HAL Id: dumas-01910229
https://dumas.ccsd.cnrs.fr/dumas-01910229
Submitted on 31 Oct 2018
HAL is a multi-disciplinary open access
archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
L’amélioration autour de l’assurance qualité produit : du
développement de la culture qualité à l’optimisation de
la gestion des non-conformités
Alexandre Beillier
To cite this version:
Alexandre Beillier. L’amélioration autour de l’assurance qualité produit : du développement de la culture qualité à l’optimisation de la gestion des non-conformités. Sciences pharmaceutiques. 2018. �dumas-01910229�
HAL Id: dumas-01910229
https://dumas.ccsd.cnrs.fr/dumas-01910229
Submitted on 31 Oct 2018
HAL is a multi-disciplinary open access
archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
L’amélioration autour de l’assurance qualité produit : du
développement de la culture qualité a l’optimisation de
la gestion des non-conformités
Alexandre Beillier
To cite this version:
Alexandre Beillier. L’amélioration autour de l’assurance qualité produit : du développement de la culture qualité a l’optimisation de la gestion des non-conformités. Sciences pharmaceutiques. 2018. <dumas-01910229>
Université de Bordeaux
U.F.R. DES SCIENCES PHARMACEUTIQUES
Année 2018 N°104
Thèse pour l’obtention du
DIPLOME d’ETAT de DOCTEUR EN PHARMACIE
Présentée et soutenue publiquement par Alexandre BEILLIER
Né le 8 Janvier 1993 à COURBEVOIE
Le 22 Octobre 2018 à BORDEAUX
L’AMELIORATION AUTOUR DE L’ASSURANCE QUALITE PRODUIT :
DU DEVELOPPEMENT DE LA CULTURE QUALITE A L’OPTIMISATION
DE LA GESTION DES NON-CONFORMITES
Directeur de thèse Madame Marion EHLINGER
Membres du Jury :
Monsieur Luc GRISLAIN Professeur des universités Président Madame Marion EHLINGER Docteur en pharmacie Directeur Madame Catherine HEUREUDE Professeur des universités Juge
2
« Regarde vers le ciel et dis-leur je t'aime »
Prêtre de la paroisse de Villebernier Grand-mère, Papi, pour vous, qui m’avez quitté en cette année 2018, et n’avez pu voir l’accomplissement de mes études.
3
REMERCIEMENTS
Au membres du jury :
A Luc GRISLAIN, Président de thèse et Professeur des Universités,
Je vous remercie d’avoir accepté de présider cette thèse. Merci pour tous les enseignements transmis au cours de mes études. Soyez assuré de ma profonde reconnaissance.
A Marion EHLINGER, Directrice de thèse et Responsable du service Assurance Qualité Produit de Guyenne,
Je tiens à te remercier pour m’avoir fait l’honneur d’être la directrice de cette thèse. Merci pour ta confiance et pour le temps que tu as pris pour me conseiller et me former. Sois assurée de mon éternelle gratitude.
A Catherine HEUREUDE, Juge et Professeurs des Université,
Je vous remercie d’avoir accepté de juger ce travail, et d’avoir été la première à me définir la notion de qualité dans l’industrie pharmaceutique. Soyez assurée de mon profond respect A Yann BOUDIER, Juge, Pharmacien du service Assurance Qualité Produit de Guyenne, Je tiens à te remercier pour avoir accepté d’être juge de cette thèse. Merci de m’avoir formé et accompagné tout au long de mon apprentissage. Merci pour ta présence et la disponibilité dont tu as fait preuve.
A mes proches :
A mes parents et mes grands-parents : merci à vous, merci d’avoir toujours été là, de m’avoir soutenu et encouragé, de m’avoir fait devenir celui que je suis aujourd’hui.
A ma sœur Angélique : sans toi je ne serais surement pas là aujourd’hui, merci pour tout. A mes frères et sœurs : Mickael, Anne-Sophie, Alicia, Alice, Arthur et Anne-Laure ; à mes cousins : Romain et Amandine ; merci pour tous les bons moments que l’on a passés et que l’on passera ensemble.
4
A mes amis :
A mes amis de longue date : Arnaud, Pierre, Cyril, Ben et Igor, merci de toujours répondre présent quand le moment se présente.
A mes camarades de pharma : Amayelle, Sacha, Gabriel, Jeremy, Thomas et bien d’autres, merci d’avoir été mes best-binômes et mes compagnons de beuverie. Je n’oublierai jamais nos belles années d’études.
A mes collègues :
Merci à Camille TREHEL, et à mes trois collègues Corinne, Laetitia et Tatiana : Merci pour toute l’assistance que vous m’avez apportée dans la réalisation des projets et au quotidien. Merci l’ensemble des collaborateurs UPSA avec qui j’ai eu l’occasion de travailler : Merci pour avoir contribué à mon épanouissement dans le rôle qui m’a été confié et pour la participation à l’apprentissage de mon futur métier de pharmacien industriel.
A tous les intervenants ayant participé à ma formation :
Je suis reconnaissant envers l’ensemble des professeurs et intervenants des facultés de pharmacie de Bordeaux et de Chatenay-Malabry, qui ont participé à mon développement professionnel et qui m’ont inculqué la culture qualité essentielle pour travailler dans l’industrie pharmaceutique.
