perspectives pour traiter au mieux les patients. La dernière décennie a été cruciale dans la compréhension de la physiopathologie et le développement de nouveaux traitements.
Ces thérapeutiques ont révolutionné la prise en charge de la SEP et démontré une efficacité intéressante dans la réduction de la fréquence et de l’intensité des poussées, ainsi que l’apparition de symptômes invalidants. Toutefois, elles s’associent à un certain nombre de contraintes et de surveillance des effets adverses. La dispensation de ces nouvelles thérapies, bien ciblées, très techniques et “pointues”, exige de la part du pharmacien des connaissances appropriées afin qu’il soit à même de fournir les explications et les conseils adaptés.
Fort de cette connaissance il sera un acteur précieux et assuré dans la prise en charge des patients. Dans une première partie, ce travail rassemble de manière non exhaustive des éléments historiques qui ont pu définir la maladie comme elle est connue aujourd’hui, puis une définition de la SEP sera faite en détaillant la genèse et le dysfonctionnement immunologique. Dans un deuxième temps, les thérapeutiques actuelles et la prise en charge du patient seront abordées.
17
Première Partie : La
maladie de la Sclérose en
18
I. Histoire
« On ne connait bien une science que si l’on en connait l’histoire », Auguste Comte
I.1 Les premières descriptions
A l’époque antique des grecs, romains ou égyptiens, aucune description de la sclérose en plaques n’a été retrouvée, même si Galien parlait déjà de tremblement d’action paralytique « Tremor », et de tremblement de repos clonique, convulsif « Palpitation ». La plus ancienne description évocatrice de la maladie « sclérose en plaques » (SEP) proviendrait de Scandinavie, d’une femme du peuple Viking (1293-1323) présentant des troubles de la parole, de la marche avec des périodes de récupération (1).
Les documents les plus anciens relatant des troubles cliniques pouvant correspondre à la SEP datent de 1421 et concernent la vie de Sainte Lidwina de Schiedam, née dans cette ville en 1380. A l'âge de 16 ans, cette jeune fille fait une chute en patinant et se fracture une côte. Suite à une infection locale, la guérison fut longue. Peu de temps après, Lidwina présente des difficultés de marche et des douleurs lancinantes dans la face. La maladie progresse lentement avec apparition successive d'une paralysie du bras droit, d'une perte de la vue d'un côté, d'une paralysie des jambes, de plaies au siège et de difficultés de déglutition. Lidwina décède à l'âge de 53 ans, probablement d’une pyélonéphrite et d’escarres. Cinq siècles plus tard, une analyse de son squelette retrouvera les stigmates d’une paraplégie prolongée (1,2).
Un troisième exemple ancien fut celui d’Auguste d’Este (1794-1848, Angleterre), petit-fils du roi George III. Il retrace dans des passages de son journal l’historique de ses symptômes suggérant qu’il avait été touché par la sclérose en plaques (1). Le début de sa maladie s’exprimait par une névrite optique bilatérale, avec un début par des taches noires et une diplopie, une fatigue permanente et des troubles de la sensibilité. Au fil du temps, la maladie progressa lentement avec une récupération de plus en plus compliquée. Les symptômes comme une paralysie des deux membres supérieurs, des raideurs dans les jambes et des pieds, une impuissance, des dysfonctionnements urinaires commencent à apparaitre (2). Il a récupéré la marche pendant un bref moment avant que celle-ci ne s’aggrave de nouveau (3).
Pendant qu’Auguste d’Este décrivait son observation personnelle en Angleterre, le professeur et anatomiste Jean Cruveilhier met en évidence en 1835, les premières représentations des lésions médullaires antéropostérieures, dénommées sclérose en taches ou en îles (dû aux cicatrices laissées par les lésions anciennes), complétées trois années plus tard par les illustrations du docteur Robert Carswell (1838) (figure 1).
19 Figure 1 : Illustration des premières lésions mises en évidence au niveau de la moelle épinière par
Cruveilhier et Carswell (4).
Les lésions sont à la fois présentes dans les matières blanche et grise, apparaissant sur l’image comme de petites taches brunes tirée de l’ouvrage de Robert Carswell 1838.
En 1863, Eduard Rindfleisch met en évidence une inflammation de la substance blanche et évoque la possibilité qu’elle soit responsable de la démyélinisation. Il note en effet la présence d’infiltrats inflammatoires périvasculaires au sein des plaques de sclérose, l’amenant à postuler que la maladie était de nature inflammatoire (5).
Le terme "sclérose en plaques" semble avoir été utilisé pour la première fois par Vulpian dans une présentation de trois patients devant la Société Médicale des Hôpitaux de Paris en mai 1866.
20
I.2 JM Charcot et la méthode anatomoclinique
C'est le neurologue Jean-Martin Charcot et son école neurologique de la Salpétrière à Paris qui baptiseront la maladie et la feront connaître quelques dizaines d'années plus tard (1868). Il fait la première synthèse de la pathogénicité de la maladie dans ses célèbres « Leçons sur les maladies du système nerveux », ouvrage encore disponible de nos jours. Sa description des symptômes cliniques s’inspire notamment de l’une de ses servantes qui présentait trois symptômes : une élocution mal articulée (dysarthrie), des mouvements saccadés des yeux (nystagmus), et un tremblement des bras lorsqu’elle voulait prendre un objet (tremblement intentionnel).
JM Charcot avait posé le diagnostic de syphilis de la moelle épinière, mais à l'autopsie il découvrit les "petites taches" typiques de la SEP et fit ainsi la première corrélation anatomo-clinique. Il proposa le diagnostic de SEP chez des patients qui présentaient l’association de ces trois symptômes, appelée « triade de Charcot ». Il constata rapidement que la SEP pouvait se manifester par d’autres signes, notamment sur des formes bénignes, qu’il appelait formes frustes.
D’autres pathologies ont été décrites par JM Charcot, dont la sclérose latérale amyotrophique qui porte son nom, maladie de Charcot, et les lésions articulaires non douloureuses observées dans la syphilis.
Quant à la cause de la maladie, JM Charcot admettait ne pas la connaître, mais notait l'existence de maladies aiguës, surtout infectieuses, dans les antécédents de ses patients. Il avait également remarqué que le début d'une SEP était parfois précédé d'un choc émotif, d'un chagrin ou d'une vive contrariété. Quoiqu'ayant parfois observé plusieurs cas dans une même famille, JM Charcot estimait que la SEP n'était pas héréditaire. (3,6).
21
I.3 Le XXème siècle
Au 20ème siècle, la présence de plaques dans la matière blanche du cerveau était déjà connue et fut observée également dans la matière grise par JM Charcot. JD Dowson développe la notion de préservation des axones des neurones présents aux niveaux des plaques anciennes démyélinisées et confirme aussi la notion d’inflammation péri-vasculaire.