5
TABLE DES MATIERES
REMERCIEMENTS ... 3
TABLE DES MATIERES ... 5
TABLE DES FIGURES ... 8
LISTE DES TABLEAUX ... 9
LISTE DES ABREVIATIONS ... 10
INTRODUCTION ... 11
CHAPITRE PREMIER : L’ASSURANCE QUALITE PRODUIT ... 13
1 LA CERTIFICATION ET LA LIBERATION DES LOTS ... 15
1.1 La notion de lot pharmaceutique ... 15
Le lot pharmaceutique ... 15
Le numéro de lot ... 15
Le dossier de lot ... 16
1.1.3.1 Le dossier de lot de fabrication ... 16
1.1.3.2 Le dossier de lot de conditionnement ... 17
1.2 La notion de personne qualifiée ... 18
1.3 Un acte pharmaceutique : la libération ... 19
1.4 La revue des dossiers de lot ... 19
1.5 Certification et Libération selon l’annexe 16 des BPF ... 20
Processus de certification ... 20
1.5.1.1 Trois prérequis : connaissance, autorisation et identification... 21
1.5.1.2 Assurance d’un circuit maîtrisé et contrôlé ... 22
1.5.1.3 Contrôle de la conformité ... 23
1.5.1.4 Preuve d’un état validé ... 23
1.5.1.5 Evaluation des changements ... 25
1.5.1.5.1 Définition de modification et de son contrôle ... 25
1.5.1.5.2 Différents types de changement ... 25
1.5.1.5.3 Prise en considération pour la certification ... 26
1.5.1.6 Accès à toute la documentation ... 26
1.5.1.7 Partage des responsabilités ... 27
1.5.1.8 Incidence maitrisée ... 27
1.5.1.9 Traçabilité des certifications ... 27
Prise en compte des évaluations de l’application des BPF ... 28
Gestion des déviations non planifiées ... 29
Libération des lots ... 29
2 LA GESTION DES DEVIATIONS ... 30
2.1 Quelques définitions ... 30
Les déviations ... 30
2.1.1.1 La déviation au sens d’incident qualité ... 30
2.1.1.2 La déviation au sens de dérogation ... 31
Les corrections ... 31
Les CAPA ... 31
2.2 Maîtrise des déviations ... 32
6
Analyse des causes ... 32
Intégration étendue ... 33
Une réponse contre les déviations : le système CAPA ... 33
2.3 L’analyse de risque, point central de la maîtrise des déviations ... 34
Notion de risque ... 34
Gestion du risque ... 34
2.3.2.1 Processus systématique ... 34
2.3.2.2 Différentes approches ... 36
2.3.2.3 Intérêts de la gestion du risque ... 37
Approche rétrospective : l’analyse du risque d’une déviation ... 38
CHAPITRE SECOND : L’AMELIORATION CONTINUE DES PRATIQUES ... 40
1 CONTEXTE INDUSTRIEL ... 42
1.1 Les Laboratoires UPSA ... 42
Les sites de production d’Agen ... 44
Une stratégie pour la performance industrielle ... 44
1.2 Le processus de revue des dossiers établi ... 45
Le circuit de revue ... 45
Les retours de dossiers ... 47
L’évaluation des performances ... 50
1.2.3.1 La notion de « bon du premier coup » ... 50
1.2.3.2 Enregistrement des non-conformités documentaires ... 51
1.3 Une gestion maitrisée des déviations ... 53
Déroulement d’une investigation ... 53
Contenu d’une investigation ... 54
Décision de l’assurance qualité produit ... 55
Processus de suivi et de traçabilité ... 56
1.3.4.1 Partage et délai ... 56
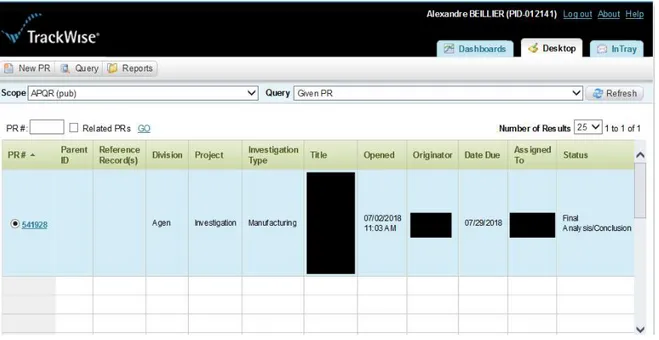
1.3.4.2 Un outil de gestion et de traçabilité : le logiciel TrackWise® : ... 57
2 DEVELOPPER LA CULTURE QUALITE ... 58
2.1 Definir la culture qualité ... 58
2.2 L’expression du besoin ... 59
2.3 Mettre en place des dialogues qualité ... 60
Le projet ... 60
Un exemple : le programme STOP® ... 60
2.3.2.1 La méthode ... 60
2.3.2.2 L’application à UPSA ... 61
2.3.2.2.1 Une formation étendue ... 61
2.3.2.2.2 Les dialogues STOP® ... 61
2.3.2.2.3 Des résultats encourageants ... 62
Un outil modèle : le PDCA ... 62
Planifier (Plan) ... 64
2.3.4.1 Contexte et Objectifs ... 64
2.3.4.2 L’équipe projet et les acteurs du dialogue qualité ... 65
2.3.4.3 Ordonnancement du projet ... 66
Développer (Do) ... 67
2.3.5.1 Thèmes définis et leur contenu ... 67
2.3.5.1.1 La documentation ... 67
2.3.5.1.2 Les IPC et les challenge tests ... 72
2.3.5.1.3 Les rejets du SIA et la réintroduction ... 74
2.3.5.1.4 Les prélèvements ... 75
2.3.5.1.5 Monitoring des conditions environnementales ... 77
7
2.3.5.1.7 Autres thèmes ... 79
2.3.5.2 Formaliser une méthodologie ... 80
2.3.5.3 Créer un mémorandum ... 81
2.3.5.4 Former les acteurs ... 82
2.3.5.5 Réalisation des dialogues qualité ... 83
Vérifier (Check) ... 83
2.3.6.1 Partage des observations ... 83
2.3.6.1.1 Partage intraservice ... 83
2.3.6.1.2 Partage interservices ... 84
2.3.6.2 Mesurer les progrès ... 84
Ajuster (Act) ... 85
2.3.7.1 Priorisation des ateliers ... 85
2.3.7.2 Réaction face aux observations ... 85
2.4 Bilan du projet ... 86
3 OPTIMISER LA GESTION DES DEVIATIONS ... 87
3.1 Challenger les limites ... 87
Une méthodologie procédurée ... 87
Une utilisation restrictive de l’outil TrackWise ... 88
3.2 Adopter une gestion différente ... 89
Objectifs de la démarche ... 89
Une première phase concluante ... 90
Réalisation de l’étude ... 91
Un logigramme pour une nouvelle gestion ... 93
Modifier le dossier de lot pour une meilleure déclaration ... 94
3.2.5.1 Description du défaut ... 95
3.2.5.1.1 Un outil modèle : Le QQOQCCP ... 95
3.2.5.1.2 Application du QQOQCCP à la description du défaut ... 97
3.2.5.2 La mise en place d’action correctives immédiates ... 99
3.2.5.3 Les conditions de redémarrage ... 100
3.2.5.4 Commentaire, traçabilité et décisionnel AQ... 101
3.3 Répondre au besoin ... 102
Les analyses de tendance ... 102
La satisfaction opérationnelle ... 102
Une application à d’autres types de défaut ... 102
CONCLUSION ... 103
BIBLIOGRAPHIE ... 104
ANNEXES ... 107
Annexe 1 : Le recto du mémorandum……….….107
Annexe 2 : Le verso du mémorandum……….108
Annexe 3 : Les 5 cas de QE or not QE « Manque tests »…...……….109
Annexe 4 : Logigramme décisionnel lors d’un défaut de marquage.…..……….112
8
TABLE DES FIGURES
Figure 1 : Schéma des étapes de production d'un médicament... 22
Figure 2 : Vue d'ensemble de la validation ... 23
Figure 3 : Deux façons d'approcher le risque ... 36
Figure 4 : Calque de la gestion d’une déviation sur le procédé classique de gestion du risque qualité de l’ICH Q9... 39
Figure 5 : L’histoire UPSA ... 42
Figure 6 : Les Localisations en France ... 43
Figure 7 : Un processus de revue des dossiers de lot ... 46
Figure 8 : Application des retours au flux de revu des dossiers ... 48
Figure 9 : Capture d'écran du logiciel TrackWise ... 57
Figure 10 : Roue de Deming - Méthode PDCA ... 63
Figure 11 : Ordonnancement du projet ... 66
9
LISTE DES TABLEAUX
Tableau 1 : Modèle de détermination de la criticité ... 38
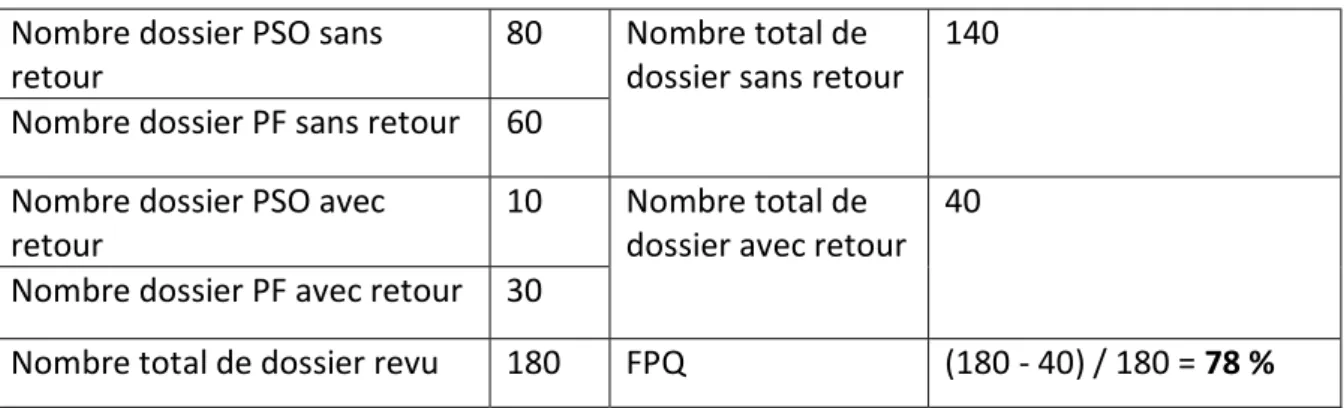
Tableau 2 : Exemple de calcul d'un RFT ... 50
Tableau 3 : Exemple de calcul d'un FPQ ... 51
Tableau 4 : Tableau de partage inter-service (1/2) ... 84
Tableau 5 : Tableau de partage inter-service (2/2) ... 85
Tableau 6 : L'outil QQOQCCP ... 96
Tableau 7 : La description du défaut ... 97
Tableau 8 : Les actions correctives immédiates ... 99
Tableau 9 : Les conditions de redémarrage ... 100
10
LISTE DES ABREVIATIONS
Principales abréviations utilisées (la liste n’est pas exhaustive) : - AE : Animateur d’Equipe
- AMM : Autorisation de Mise sur le Marché - AQ : Assurance Qualité
- AQP : Assurance Qualité Produit - BMS : Bristol-Myers-Squibb
- BPF : Bonnes Pratiques de Fabrication - BU : Business Unit
- CAPA : Corrective and Preventive Action - DDL : Dossier De Lot
- EHS : Environnement, Hygiène et Sécurité - FPQ : First Pass Quality
- LIMS : Laboratory Information Management System - MT : Manufacturing Technology
- PF : Produit Fini
- PSO : Produit Semi-Ouvré - QE : Quality Event
- QP : Qualified Person - RFT : Right First Time
- SAP : Systems, Applications and Products for data Processing (Progiciel de gestion intégré) - SN : Suivi de Nettoyage
- SOP : Standard Operating Procedure - STI : Service Technique Infrastructure - TAQ : Technicien Assurance Qualité - VDL : Vide De Ligne
11
INTRODUCTION
Un médicament n’est pas un produit de consommation comme les autres, en effet il répond à une définition formelle inscrite dans le code de la santé publique (L5111-1): « On entend par médicament toute substance ou composition présentée comme possédant des propriétés curatives ou préventives à l'égard des maladies humaines ou animales, ainsi que toute substance ou composition pouvant être utilisée chez l'homme ou chez l'animal ou pouvant leur être administrée, en vue d'établir un diagnostic médical ou de restaurer, corriger ou modifier leurs fonctions physiologiques en exerçant une action pharmacologique, immunologique ou métabolique. » (1)
Destiné à servir la santé publique le médicament se doit de disposer de trois composantes intrinsèques inaltérables : la qualité, la sécurité et l’efficacité. Ces trois composantes, démontrées avant l’obtention d’une autorisation de mise sur le marché (AMM), doivent être garanties depuis l’acquisition des matières premières jusqu’à la délivrance et l’utilisation du médicament par le patient. A cet égard, les médicaments sont soumis à une surveillance stricte de la part des autorités, qui opposent plusieurs textes définissant les exigences à respecter.
Parmi ces textes se trouve le guide des bonnes pratiques de fabrication (BPF). Ce guide expose les dispositions générales à mettre en œuvre dans un établissement pharmaceutique se livrant, en vue de leur vente en gros, de leur cession à titre gratuit ou de leur expérimentation à des opérations de fabrication de médicaments.
Un site de production pharmaceutique se compose, dans une majorité des cas, de différents services, permettant l'ensemble des opérations de transformation des matières premières en produits finis. La fabrication, le conditionnement, la maintenance et la logistique sont les services fondamentaux de la production, mais pour répondre aux exigences de qualité et de sécurité d’un médicament un service indispensable intervient à tous les niveaux : il s’agit de l’Assurance Qualité.