En 1921, Hortega et Penfield mettent en évidence un nouveau type de cellule gliale chargé de produire la gaine de myéline entourant les axones, ce sont les oligodendrocytes. Des capacités de remyélinisation ont été repérées au niveau du système nerveux après une démyélinisation induite expérimentalement au niveau de la moelle épinière chez le chat en 1961 par Richard et Mary Bunge. (5)
Pendant la première moitié du 20ème siècle, ont été développées de nombreuses expériences d’inoculation, allant de la simple injection de liquide céphalo rachidien (LCR) à des animaux à des expériences complexes durant la seconde guerre mondiale. L’injection de LCR de patients SEP à des animaux sains était censée déclencher les symptômes de la SEP. La recherche d’un agent infectieux (bactérien, viral, fongique) causal reste toujours inconnue à ce jour (3,6).
En parallèle de l’hypothèse d’une origine infectieuse de la maladie, la deuxième moitié du 20ème siècle fut marquée par le concept d’auto-immunité en tant que mécanisme physiopathologique. A cette période, EA Kabat (1942) rapporte une augmentation des gammaglobulines dans le LCR, et Laterre (1964) repère des bandes oligoclonales à l’électrophorèse. L’Encéphalite Allergique Expérimentale, un modèle expérimental animal, est monté à cette période et sera rapidement protocolisé (5) (voir l’Annexe 5 pour plus d’informations).
22
I.4 The Decade of the brain (1990-2000)
La fin du 20ème siècle est marquée par le perfectionnement des essais thérapeutiques et par l’essor de nouveaux outils diagnostiques tels que les potentiels évoqués visuels, auditifs, sensitifs, l’examen du liquide céphalo-rachidien par la recherche qualitative de bandes oligoclonales et quantitatives d’immunoglobine G (IgG), et l’imagerie par résonance magnétique (IRM).
Les années 1970-80, suite aux affaires du Stalinon en 1954 puis de la Thalidomide en 1957, ont conduit à renforcer la notion du « rapport efficacité / tolérance » et à prouver l’efficacité en éliminant l’effet placebo. C’est le développement des « essais cliniques randomisés en double aveugle » et l’évolution vers une médecine « basée sur les preuves » (EBM) dans laquelle les maladies sont définies d’abord par des considérations statistiques.
Durant cette période, les traitements les plus utilisés aujourd’hui pour traiter la maladie ont vu le jour. La première Autorisation de Mise sur le Marché (AMM) européenne est délivrée pour le Betaféron dans les formes rémittentes en 1995 après une période préalable d’Autorisation Temporaire d’Utilisation (ATU). Cette même année, on verra les premières publications de l’acétate de glatiramere, avec une AMM en 2002.
L’Avonex est également prescrit dans les formes rémittentes en décembre 1997.
Le Rebif sera mis sur le marché en décembre 1998 avec le dosage de 22μg, en août 2000 pour le dosage de 44μg.
Une évolution parallèle des critères diagnostiques et des critères d’évaluation des traitements se met en place pour s’adapter à cette finalité thérapeutique. Les critères cliniques de WI Mc Donald (2001) seront adoptés et mieux adaptés aux protocoles.
A ce jour, même si la pathogénie reste encore mal connue, la prise en charge se fonde sur une approche pluridisciplinaire.
23
II. Epidémiologie et histoire naturelle
La Sclérose en Plaque est la maladie démyélinisante la plus fréquente qui touche plus de 2,3 millions de personnes à travers le monde, avec plus de patients qu’en 2008 d’après les dernières analyses de la MS International Federation en 2008. La prévalence varie considérablement, élevée en Amérique du Nord et en Europe (>100/100 000 habitants) et faible en Asie de l’est et en Afrique Sub-saharienne (2/100 000 habitants) (7).
Egalement, la médiane de prévalence a augmenté de 30/100 000 habitants en 2008 à 33/100 000 habitants en 2013 (7).
Figure 2: Prévalence mondiale de SEP en 2013 (7)
La race caucasienne paraît plus exposée à la maladie que les noirs d'origine africaine, les asiatiques, les aborigènes d'Australie ou les esquimaux. Les femmes sont deux fois plus touchées que les hommes (8,9).
Cette augmentation de l’incidence chez les femmes semble être due à des différences physiologiques, notamment hormonales. En effet, les faibles taux d’œstrogènes retrouvés au cours de la vie d’une femme, semble augmenter la sécrétion de cytokines Th1 pro-inflammatoires, alors qu’un taux élevé d’œstrogènes, comme au cours de la grossesse, et la testostérone à taux élevé chez l’homme favorisent la voie TH2 antiinflammatoire (10,11).
La maladie débute chez l’adulte jeune entre 20 et 40 ans dans 70% des cas. Elle commence rarement avant 20 ans (10%) ou après 40 ans (20%). L’espérance de vie lorsqu’on est atteint de SEP est réduite de 6 à 14 ans (12).Une large étude française SURVIMUS, qui a inclut 27 603 patients atteints de SEP a montré que durant les premières 20 années de la maladie, le taux de survie était
24 très proche que la population normale. L’augmentation de la mortalité a été évaluée après cette période, avec une réduction de l’espérance de vie de 6 à 7 ans (12).
II.1 La prévalence en France
Une première étude en 2003 sur les données d’ALD (Affection de Longue Durée) de la Mutualité Sociale Agricole (MSA) avait rapporté une prévalence de 65 cas de SEP pour 100 000 habitants (96.3 pour 100 000 habitants chez les femmes et 41.9 habitants pour 100 000 habitants chez les hommes) (13).
La MSA ne couvre que 7% de la population française et reconnaît 2 667 patients ayant une ALD-SEP, elle n’est donc pas représentative.
Ainsi, une autre étude a été menée à partir des données d’ALD-SEP de la Caisse Nationale d’Assurance Maladie des Travailleurs Salariés (CNAMTS) a estimé la prévalence à 94,7 cas de SEP pour 100 000 habitants en 2004. Elle couvre 87% de la population française soit 52 millions d’habitants.
Une étude parallèle menée en 2004 par le réseau LORSEP (Lorraine), impliquant tous les patients atteints de SEP a montré que la prévalence était estimée à 120 cas pour 100 000 habitants (13).
25 Cette répartition de la SEP en France peut s’expliquer par divers facteurs socio-économiques, environnementaux et génétiques.
Ces études montrent que l’utilisation de bases de données différentes au sein d’un même pays peut conduire à des estimations discordantes. On peut expliquer ces différences par l’hétérogénéité des populations étudiées : population agricole, plus âgée et moins féminine que la population urbaine, plus jeune, et plus mixte. Il est donc difficile d’établir une carte précise de la prévalence de la SEP (13).