12 L’assurance qualité permet, de par son indépendance, de garantir que l’ensemble des opérations s’opère dans le respect des bonnes pratiques de fabrication (BPF), dans le respect des directives, et dans le respect des procédures mises en place. L’assurance qualité assure également la conformité des produits à leur dossier d’autorisation de mise sur le marché. Dans les grosses structures fabricantes l’assurance qualité se décline en plusieurs entités, dont une de première proximité vis-à-vis des produits fabriqués et des patients : l’assurance qualité produit (AQP). L’assurance qualité produit, de par ses attributions, se situe au dernier échelon du système global de management de la qualité appliqué à la fabrication, ce qui impose la préoccupation d’une maitrise continue et croissante de la qualité et une responsabilité dans les décisions.
Nous verrons dans une première partie deux des missions s’inscrivant dans l’assurance de la qualité des produits au sein d’un laboratoire pharmaceutique : La certification et la libération ; la gestion des déviations. Dans une seconde partie nous traiterons d’un cas pratique : la mise en place de démarches d’amélioration continue visant le développement de la culture qualité et l’optimisation de la gestion des non-conformités.
13
14 L’assurance qualité produit, également appelée assurance qualité production ou assurance qualité opérationnelle, est le service responsable de la mise sur le marché des produits fabriqués.
Cette responsabilité octroie au service AQP un pouvoir décisionnel important et requiert sa participation à l’ensemble des sujets qui touchent la production et impactent potentiellement la qualité, la sécurité ou l’efficacité des produits.
L’AQP dans la plupart des laboratoires pharmaceutiques, intervient dans de nombreuses missions telles que la certification et la libération des lots, la gestion des déviations et leur investigation, le choix des actions correctives et préventives (CAPA), les réclamations, le contrôle du changement (change control) ou encore les audits et inspections.
Dans cette première partie nous allons aborder, sous un regard réglementaire, deux des principales missions que sont la certification et la libération des lots et la gestion des déviations.
15
1
LA CERTIFICATION ET LA LIBERATION DES LOTS
1.1 LA NOTION DE LOT PHARMACEUTIQUE
Le lot pharmaceutique
Un lot se définit comme une quantité déterminée d’une matière première, d’un article de conditionnement ou d’un produit, fabriquée en une opération ou en une série d’opérations, telle qu’elle puisse présenter des caractéristiques d’uniformité avec une qualité homogène et continue. (2)
Le numéro de lot
Chaque lot, que ce soit un lot de matière première, d’article de conditionnement ou de produit fini possède un identifiant unique : il s’agit du numéro de lot. Ce numéro de lot est de manière générale une combinaison caractéristique de chiffres et/ou de lettres qui identifie spécifiquement un lot et à partir de laquelle la traçabilité de la production et de la distribution peut être établie. (2)
La notion d’unicité du numéro de lot est une notion indispensable, elle permet de discriminer un lot de façon formelle en excluant toute confusion sur l’identité ou la nature de ce lot. Lors de la fabrication d’un produit pharmaceutique il y aura autant de numéro de lot que d’étapes distinctes de fabrication. Par exemple dans le cas de la fabrication d’un comprimé pelliculé en plusieurs opérations il y aura :
Un numéro de lot pour la poudre issue du mélange des matières premières ; Un numéro de lot pour les comprimés nus issus de la compression de la poudre ; Un numéro de lot pour les comprimés pelliculés issus du pelliculage des comprimés
16
Le dossier de lot
Le dossier de lot, qu’il soit en format papier ou en format électronique, est un dossier qui regroupe l’ensemble des éléments (données et documents) caractéristiques d’un lot. Il permet l’enregistrement en temps réel de toutes les données de la production du lot : les matières utilisées, l’enchainement de toutes les opérations réalisées, les personnes qui interviennent, les locaux et équipements qui sont utilisés, l’historique de chaque étape, le résultat de chaque contrôle.
Le dossier de lot reprend généralement une part des spécifications des produits et des informations issues du dossier d’autorisation de mise sur le marché (AMM) et est donc une source d'informations pour les personnes le complétant. Il doit être renseigné dans le respect des règles de « Data Integrity » [intégrité des données] car il constitue une preuve que la composition du lot et sa confection sont conformes aux exigences réglementaires. (3)
Le dossier de lot est un véritable outil de la traçabilité. C’est un document règlementé qui doit répondre aux exigences des bonnes pratiques de fabrication : Selon les BPF, un dossier de lot doit comprendre deux dossiers, l’un de fabrication et l’autre de conditionnement. Les informations obligatoires devant composer ces deux dossiers sont décrites dans la première partie du guide des BPF.
1.1.3.1 Le dossier de lot de fabrication (4)
Le dossier de fabrication de lot doit être constitué pour chaque lot fabriqué. Il doit être basé sur les éléments correspondants de la formule de fabrication et des instructions de fabrication approuvées. Il doit contenir les informations suivantes :
le nom et le numéro de lot du produit ;
les dates et heures de début, de chaque étape intermédiaire importante et de la fin de la production ;
les initiales de(s) l’opérateur(s) réalisant les étapes critiques de la fabrication et, le cas échéant, de toute personne ayant vérifiée ces opérations ;
17 le numéro de lot et/ou le numéro d’analyse, les quantités de chaque matière première réellement pesée (y compris le numéro de lot et la quantité de tout produit récupéré ou retraité qui a été ajouté) ;
toute opération de fabrication ou tout événement d’importance et les principaux équipements utilisés ;
un relevé des contrôles en cours de fabrication, les initiales de(s) la personne(s) les ayant réalisés et les résultats obtenus ;
le rendement obtenu à différentes étapes intermédiaires clé de la fabrication ;
des notes détaillées portant sur tout problème particulier, même de détail et une autorisation signée pour chaque déviation à la formule et aux instructions de fabrication ;
l’approbation par la personne responsable des opérations de fabrication.
1.1.3.2 Le dossier de lot de conditionnement (4)
Le dossier de conditionnement du lot doit être constitué pour chaque lot ou partie de lot conditionné. Il doit se baser sur les éléments correspondants des instructions de conditionnement. Il doit contenir les informations suivantes :
le nom et le numéro de lot du produit ;
les dates et heures des opérations de conditionnement ;
l’identification (initiales) de(s) l’opérateur(s) réalisant les étapes critiques du conditionnement et, le cas échéant, le nom de toute personne ayant vérifiée ces opérations ;
les enregistrements des vérifications portant sur l’identité et la conformité aux instructions de conditionnement, y compris les résultats des contrôles en cours de conditionnement ;
les informations sur les opérations de conditionnement réalisées, y compris les références aux équipements et aux lignes de conditionnement utilisées ;
si possible, des échantillons des articles de conditionnement imprimés, y compris les modèles des codes de lot, des dates de péremption et de toute surimpression ;
18 des notes détaillées portant sur tout problème particulier ou évènement inhabituel, avec une autorisation signée pour chaque déviation aux instructions de conditionnement ;
les quantités et le numéro de référence ou marque d’identification de tous les articles de conditionnement imprimés ainsi que des produits vrac fournis, utilisés, détruits ou retournés en stock et les quantités de produit obtenu, avec le bilan comparatif. Lorsque des systèmes de contrôles électroniques robustes sont en place durant le conditionnement, il peut être justifié de ne pas inclure cette information ;
l’approbation par la personne responsable des opérations de conditionnement.