II.2 L’incidence en France
Une première étude à l’échelle nationale a estimé le nombre de nouveaux cas à 7,91 pour 100 000 personnes en 1999. Les taux d’incidence par département montraient des disparités avec l’existence d’un gradient Nord Est – Sud-Ouest avec une incidence supérieure à 9,12 cas pour 100 000 au Nord-Est et inférieure à 6,39 pour 100 000 au sud-ouest. (13)
Une étude récente révèle une augmentation de l’incidence de la SEP en Lorraine, puisqu’elle passe de 3,7 à 7 cas pour 100 000 habitants entre 1990 et 2000. Cette tendance se confirme chez les femmes où l’incidence passe de 4,5 à 9.8 cas pour 100 000 entre 1990 et 2002 tandis que le taux ne change pas chez les hommes (13)
La SEP affecte aujourd’hui entre 70 000 et 90 000 patients en France (soit environ une personne sur 1000), avec une incidence annuelle probable de 4 à 6 pour 100 000 habitants (soit environ 2 500 à 4 000 nouveaux cas par an) (14).
Cette augmentation de prévalence est probablement due à une augmentation de l’espérance de vie des patients ou de l’accès plus facile au diagnostic (plus de neurologues et d’outils diagnostics comme l’IRM), avec un impact sur l’incidence (nombre de nouveaux cas) et sur la prévalence due au diagnostic plus précoce.
26
III. Étiologie
III.1 Facteurs génétiques
La SEP n’est pas une maladie héréditaire, mais il existe une susceptibilité génétique, c’est-à-dire des facteurs génétiques favorables à son apparition. Le développement de la SEP est multifactoriel, dépend donc des facteurs génétiques et environnementaux que nous verrons plus en détail par la suite (15).
La maladie montre environ 30% de taux de concordance chez les vrais jumeaux, "génétiquement" identiques, contre seulement moins de 5% chez les faux-jumeaux (situation hétérozygote). Ce dernier taux est semblable à celui observé chez les frères et sœurs ainsi que chez les enfants de sujets atteints de SEP (17) (Figure 3).
Ce qu’il faut retenir, c’est que plus le partage de matériel génétique est important avec le membre de la famille atteint, plus le risque de développer la SEP augmente. Puis, le risque décroît, jumeaux hétérozygotes, frères et sœurs, demi-frères. Lorsque le membre de la famille qui est atteint de la maladie est un enfant adopté, alors les autres membres de la famille ne présentent pas plus de risque que la population générale. Ces données montrent bien qu’il existe une augmentation du risque dans les familles où un des membres est atteint, mais cette augmentation du risque est relative et une fois de plus doit être confrontée aux facteurs d’environnement(18).
Cependant l’hérédité seule n’est pas déclencheuse d’une SEP, mais va augmenter le risque de développement pathologique dans les familles.
Figure 4 : Risque de développer la sclérose en plaques suivant le degré de parenté avec un individu atteint (16)
27 Lorsque l'on tente de comparer la susceptibilité héréditaire et l'influence environnementale, il est donc bien difficile de faire la part des choses. Les études menées sur les enfants adoptés et les demi-frères et sœurs suggèrent que les facteurs génétiques sont plus importants que l'influence de l'environnement.
Depuis les années ’70’, où une association entre l’haplotype HLA (Human Leukocyte Antigen) et le risque de SEP a été identifié, plusieurs études génétiques GWAS (Genome Wide Association Studies) ont été engagées et des gènes ont été clairement associés avec une susceptibilité pour la SEP.
Ces différentes variations génétiques pourraient non seulement influencer le risque de développer la SEP, mais également jouer un rôle dans l’hétérogénéité de l’évolution clinique (19).
III.1.1. Rôle de l’immunogénétique HLA
Les antigènes peptidiques, pour être reconnus par les lymphocytes T, doivent au préalable être rendus accessibles à un récepteur pour l’antigène présent à la surface du lymphocyte T (TCR). Cette fonction de présentation de l’antigène (peptide) est assurée par les molécules du Complexe Majeur d’Histocompatibilité (CMH). Ce sont des glycoprotéines de membrane qui assurent au système immunitaire une différenciation soi-non soi. Les gènes codant pour ces protéines sont localisés sur le bras court du chromosome 6. (20)
Le CMH est subdivisé en 3 régions :
La région CMH de classe I comprend 3 gènes HLA de classe I, HLA-A, HLA-B, HLA-C.
La région CMH de classe II comprend 3 paires de gènes HLA de classe II, HLA-DP (gènes
DPA et DPB), HLA-DQ (DQA et DQB) et HLA-DR (DRA et DRB1).
La région III, située entre les régions I et II, ne renferme pas de gènes intervenant dans la présentation antigénique. Elle contient des gènes codant pour des protéines du système du complément (C2, C4, facteur B), pour le TNF et pour les lymphotoxines (20)
28 Figure 5 : Localisation de la région HLA sur le chromosome 6 (21)
Il a ainsi été bien démontré que les allèles de HLADR15 (plus particulièrement HLADRB1*1501) augmentent le risque relatif de SEP de 2 à 4 fois. Cette susceptibilité est liée au fait que cet allèle HLA est capable de lier un auto-antigène de la myéline avec une affinité suffisante pour développer une réponse des cellules T auto-immunes (19).
HLADRB1*15 a été associé de manière significative avec un phénotype féminin et un début précoce dans plusieurs études, et ce, dans toutes les formes cliniques (RR, SP ou SEP-PP). La fréquence de cet haplotype est effectivement beaucoup plus élevée chez les patientes atteintes de SEP que chez les sujets masculins.
Par contre, l’haplotype HLADRB1*04 pourrait présenter un effet protecteur vis-à-vis de l’inflammation et diminuer le risque de poussée.
Les allèles HLA pourraient dès lors influencer le risque et la durée de la phase inflammatoire de la maladie. Leur influence est très probable, bien que non encore clairement démontré (19).
III.1.2. Les régions non HLA
Depuis 2007, grâce à la publication d’études à très grande échelle et le repérage de nouveaux polymorphismes (Single Nucleotide Polymorphisms ou SNPs), de nombreux facteurs génétiques de prédisposition à la SEP ont pu être identifiés.
D’autres gènes de la réponse immunitaire sont maintenant connus avec une grande certitude, le récepteur à l’interleukine 2, le récepteur à l’interleukine 7, une tyrosine kinase (TYK2), un récepteur du Tumor Necrosis Factor (TNF) (18). La liste pourrait s’allonger en fonction du degré de reproduction (réplication) des études génétiques. Ces gènes codent tous pour des protéines de
29 l’immunité. Le système HLA possède probablement 40 % de l’effet génétique à lui seul. Les autres gènes ne joueraient qu’un faible rôle dans la susceptibilité puisque le risque pour les porteurs des allèles de susceptibilité n’est augmenté que d’un facteur de 1,3 à 1,5. Cela montre donc qu’il existe une multitude de gènes qui ont chacun un faible rôle (18).
Certains gènes qui ont été impliqués ne sont pas directement liés à la réaction immunitaire et par exemple le gène KIF1b est une protéine de l’axone, ce qui argumente le fait que la SEP est une réaction immunitaire contre les composants du système nerveux à la fois sur la myéline mais aussi sur l’axone (22).
III.1.3. Les facteurs infectieux
Depuis longtemps, de nombreux agents infectieux notamment neurotropes ont été suspectés dans la physiopathologie de la sclérose en plaques dont les virus d’Epstein-Barr (EBV), Herpès 6 (HHV-6), du Virus Varicello-Zonateux (VZV) et de rétrovirus endogènes (HERVs). L’analyse d’échantillons de LCR et du sang de patients atteints de SEP a permis de retrouver de manière plus importante que chez les non malades de l’ADN, de l’ARN et des anticorps dirigés contre des antigènes d’agents infectieux tels que le virus de l’Epstein Barr, l’herpès virus (HHV-6), le rétrovirus humain endogène (HERVs), et la bactérie Chlamydia pneumoniae. Ces virus pourraient être à la fois impliqués dans le déclenchement et l’évolution de la sclérose en plaques (23).
Parmi les virus incriminés dans le développement de la SEP, le virus EBV est le virus le plus souvent évoqué pour son implication possible dans la pathogenèse de la SEP. Il infecte plus de 90 % de la population. La plupart du temps, la primo-infection, asymptomatique, a lieu dans la petite enfance.
De multiples études sont en faveur de l’association entre virus EBV et la SEP. L’étude des militaires américains est intéressante sur le plan méthodologique, permet de conserver des échantillons d’une grande cohorte de militaires et ainsi d’étudier à travers le temps le sérum des sujets atteints d’une pathologie retenue, ici la SEP. Dans cette étude, il est bien démontré, que les sujets avec un taux élevé d’anticorps anti-EBV ont plus de risque de développer la SEP (24).
Toutefois, l’impact du virus EBV sur l’émergence de la maladie ne serait pas linéaire, il existerait un effet protecteur de l’infection par EBV, si elle est contractée précocement dans la vie, par exemple avant six ans. Alors que le contact avec le virus à un âge plus tardif, notamment à l’adolescence, serait un facteur de susceptibilité. Il existe des bases physiopathologiques pour incriminer le virus EBV et notamment des études comparatives structurales par cristallographie des associations peptide-HLA suggèrent des similitudes avec la protéine basique de la myéline (18). Cela accrédite l’hypothèse que la SEP pourrait se développer dès lors qu’un agent infectieux présente dans sa structure un motif proche d’une protéine de la myéline ; ce qui, en quelque sorte, induirait une confusion du système immunitaire qui développe des cellules T pour neutraliser le virus EBV, mais qui par similitude attaqueraient la myéline.
Cependant, malgré de nombreuses recherches, il n’y a pas d’arguments, à l’heure actuelle, permettant d’affirmer que la SEP est une maladie infectieuse au même titre que la grippe ou le sida (25).
30
III.1.3.1. Le mimétisme moléculaire
La théorie du mimétisme moléculaire repose sur le fait que certains des antigènes des agents pathogènes exogènes (viraux ou bactériens) présentent des séquences peptidiques homologues avec des protéines du soi. Ainsi, se déclenche une réponse immunitaire vis-à-vis de l’antigène exogène mais aussi vis-à-vis des protéines du soi qui lui sont similaires (25).
Dans le cas de la SEP, cette théorie repose sur l’existence de réactions croisées entre des antigènes myéliniques MBP (Myelin Basic Protein) et des antigènes divers activant des CD4+. Des réactions croisées sont aussi observées avec HHV6, HSV ou EBV. Ces virus peuvent enclencher un processus autoimmunitaire contre la myéline car leur enveloppe présente des séquences d’acides aminés qui se retrouvent également dans la structure de la MBP (24,26).
Ainsi, les cellules T spécifiques de l’antigène de l’agent infectieux et un des peptides de la myéline pourraient être activés en périphérie lors d’une infection, ce qui les rendrait capables de traverser la BHE et d’entraîner une inflammation au sein du système nerveux central (SNC) (27).
31
III.2 Facteurs environnementaux
III.2.1. HygièneAu début de la décennie, le professeur J-F Bach a remarqué l’inversion des probabilités de développer une maladie infectieuse et auto-immune dans nos sociétés occidentales.
Il existe une chute vertigineuse des parasitoses, tuberculose, etc., mais il y a aussi augmentation du risque de diabète de type I, SEP, etc. La théorie de l’hygiène consiste à dire que bien que permettant une augmentation de l’espérance de vie, une meilleure hygiène, voire une hygiène trop importante dès le plus jeune âge, pourrait favoriser les maladies auto-immunes (figure 6). (18,28).
Les maladies auto-immunes (MAI) représentent la troisième cause de morbi-mortalité dans les pays industrialisés, derrière le cancer et les maladies cardiovasculaires (29).
32 Des données immunologiques soutiennent cette hypothèse : notamment, une étude a rapporté que les Argentins atteints de parasitoses intestinales produisent plus de lymphocytes T régulateurs (30). Ceci est en lien avec l’hypothèse du microbiome intestinal qui permettrait de pencher la balance immunitaire vers une réponse anti-inflammatoire (31).
III.2.2. Tabac
Les mécanismes par lesquels le tabac favorise la SEP sont mal connus, multiples et complexes car le tabac renferme plus de 1000 composés chimiques (32).
La nicotine agirait sur la perméabilité de la BHE en augmentant la concentration plasmatique de monoxyde d’azote (NO), et aurait pour conséquence l’influx de lymphocytes auto-réactifs au niveau du SNC. Les composés cyanidiques contenus dans la cigarette, dont l’oxyde nitrique serait toxique pour la myéline. La fumée de cigarette contient des oxydants, des éléments carcinogènes et mutagènes. Cette fumée provoquerait au niveau pulmonaire des réactions de stress oxydatif et proinflammatoire, qui réactiveraient les cellules mémoires auto-immunes (32).
D’après une méta-analyse qui reprend les résultats des 6 études prospectives et rétrospectives les plus pertinentes sur le sujet, il y aurait un risque de 1,2 à 1,5 fois plus important de développer une SEP chez les fumeurs que les non-fumeurs (33).