1.2 LA NOTION DE PERSONNE QUALIFIEE
La Personne Qualifiée (ou « Qualified Person », QP) est un terme technique utilisé dans la règlementation pharmaceutique Européenne. Ce terme désigne une personne ayant acquis des connaissances scientifiques théoriques et pratiques reconnus et suffisantes pour endosser la responsabilité pharmaceutique d’un produit de santé.
Lorsque l’opération de libération à lieu en France, le rôle de QP est dévolu au Pharmacien Responsable (PR) ou au pharmacien délégué ou au pharmacien adjoint, par délégation du PR, formé et habilité à la certification de lot. (5)
19
1.3 UN ACTE PHARMACEUTIQUE : LA LIBERATION
La libération est la décision de vente ou de mise sur le marché des lots de médicaments. La libération contient l’étape préalable de certification, c’est un acte pharmaceutique qui doit obligatoirement être réalisé par la personne qualifiée.
« Les médicaments ne sont ni vendus ni distribués tant qu’une personne qualifiée n’a pas certifié que chaque lot de production a été produit et contrôlé conformément aux exigences
de l’autorisation de mise sur le marché et de toute autre réglementation portant sur la production, le contrôle et la libération des médicaments. »
Guide des bonnes pratiques de fabrication, partie 1, chapitre 1.
1.4 LA REVUE DES DOSSIERS DE LOT
Le guide des bonnes pratiques de fabrication décrit, à travers le contenu du dossier de lot et le processus de libération, des règles concernant la revue et l’approbation des dossiers de lot. Il appartient au fabricant, dans le respect de ces règles, de définir son propre processus opérationnel de revue permettant de s’assurer de la maîtrise qualitative du produit et de la documentation (du dossier de lot) s’y afférant.
Ce système de revue doit s’appuyer sur des procédures écrites, devant être établies préalablement et suivies pour la revue et l’approbation des dossiers de production, afin de déterminer la conformité aux spécifications établies avant la libération ou la distribution du lot. Tout rapport relatif à un écart, une enquête, ou une non-conformité aux spécifications doit être revu comme partie intégrante du dossier de lot, avant que le lot ne soit libéré.
20
1.5 CERTIFICATION ET LIBERATION SELON L’ANNEXE 16 DES BPF (4)
Les évolutions constantes de l’univers pharmaceutique (mondialisation de la chaîne d’approvisionnement, harmonisations des législations européennes, évolutions réglementaires, nouvelles approches dans les stratégies de contrôle…), nécessitent d’adapter les processus de certification et de libération des lots, en tenant compte des nouveaux contextes et des nouvelles contraintes. (6)
Dans un effort d’adaptation à ces évolutions, au 30/12/2016, le directeur général de l’ANSM Dominique Martin a modifié les bonnes pratiques de fabrication : les annexes 15 et 16 ont remplacé les lignes directrices 15 et 16. (4)
L’annexe 16 des BPF, au titre de « Certification par une personne qualifiée et libération des lots », donne, hors dispositions de base applicables à la libération des lots d’un produit définies par son AMM, des indications concernant le processus de certification et de libération.
Processus de certification
Avant la libération des lots de production pour la vente et la distribution, la personne qualifiée doit procéder à leur certification.
Le processus de certification est un processus très codifié qui présente de nombreuse exigences vis-à-vis de la personne qualifié, du produit, de l’environnement de production ainsi que du système qualité pharmaceutique dans sa globalité. C’est à la QP que revient la responsabilité de s’assurer que ces nombreuses exigences sont garanties, toutefois concernant certaines tâches, des délégations à du personnel ou à des Tiers ayant reçu une formation appropriée sont envisageables.
21
1.5.1.1 Trois prérequis : connaissance, autorisation et identification
La personne qualifiée doit posséder des connaissances approfondies des étapes sur lesquelles elle engage sa responsabilité. Cette connaissance s’acquiert par une formation complète sur les produits et sur les processus de production. Dans le cadre d’un développement professionnel continue, la personne qualifiée doit s’assurer d’une constante prise de connaissance sur les avancées techniques et sur toutes les modifications apportées aux BPF. La personne qualifiée doit s’assurer que l’autorisation d’ouverture de l’établissement pharmaceutique auquel elle est rattachée intègre l’autorisation de sa certification et que toutes les obligations et exigences législatives sont respectées.Dans l’éventualité d’un défaut qualité nécessitant une enquête ou un rappel de lot, toutes les Personnes Qualifiées impliquées dans la certification (ainsi que tous les enregistrements pertinents) doivent être aisément identifiables.
22
1.5.1.2 Assurance d’un circuit maîtrisé et contrôlé
Il incombe à la QP qui procède à la certification du produit fini de s’assurer que toutes les étapes requises sont effectuées dans le respect des systèmes de qualité pharmaceutiques reconnus afin de garantir la conformité du lot avec les BPF, l’AMM et toute autre obligation légale au sein de l’État membre dans lequel a lieu la certification. A cet effet, la QP doit disposer de la garantie que tous les sites et leurs activités qui sont impliquées avec la production du médicaments sont référencés dans l’AMM et conforme avec leurs spécifications.
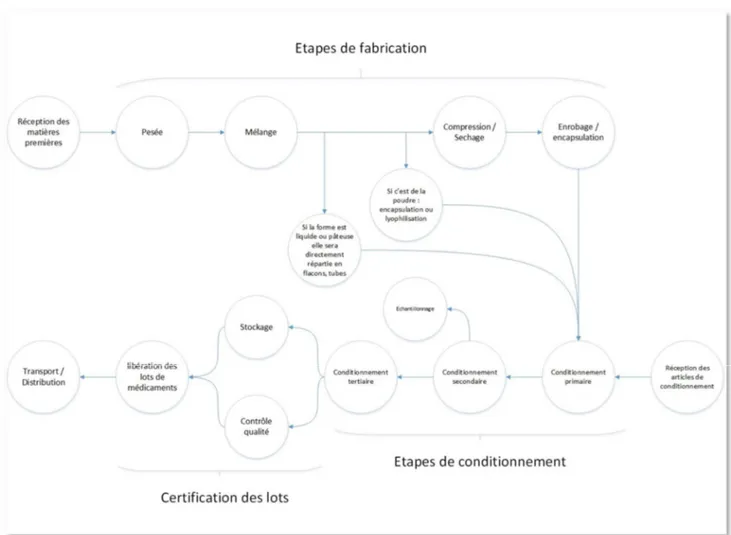
Figure 1 : Schéma des étapes de production d'un médicament (7)
L’ensemble des conditions de réceptions et de mise en œuvre des composants du produit, de contrôle, de stockage et de transport ainsi que le processus d’échantillonnage doivent être pris en considération.