D’après une étude suédoise, il y aurait un risque d’avoir une SEP augmenté de 40 % chez les fumeuses et de 80 % chez les fumeurs. Ce risque apparaitrait pour une consommation de moins de 5 paquets années (nombre de paquets fumés par jour x nombre d’année de tabagisme) et serait dose dépendante, c’est-à-dire que le risque de SEP augmente avec le nombre de paquets de tabac consommés. L’augmentation du risque de SEP associée au tabagisme persisterait jusqu’à 5 années après son arrêt (33). Cette même étude montre que l’utilisation du tabac à priser n’augmenterait pas le risque de SEP ce qui tendrait à prouver que la nicotine n’est pas la substance responsable (32). Concernant le rôle du tabagisme sur l’évolution de la SEP, le suivi sur 6 ans de 122 patients atteints de SEP dont 76 étaient fumeurs, révélait un risque plus important d’évoluer vers une forme progressive chez les fumeurs (p=0,006). Ce risque serait d’autant plus important et précoce que le tabagisme avait débuté tôt avant l’âge de 15 ans (p=0,005).
Le risque de passage en forme secondairement progressive serait multiplié par 3 chez les fumeurs (35).
33
III.2.3. Vitamine D
Il n’y a pas que dans la SEP que la Vitamine D a été impliquée, il y a des données dans d’autres maladies auto-immunes, notamment le lupus, le diabète de type 1 et la polyarthrite rhumatoïde (36). Il n’y a ce jour pas de données suffisantes pour conseiller aux patients de prendre de la vitamine D (36).
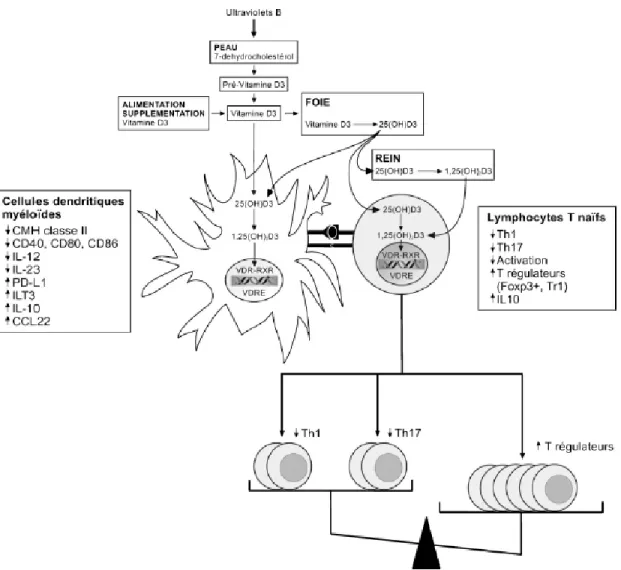
Figure 7 : Cibles de la Vitamine D dans le système immunitaire (37)
La vitamine D provient de l’alimentation et de la supplémentation, mais surtout de l’exposition aux ultraviolets B qui entraînent au niveau de l’épiderme la conversion du 7-déhydrocholestérol en pré-vitamine D3, rapidement convertie en pré-vitamine D3 native. La pré-vitamine D native subit une double hydroxylation pour devenir biologiquement active, au niveau du foie et du rein respectivement. Les cellules dendritiques expriment le récepteur nucléaire à la vitamine D (VDR) à l’état basal, les lymphocytes T et B essentiellement à l’état activé. Les macrophages et les cellules dendritiques
34 expriment les enzymes nécessaires aux deux étapes d’hydroxylation de la vitamine D native, alors que les lymphocytes T activés et les lymphocytes B n’expriment que la 1α-hydroxylase.
De façon schématique, les cellules dendritiques, « sentinelles » du système immunitaire, capturent l’antigène en périphérie, migrent vers les organes lymphoïdes secondaires, où elles initient la réponse immunitaire primaire en présentant l’antigène aux lymphocytes T naïfs. Pendant leur migration, elles subissent un processus de maturation augmentant leurs propriétés immunostimulatrices (38).
Les cellules dendritiques myéloïdes (M-DC) sont les cellules présentatrices d’antigène les plus efficaces mais peuvent être, selon les conditions, immunogènes ou tolérogènes. C’est en partie via cette action sur les cellules dendritiques myéloïdes M-DC que le calcitriol induit, à partir de lymphocytes T naïfs, la différenciation de lymphocytes T régulateurs, tels que les lymphocytes T régulateurs exprimant Foxp3, mais également les lymphocytes T régulateurs de type 1 sécréteurs d’IL-10 (Tr1), et va diminuer la production des lymphocytes Th1 et Th17 (38).
Les mécanismes impliqués dans la différenciation en lymphocytes T régulateurs induite par les M-DC régulatrices sont vraisemblablement multiples. (36,37)
35
III.2.4. Migration
Différentes études sur les immigrants d’Afrique du Sud, d’Israël, d’Hawaï, et d’Angleterre ont montré une corrélation entre le risque de SEP et le lieu de vie durant l’enfance.
Ainsi, une migration durant l’enfance d’une région à haut risque à une région à faible risque diminuerait le risque d’avoir une SEP, et à l’inverse, un mouvement d’une zone à faible risque à une zone à haute prévalence augmenterait le risque de SEP en comparaison avec la population d’origine qui n’a pas migrée (16).
III.2.5. Alimentation
De nombreuses études sur le régime riche en graisses saturées ont été menées en SEP.
Une alimentation riche en gras saturés diminue la fluidité des membranes, mène à la production de cholestérol et contribue à la formation de molécules inflammatoires, des facteurs nuisibles dans la sclérose en plaques. En 2003, Swank et Goodwin ont rapporté que la restriction en gras saturés à moins de 20g/jour induisait la rémission de la maladie et produisait des effets bénéfiques chez les patients atteints de sclérose en plaques. (38)
L’hypothèse d’un lien entre la consommation de lait et la sclérose en plaques a émergé dans le milieu des années 1970. Plus tard, des études épidémiologiques ont soutenu cette hypothèse (40,41). Une étude a montré la relation entre la prévalence de la SEP et la consommation de produits laitiers dans 27 pays et 29 populations dans le monde. Une bonne corrélation entre le lait de vache et la prévalence de la SEP (p = 0,836) a été trouvée; cette corrélation était hautement significative (p <0,001). Une corrélation faible mais néanmoins significative a été obtenue avec la consommation de crème ou de beurre (p = 0,619 et p = 0,504, respectivement). Aucune corrélation n'a été trouvée
pour le fromage.
Ces résultats suggèrent que le lait de vache pourrait contenir des facteurs - qui ne sont plus présents dans le lait transformé - influençant l'aspect clinique de la SEP (41).
Récemment, le régime hyper sodé fait l’objet d’un postulat dans l’induction du développement de maladies auto-immunes, via l’induction des cellules T17 (42)
36
III.2.6. Obésité et régime
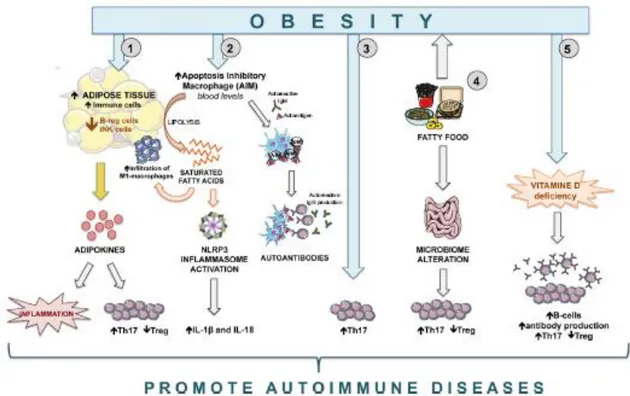
Il a été récemment reporté dans différents modèles de souris que l’obésité pourrait induire une sur-expression des lymphocytes Th17, avec possible augmentation des maladies auto-inflammatoires comme la SEP ou même les colites (43).
Figure 8 : Obésité : principaux mécanismes pouvant engendrer un dysfonctionnement immunitaire (43)
L’augmentation du tissu adipeux induit l’augmentation d’adipokines, responsable d’une réponse pro-inflammatoire et d’une rupture de la balance Treg/Th17, en faveur d’une sur production de Lymphocyte Th17.
Le régime des pays occidentaux, pourrait engendrer un dérèglement de la flore intestinale. Ce dérèglement engendrerait une profonde modulation de la réponse immunitaire du microbiote intestinal et de la balance Th17/Treg, en faveur encore une fois des Lymphocytes Th17. L’obésité induit une déficience en vitamine D et donc une augmentation de Lymphocytes TH17, de lymphocytes B d’anticorps et une diminution de lymphocytes T reg.
Plusieurs études ont investigué l’impact de l’obésité durant l’enfance et l’adolescence sur le développement de SEP.
Pour commencer, deux grandes études, l'une sur deux cohortes de plus de 200 000 femmes américaines (44) et l'autre, une étude cas-témoin suédoise (45), ont rapporté que le risque de développer une SEP chez les sujets jeunes ayant un IMC ≥ 30 kg / m2 étaient doublé par rapport
37 aux sujets de même âge et de poids normal. Il faut quand même préciser que ces études ont présenté des biais, par contre cette tendance a été confirmée par la suite dans des études ultérieures (46-47). Effectivement, dans une étude de cohorte prospective, Munger et al. (46) ont trouvé que L'IMC entre 7 et 13 ans était associé à une augmentation significative de 1,61 à 1,95 du risque SEP chez les filles.
De même, une autre étude a identifié un risque plus élevé de SEP et de syndrome cliniquement isolé chez les filles adolescentes atteintes d’obésités morbides (IMC ≥ 35 kg / m2) avec un Odd’s ratio (OR) = 2,57. (47)
L’obésité agit aussi sur nos gènes. Fait intéressant, une étude a étudié les interactions entre génotype HLA et état de l'IMC. De nombreux allèles ont été identifiés pour la prédisposition à la SEP, l’allèle HLA-DRB1*15 conférant un risque trois fois plus élevé, et l’allèle HLA-A*02 étant protecteur avec un risque deux fois plus faible. En utilisant deux études cas-témoins, les auteurs ont montré que les sujets avec un IMC ≤ 27 kg / m2 et les deux génotypes de risque (porteur de DRB1*15 et absence de A*02) affichaient un OR = 5.1-5.7 alors que le même génotype pour les sujets avec un IMC ≥ 27 kg / m2 rendu un OR = 13,8-16,2 dans les deux cohortes. (48)
L’adiponectine et la leptine sont des hormones retrouvées en grande quantité chez le sujet obèse avec un rôle important dans la balance inflammatoire. Chez un sujet obèse, il y a un fort taux de leptine, de visfatine et un taux faible d’adiponectine, profil qui est également observé chez les patients obèses atteints de SEP. Ce profil hormonal a été corrélé à des niveaux plus élevés des médiateurs de l’inflammation (CRP, TNFα, IL-1β) et une diminution des cellules Foxp3 Treg. (43) Piccio et al. ont montré que les souris déficientes en adiponectine développaient une maladie plus grave sur le plan clinique et histologique, avec des quantités plus élevées d'IFNγ, IL-17, TNFα et IL-6, et moins de cellules Treg que les souris de type témoins (49).
La leptine a fait des preuves dans l’induction de la SEP chez la souris. En effet, Matarese et al. ont étudié le rôle de la leptine dans plusieurs expériences sur des modèles murins atteints de SEP. Il a été rapporté, sur les observations des souris déficientes en leptine, qu’elle était nécessaire à l’induction et à la progression de l’EAE en transformant la réponse immunitaire Th2 en Th1 [43]. Le mécanisme physiopathologique est compliqué mais engendre un profil proinflammatoire Th1 et une diminution des T reg ce qui supposerait avoir un impact sur la maladie.
Malgré des rapports qu’un taux élevé de cholestérol, l’hypertension artérielle, les problèmes cardiovasculaires, le diabète et l’obésité peuvent augmenter la progression et la sévérité de la pathologie, l’absence de données robustes ne permet pas de corréler directement le régime alimentaire et l’obésité à la SEP (50).
38
III.2.7. Vaccin HBV
La vaccination se définie par l’administration artificielle à l’individu de l’agent infectieux contre lequel on veut le protéger. Cet agent infectieux peut être inactivé ou vivant mais atténué. Ainsi, son système immunitaire développera une réponse immune vis-à-vis l’agent infectieux avec anticorps et cellules spécifiques. L’objectif est de protéger le sujet vis-à-vis de l’exposition naturelle à l’agent infectieux.
Les médecins ont toujours redouté que les vaccinations n’aggravent les maladies inflammatoires et, plus particulièrement, les maladies auto-immunes. Ce débat a été relancé à la suite de la campagne de vaccination de masse contre l’hépatite B pratiquée chez l’adulte en France, au milieu des années 1990.
Trois études, menées aux USA et au Canada, avec des résultats concordants, ont été rapportées dans des revues médicales prestigieuses : il n’y a pas d’augmentation du risque de début de SEP suite à une vaccination contre l’hépatite B.