23
1.5.1.3 Contrôle de la conformité
La personne qualifiée doit s’assurer que les résultats de l’ensemble des contrôles appliqués jusqu’au produit fini sont conformes aux spécifications décrites dans le dossier d’AMM, aux spécifications décrites dans les procédures internes ainsi qu’aux autres textes règlementaires et législatifs (Pharmacopée, DGCCRF, etc.).
Dans cet ensemble de contrôles sont compris les contrôles réalisés sur les matières premières, sur le produit semi-ouvré, sur les articles de conditionnement, sur le produit dans son conditionnement primaire, secondaire et tertiaire, ainsi que l’ensemble des contrôles de l’environnement de production (contrôles des paramètres environnementaux, contrôles microbiologiques et particulaires, contrôles techniques des équipements).
Quelle que soit la nature des contrôles (contrôle en cours de process ou contrôle réalisé par le laboratoire de contrôle qualité) les enregistrements et les vérifications doivent être complets et approuvés par le personnel approprié.
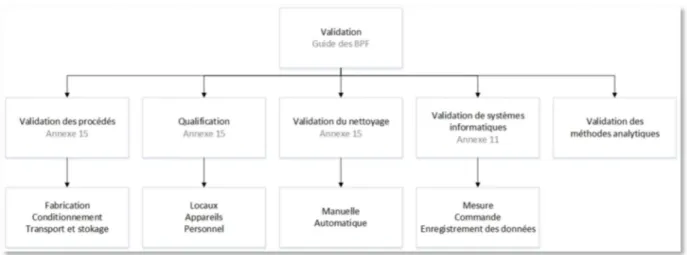
1.5.1.4 Preuve d’un état validé
24 La validation est l’établissement de la preuve documentée que la mise en œuvre ou l’utilisation de tout processus, procédure, matériel, matière première, article de conditionnement ou produit, activité ou système permet réellement d’atteindre les résultats escomptés.
La qualification fait partie de la validation, mais les étapes de qualification à elles seules ne constituent pas une validation de procédé. La qualification est l’action de prouver et de documenter qu'un équipement ou ses systèmes auxiliaires sont installés convenablement, travaillent correctement et conduisent réellement aux résultats attendus. Concernant la qualification du personnel il s’agit de la démonstration que le personnel a les capacités d’exercer l’activité qui lui est confiée grâce à une formation initiale, une formation complémentaire et / ou une expérience appropriées.
La qualification et la validation sont des composantes indissociables d’une assurance de la qualité. Tout fabricant doit fournir un travail de validation pour démontrer que les aspects critiques des opérations sont contrôlés afin de disposer des produits de qualité, de façon efficace, reproductible et conforme aux critères d’acceptation prédéfinis.
La QP n’a pas l’obligation d’interagir dans ce processus de qualification/validation mais elle doit néanmoins s’assurer que les système est maitrisé afin de produire et de préserver un état validé, et que le personnel est formé et qualifié de manière appropriée.
25
1.5.1.5 Evaluation des changements (9)
Dans la réalité quotidienne du fonctionnement normal d’une usine, il arrive que des modifications par rapport aux référentiels soient appliquées. Ces modifications ou changement peuvent avoir un impact critique, majeure ou mineure sur la qualité des produits, il convient donc d’évaluer cet impact afin de prendre les mesures adéquates pour maintenir un niveau approprié de contrôle.
1.5.1.5.1 Définition de modification et de son contrôle
La modification se définie comme tout changement prévu, permanent et planifié d’un ou plusieurs éléments couverts directement ou indirectement par les BPF, l’AMM, le Code de la Santé Publique et tout autre document issu des autorités de tutelle.
Le système de maîtrise des changements ou système de Change Control est un système formel par lequel des représentants qualifiés des disciplines concernées examinent les changements proposés ou effectifs susceptibles de modifier le statut validé des installations, systèmes, équipements ou procédés. L’objectif est de déterminer les mesures pouvant s’avérer nécessaires pour garantir et démontrer que la validité du système perdure.
1.5.1.5.2 Différents types de changement Le changement appliqué au produit
Le premier type de changement auquel on peut penser est le changement lié au produit. Il peut s’agir du changement d’une matière de fabrication, de fournisseur d’un composant, d’une modifications d’une recette au niveau quantitatif ou qualitatif, du changement d’un paramètre lié à la conception, de l’augmentation des volumes de production, etc.
26 Le changement de processus
Le changement de processus peut consister à la mise en place de nouveaux procédés de fabrication, à l’amélioration des procédures en vigueur, au lancement de projets qu’ils soient volontaires ou imposés par une exigence réglementaire, etc.
Le changement de structure
Afin de lutter contre la dérive vers l’inefficacité d’un système ou de viser son optimisation il peut être intéressant d’introduire de nouveaux outils matériels ou immatériels, de nouveaux équipements, de restructurer une unité, de créer de nouveaux postes, etc.
1.5.1.5.3 Prise en considération pour la certification
La QP doit s’assurer que l’incidence de tous les changements sur la fabrication ou le contrôle du produit a fait l’objet d’une évaluation. La certification de lots impliqués dans un change control ne peut être effectuée dès lors qu’un rapport sur l’incidence n’a pas été formalisé et approuvé par les personnes compétentes concernées.
1.5.1.6 Accès à toute la documentation
La QP doit se voir garantir un accès sans restriction à l’ensemble de la documentation issus de la chaîne d’approvisionnement de la substance active et du médicament jusqu’à l’étape de certification. Les rapports d’audits et d’inspections sont tenus à la disposition de la QP afin qu’elle puisse avoir connaissance de toutes informations obtenues concernant la compliance aux BPF sur les points sur lesquels elle engage sa responsabilité. La Personne Qualifiée doit également avoir accès aux détails nécessaires de l’AMM des produits qu’elle certifie.