L’AFSSAPS (Agence Française de Sécurité Sanitaire des Produits de Santé, ancienne ANSM) en 2010 a produit un travail de synthèse dans le cadre d’une réflexion collective et validée. Les conclusions sont, d’un point de vue épidémiologique, sans ambigüité : il n’y a pas d’augmentation du risque de développer une sclérose en plaques après une vaccination contre l’hépatite B (51).
39
IV. Physiopathologie
La SEP est une maladie inflammatoire et démyélinisante du SNC. Les plaques de démyélinisation, qui sont une atteinte de la myéline au niveau de la Substance blanche du système nerveux sont responsables d’une altération de la conduction nerveuse entrainant ainsi un handicap fonction de la localisation de ces plaques. Dans un second temps il peut y avoir une dégénérescence axonale. (Figure 9)
Figure 9. Physiopathologie de la sclérose en plaques (52)
Ces plaques sont caractérisées par la présence d’un infiltrat inflammatoire essentiellement composé de macrophages et de LT. Il existe 4 profils de plaques.
40
IV.1. Anatomopathologie : les plaques de démyélinisation
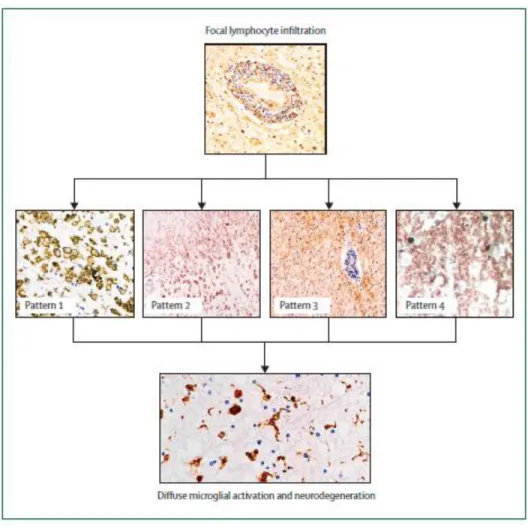
Figure 10 : Les 4 différents profils de plaques (16)
La composition cellulaire et les molécules impliquées peuvent être différentes d’un patient à un autre et être décrites en quatre profils. C’est ce qui a été présenté à l’issue d’une étude des lésions activées prélevées à partir de tissus humains post-mortem (53).
Ces profils prennent en compte la présence et la quantité de cellules immunitaires, de complément et d’anticorps, ainsi que la perte myélinique et l’apoptose oligodendrocytaire.
Les différents profils sont décrits ci-dessous.
Profil 1 : retrouvées dans 15% des patients, il correspond à une prépondérance de lymphocytes T et de macrophages activés, avec comme molécules effectrices le TNFα, les radicaux libres et l’IFN-γ.
Profil 2 : retrouvées dans environ 58% des biopsies, il correspond à la prédominance d’anticorps et de complément. Des anticorps anti-MOG et anti-MBP sont retrouvés au sein
41 des lésions. Le mécanisme de destruction de la myéline semble médié par l’immunité humorale.
Profil 3 : trouvées dans 26% des patients, on y retrouve une démyélinisation active avec une atteinte oligodendrocytaire. Il y a une absence de remyélinisation, d’immunoglobuline et de dépôt du complément .
Profil 4 : Lésions extrêmement rares, trouvées dans seulement 1%. Elles sont caractérisées
par une atteinte dégénérative des oligodendrocytes. Ce profil est surtout présent dans les formes progressives de SEP.
L’hétérogénéité des plaques illustrent bien les différents mécanismes de démyélinisation qui existent en SEP. (53,54)
42
IV.2. Aspects cliniques
IV.2.1. Phénotypes cliniques de la SEP
Les classifications évolutives de la SEP reposent sur la notion de poussées et de progression. Elle est classée selon trois formes évolutives principales:
La SEP récurrente-rémittente (SEP-RR) (85%) : Elle débute vers 30 ans et est composée exclusivement de poussées, qui peuvent laisser des séquelles qui restent stables entre deux épisodes. Les poussées sont l’apparition clinique des plaques aigues. Dès que la perte axonale dépasse les mécanismes de compensation du SNC (remyélinisation spontanée, redistribution des canaux sodiques), la SEP-RR évolue vers une SEP-SP.
La SEP secondairement progressive (SEP-SP) : Ce type de SEP est l’évolution tardive de la
forme précédente, une phase de progression succédant à la phase rémittente. Elle peut toucher en théorie tous les patients initialement rémittents, après une période plus ou moins
longue de 15 à 20 ans en moyenne .
La SEP progressive primaire (SEP-PP) (15%) : Cette forme est plus grave, et débute en moyenne un peu plus tardivement vers 40 ans. C’est une forme progressive d’emblée, ou la progression est présente dès le début sans aucune amélioration et sans poussée ni rémission.
Figure11 : Évolution du handicap pour les trois formes de sclérose en plaques au cours du temps (5).
43
IV.2.2. Les poussées et le Syndrome Clinique Isolé (SCI)
La définition d’une poussée est l’apparition d’un nouveau symptôme pour une période minimale de 24 heures en l’absence de fièvre. Pour être distinctes, deux poussées doivent être séparées par un intervalle d'au moins 30 jours.
Il faut au moins deux poussées pour poser un diagnostic de SEP. Après un seul épisode de symptômes neurologiques, on ne peut pas dire qu’il s’agit de la SEP, alors on appelle cette première poussée un syndrome clinique isolé (SCI). (8)
Le SCI est un premier épisode de démyélinisation d’origine inflammatoire touchant le système nerveux, autrement dit une poussée que l’on observe pour la première fois et qui peut évoluer en SEP.
Pour poser un diagnostic de SCI, les symptômes du sujet atteint doivent être caractéristiques d’une poussée de SEP – qu’il s’agisse d’une névrite optique, de symptômes révélateurs d’une atteinte du tronc cérébral ou du cervelet, ou d’une myélite – et le neurologue doit constater des signes tout aussi caractéristiques d’une poussée à l’examen neurologique. Au cours de cet épisode, le sujet atteint peut présenter un seul trouble neurologique (atteinte monofocale), ou plus d’un trouble neurologique à la fois (atteinte multifocale).
Les sujets avec un diagnostic de SCI peuvent ou non développer ultérieurement une SEP. Les études ont démontré que lorsque le SCI est accompagné de lésions visibles à l’IRM qui sont semblables à celles que l’on observe en SEP, le risque d’avoir un second épisode et d’en arriver à un diagnostic de SEP est plus élevé (environ 80 % en 10 ans). Lorsque le SCI n’est pas accompagné de lésions typiques de la SEP, le risque d’être atteint de la maladie demeure relativement faible (environ 20 % en 10 ans). (59)
IV.2.3. Le syndrome radiologiquement isolé
Le syndrome radiologiquement isolé (SRI) concerne des patients qui présentent des hypersignaux de la substance blanche visibles lors d’IRM cérébrales réalisées pour des raisons médicales autres que des maladies inflammatoires du SNC (traumatismes crâniens, endocrinopathies…). Si l’on observe au moins deux lésions présentes dans des zones différentes du SNC (critère de dissémination spatiale), on parlera alors de SRI. Et si l’analyse du LCR révèle des bandes oligoclonales, et qu’une IRM de contrôle montre la présence de nouvelles lésions hyperintenses en T2 ou des prises de contraste après injection de gadolinium, le RIS est qualifié de RIS à haut risque de développer un évènement clinique et ainsi d’évoluer en SCI et SEP (57).
44 Figure 12 : Evolution différentielle de la SEP (58)
L’inflammation est très présente lors de la phase rémittente puis diminue lors de la phase progressive (ou la perte axonale prédomine). Ainsi, comme on peut le voir sur ce schéma, même si lors de la phase rémittente, il y a des périodes de poussées et de rémissions (repos), les lésions inflammatoires se développent et évoluent presque continuellement. Inversement, le volume cérébral diminue progressivement tout au long de la maladie. Dès que la perte axonale dépasse un seuil critique au-delà duquel les mécanismes de compensation du SNC (remyélinisation spontanée, redistribution des canaux sodiques) ne sont plus efficaces, la SEP-RR évolue vers une SEP-SP. A ce stade, le volume cérébrale diminue beaucoup plus rapidement qui va engendrer un handicap élevé et irréversible. (58)
45
IV.2.4. L'échelle Expanded Disability Status Scale (EDSS)
L'échelle Expanded Disability Status Scale (EDSS), bien que critiquée, reste aujourd’hui l’échelle de référence de cotation clinique de base à tous les neurologues pour évaluer le handicap lié à la
SEP des patients.
Elle a été proposée par en 1983 par l’américain et neurologiste JF Kurtzke.
L'examen neurologique standardisé évalue huit systèmes ou paramètres fonctionnels :
Fonction pyramidale
Fonction cérébelleuse
Fonction sensitive
Fonction du tronc cérébral
Fonction urinaire et du transit intestinal
Fonction visuelle
Fonction mentale
Autres fonctions
Pour le détail de la grille d’évaluation de chaque système fonctionnel voir l’annexe 2.
Le score global de l'échelle se mesure sur une échelle de 0 à 10, et permet d’évaluer le handicap générée par la SEP. (55)
46
IV.3 Désordres auto-immuns
Les progrès faits sur la compréhension des mécanismes de la physiopathologie de la SEP ont permis d’avoir une vision plus claire des acteurs en jeu. Il faut retenir que la plupart des cellules de la cascade immunitaire peuvent être impliquées dans cette maladie. Il n’en reste pas moins qu’il existe des acteurs majeurs, tels les lymphocytes T CD4, T régulateurs, les lymphocytes T CD8, les lymphocytes B ou les macrophages que nous allons voir par la suite dans cette partie (18).
Sur un plan mécanistique, la SEP est considérée comme une maladie auto-immune. Les premiers arguments impliquant le système immunitaire dans le développement de la SEP proviennent des modèles animaux d’encéphalite auto-immune expérimentale (EAE). En effet, l’injection d’extraits de cerveaux de lapin chez le singe est suivie, chez certains sujets, par une infiltration de cellules immunitaires et par une démyélinisation périvasculaire dans le SNC ressemblant à ce qui est observé dans la maladie humaine. (27)
La nature multifactorielle de la SEP, impliquant potentiellement différentes interactions gène-environnement comme déclencheurs, suit un processus inflammatoire multicellulaire complexe qui évolue tout au long de la maladie et qui commence en dehors du SNC (58).
Nous allons voir dans un premier temps les acteurs majeurs de cette maladie, puis les différentes étapes qui définissent la SEP comme la communauté scientifique la connait aujourd’hui.
IV.3.1 Les lymphocytes T (LT)
L’implication des LT dans la SEP est depuis longtemps démontrée par les données expérimentales obtenues chez l’homme et dans le modèle animal de la maladie. En effet, ces cellules sont présentes au sein même des lésions du SNC avec une répartition oligoclonale évoquant une sélection antigène-dépendante (60,61).
La SEP est classiquement considérée comme une maladie médiée principalement par les LT CD4+, notamment grâce aux nombreuses études développées sur le modèle de l’EAE (62). Comme la figure 14 l’illustre, en fonction des différents médiateurs cytokiniques, le LT CD4+ naïf peut se différencier en 3 principaux sous types de populations helper :
Lymphocyte CD4+ Th1 (profil pro inflammatoire)
Lymphocyte CD4+ Th17 (profil pro inflammatoire)
Lymphocyte CD4+ Th2 (profil anti inflammatoire)
47 Figure 14 : Différenciation des LT CD4+ naïfs en fonction des cytokines (27)
(les facteurs de transcription sont en italique)
IV.3.2 Implication des LT CD4+ helper Th1 et Th17
Chez l’homme, il a été montré que l’augmentation de l’activité de la maladie était corrélée à une augmentation d’expression d’IFN-γ et d’interleukine 12 (IL-12) dans le SNC et le liquide céphalorachidien (LCR) de patients. De plus, l’administration d’IFN-γ exacerbe la maladie (63,64). L’implication de ces LT CD4+ est renforcée par le lien génétique entre la SEP et les molécules CMH II présentes à la surface des cellules présentatrices d’antigènes (CPA) (65).
Ces cellules de type Th1 activées vont alors sécréter d’importante quantité d’IFN-γ, d’IL2 et TNF α, cytokines proinflammatoires.
Les cellules Th1 sont mobilisées préférentiellement pour éliminer les agents pathogènes intracellulaires (27).
Dans les années 1990, certaines observations ont suggéré que les LTh1 n’étaient peut-être pas la seule sous-population de LT CD4+ impliquée dans la maladie. Des expériences ont montré que les souris déficientes en IFN-γ induisaient toujours l’EAE (27).
Une étude a montré que des cellules Th17 spécifiques de la PLP (Proteolopid protein) induisaient un phénotype d’EAE et étaient présents dans le SNC des animaux en EAE en phase aiguë. La présence d’IL6 produite par les cellules de l’immunité innée, associée au TGFβ, permet l’orientation vers la voie Th 17. Les cellules Th 17 produisent ainsi les IL17, 21 et 22 (27).
Au niveau anatomopathologique, la quantité de LT producteurs d’IL-17 est augmentée dans les lésions actives ou en bordure des lésions chroniques actives (80 % des LT produisent de l’IL-17, contre 20 % dans les lésions inactives). On retrouve aussi de l’IL-23 au niveau des lésions actives et