27
1.5.1.7 Partage des responsabilités
Plusieurs sites peuvent être impliqués dans les différentes étapes de fabrication et de conditionnement, de contrôle et de stockage du lot avant qu’il ne soit soumis au processus de certification. Chacun de ces sites doit disposer d’une QP, ce qui permet, si cela est souhaité et formalisé, le partage de la responsabilité.
1.5.1.8 Incidence maitrisée
Toutes les investigations qui portent sur le lot à certifier (notamment les investigations liées à des déviations) doivent être menées jusqu’à un niveau suffisant pour appuyer la certification. L’impact des réclamations ou des rappels en cours doit être mesuré afin de démontrer que les conditions requises pour la certification sont toujours présentes.
1.5.1.9 Traçabilité des certifications
Lorsque toutes les conditions nécessaires sont respectées, survient alors la dernière étape du processus qui est l’enregistrement par la QP de sa certification du lot de médicament sur un document/registre dédié.
Ce registre doit indiquer que chaque lot de production satisfait aux dispositions de l’article 51 de la directive 2001/83/CE, telle que modifiée, ou de l’article 55 de la directive 2001/82/CE. Le document doit être tenu à jour au fur et à mesure des opérations effectuées et mis à la disposition des agents de l’autorité compétente pendant la période spécifiée dans les dispositions des États membres concernés, et en tout état de cause, au minimum durant cinq ans.
28
Prise en compte des évaluations de l’application des BPF
Dans certains cas, la QP se basera sur le bon fonctionnement du système de qualité pharmaceutique des sites impliqués dans la fabrication du produit, qui peut découler des audits réalisés par des tierces parties.
Une attention particulière doit alors être accordée à l’approbation des rapports d’audit : Le rapport d’audit doit porter sur les exigences générales de BPF, telles que, par
exemple, le système de gestion de la qualité, toutes les procédures appropriées relatives à la production et au contrôle de la qualité ayant trait au produit fourni ; Il convient de déterminer si la fabrication et le contrôle de la qualité de la substance
active et du médicament sont conformes aux BPF ;
Dans le cas d’activités externalisées, la conformité avec l’AMM doit être vérifiée ; La QP doit s’assurer qu’une évaluation et une approbation écrites finales des rapports
d’audit ont été effectuées. La QP doit avoir accès à l’ensemble de la documentation qui facilite l’examen des conclusions de l’audit ;
Les activités externalisées ayant une incidence critique sur la qualité du produit doivent être définies dans le respect des principes de la gestion du risque qualité ; Des audits répétés doivent être effectués conformément aux principes de la gestion
du risque qualité.
En effet, dans un contexte industriel, la QP ne peut généralement pas à elle seule s’impliquer étroitement dans chaque étape de la fabrication et dans chacune des exigences requises pour la certification. De ce fait, la QP chargée de certifier un lot de produit fini peut être amenée à se baser en partie sur l’avis et les décisions d’autres personnes, soit par connaissance personnelle, soit par la confirmation par d’autres personnes qualifiées dans le cadre d’un système de la qualité qu’elle a préalablement accepté.
29
Gestion des déviations non planifiées
La QP peut envisager la certification d’un lot lorsqu’une déviation inattendue relative au procédé de fabrication et/ou aux méthodes de contrôle analytique est survenue, uniquement si les spécifications du produit enregistrées sont respectées.
La déviation doit faire l’objet d’une enquête approfondie et la cause principale doit être corrigée. Le processus de gestion du risque qualité doit inclure l’évaluation de l’incidence potentielle de la déviation sur la qualité, la sécurité ou l’efficacité du ou des lots concernés. La soumission d’une modification de l’AMM peut être requise pour la poursuite de la fabrication du produit.
Libération des lots
Les lots de médicaments ne peuvent être libérés pour la vente ou la distribution sur le marché qu’après leur certification par une Personne Qualifiée. Jusqu’à ce qu’un lot soit certifié, il doit rester sur le site de fabrication ou être expédié en quarantaine vers un autre site approuvé à cette fin.
Des dispositifs de protection permettant de garantir que les lots non certifiés ne sont pas transférés vers un stock destiné à la vente doivent être mis en œuvre ; ils peuvent être de nature physique (recours à la séparation et à l’étiquetage des lots) ou de nature électronique avec l’utilisation de systèmes informatisés validés.
Lorsque des lots non certifiés sont déplacés d’un site autorisé vers un autre, les dispositifs de protection empêchant toute libération anticipée doivent être maintenus. Une notification formelle et sans ambigüité de la certification par la QP est requise pour permettre le transfert dans un stock destiné à la vente.
30
2
LA GESTION DES DEVIATIONS
2.1 QUELQUES DEFINITIONS
Les déviations
Une déviation se définie comme un écart par rapport à une procédure d'utilisation normalisée (SOP : standard operating procédure) ou un écart par rapport à une norme établie. (2)
2.1.1.1 La déviation au sens d’incident qualité
Un incident qualité est un évènement qui sort du cadre d'exploitation normale d'un service et qui entraîne ou peut entraîner une interruption ou une baisse de la qualité de ce service. (10) Une déviation au sens d’un incident qualité signifie donc l’apparition d’un écart par rapport aux critères d’acceptation définis : il s’agit d’une non-conformité.
Exemple 1 : l’encrassement d’une tête de marquage jet d’encre à l’origine d’un défaut de marquage.
Exemple 2 : le rendement de production n’est pas conforme, il est en dehors des tolérances définies.
31
2.1.1.2 La déviation au sens de dérogation
Dans certains cas, une déviation prendra le sens d’une dérogation. Il s’agira d’une autorisation de s'écarter des exigences spécifiées à l'origine, pour un produit ou un service, avant sa réalisation. Une telle dérogation est généralement accordée pour une quantité de produits et de services ou une durée limitée, et pour une utilisation spécifique. (11)
Exemple : à la suite d’un bogue informatique une déviation de procédure autorise de fonctionner en mode dégradé, les enregistrements pourront se faire manuellement et non plus électroniquement.
Les corrections
Une correction est une action visant à éliminer une non-conformité détectée (11). Il peut s’agir d’une destruction ou d’un retraitement (tri, reprise).
Les CAPA
Une action préventive est une action visant à éliminer la cause d'une non-conformité potentielle ou d'une autre situation potentielle indésirable, tandis qu’une action corrective est une action visant à éliminer la cause d’une non-conformité avérée et à éviter qu’elle ne réapparaisse.
Autrement dit, l’action corrective est entreprise pour empêcher la réapparition (récurrence) alors que l’action préventive est entreprise pour empêcher l'apparition (occurrence). (11)
32
2.2 MAITRISE DES DEVIATIONS (4)
Un système qualité pharmaceutique approprié pour la fabrication de médicament doit garantir que les résultats de la surveillance des produits et des procédés sont pris en considération dans l’investigation des déviations, et en vue de mettre en place des actions préventives pour éviter de potentielles déviations dans le futur.
Gestion et traçabilité procédurée
Des politiques, procédures, des protocoles et des rapports écrits, ainsi que, le cas échéant, les enregistrements des actions décidées ou des conclusions doivent être établis pour les investigations des déviations et des non conformités.
Toutes les déviations significatives sont enregistrées de façon détaillées et examinées, dans le but d’en déterminer la cause et de mettre en œuvre des actions correctives et préventives appropriées.
Analyse des causes
Un niveau approprié d’analyse des causes principales doit être appliqué pendant l’investigation des déviations, des défauts potentiels de produit et autres problèmes. Ceci peut être déterminé en utilisant les principes de la gestion du risque qualité.
Dans les cas où la véritable cause principale du problème ne peut être trouvée, l’attention doit être portée sur l’identification des causes les plus probables en vue de les traiter.
Lorsqu’une erreur humaine est suspectée ou identifiée comme étant la cause, cela doit être justifié, après avoir pris le soin de s’assurer que des erreurs ou problèmes liés au procédé, aux procédures ou au système n’ont pas été négligés, le cas échéant.
33
Intégration étendue
Les déviations et leur gestion impliquent l’ensemble des services d’un site de production ayant un rapport direct ou indirect avec le produit, c’est pourquoi les déviations et leur gestion s’intègrent à de nombreuses activités quels que soient leur lien et leur criticité sur la qualité du produit, tel que :
le contrôle de la qualité ;
le processus de qualification et de validation ; la revue annuelle qualité produit ;
le processus de certification des lots ; le processus de contrôle du changement ; le processus d’audit et d’inspection ; Etc.
Maitriser la gestion des déviations est une nécessité car en plus d’accroitre la connaissance sur le produit, les équipements et les procédés, cela permet, en aval de la clôture d’un évènement, d’améliorer et de faciliter de nombreux processus.
Une réponse contre les déviations : le système CAPA
L’entreprise pharmaceutique doit bénéficier d’un système de CAPA issues des investigations sur les non-conformités et les déviations. Des actions correctives et / ou actions préventives appropriées doivent être identifiées et mises en place systématiquement. Leur efficacité doit être surveillée et évaluée (CAPA de revue d’efficacité), conformément aux principes de gestion du risque qualité.
Les moyens mis en œuvre doivent être mesurés et adaptés, qu’ils soient préventifs et concerner la conception des équipements, la maîtrise du l’environnement de travail, la formalisation et la standardisation des dossiers de lot pour faciliter leur utilisation et contribuer à la limitation des écarts, ou correctif : il s’agirait alors des opérations d’investigation, des décisions sur la / les correction(s) à effectuer et la mise en place d’actions
34 correctives les plus pertinentes. La méthodologie CAPA doit faire l’objet de procédures standardisées, qui tiennent compte des étapes critiques et des différentes sources de déviations.
Les CAPA sont une nécessité, en plus de l’apport de garantie sur la qualité future, elle permette l’amélioration du produit et du procédé et améliore leur compréhension.
2.3 L’analyse de risque, point central de la maîtrise des déviations
Notion de risque
Le risque est la prise en compte d'une exposition à un danger, un préjudice ou autre événement dommageable, inhérent à une situation ou une activité. Autrement dit, le risque est défini par la probabilité de survenue (occurrence) de cet événement (incident ou accident) et par l'ampleur de ses conséquences (gravité) sur une cible donnée (qualité produit, sécurité patient). (12)
Nous pouvons noter que la notion de risque se distingue de celle du danger, en effet un danger est une source potentielle de dommage (conséquence défavorable sur la sureté). La définition de danger n’intègre pas la dimension de probabilité d’apparition ni celle de gravité.
Gestion du risque
2.3.2.1 Processus systématique
La gestion du risque qualité fait partie intégrante d’un système qualité pharmaceutique efficace, c’est un processus systématique d’évaluation, de maîtrise, de communication et d’examen des risques qualité du médicament tout au long du cycle de vie du produit.
35 Il est important de comprendre que la qualité du produit doit être maintenue pendant tout le cycle de vie du produit afin que les spécifications importantes pour la qualité du médicament restent conformes à celles déterminées lors des études cliniques. Une approche efficace de la gestion du risque qualité peut permettre de garantir un haut niveau de confiance de la qualité du médicament pour le patient en donnant des moyens d’identification et de maîtrise des dommages potentiels pendant la fabrication.
La gestion du risque qualité se doit de garantir deux points essentiels :
L’évaluation du risque qualité est basée sur la connaissance scientifique, l’expérience du procédé et, au final, s’assure de la protection du patient ;
le degré d’effort, de formalisation et de documentation du processus de gestion du risque qualité est proportionné au niveau de risque considéré.
Des exemples de processus et d’application de la gestion du risque qualité sont présentés, entre autres, dans l’ICH Q9 en partie III du guide des bonnes pratiques de fabrication. L’objectif de ce document est d’indiquer une méthodologie type pour gérer le risque et de proposer des outils pour identifier, par une approche structurée et rigoureuse, les caractéristiques ayant un impact sur la qualité du médicament, les points faibles d’un procédé, ceci afin d’augmenter la connaissance sur les produits et les processus et, in fine, de diminuer la probabilité d’occurrence et/ou la sévérité d’un risque et donc d’améliorer ainsi la qualité du produit.
36
2.3.2.2 Différentes approches
On peut approcher les risques sous trois aspects :
Les évènements qui les déclenchent : évènements causals. Les facteurs humains de tout type (volontaires ou non) et les causes accidentelles technique (aléa pur) sont les deux causes principales ;
La façon dont ils se manifestent : les modes de défaillance et leurs effets ; Les conséquences qu’ils entraînent.
Figure 3 : Deux façons d'approcher le risque
Le gestion du risque peut être appliquée de façon prospective, par exemple dans le cadre de projet, de contrôle du changement, de la validation de procédés, etc, ou de façon rétrospective comme pour l’identification des causes d’une déviations, l’analyse de l’incidence, la recherche de solutions (corrections), la réduction des récurrences (actions correctives) et l’éviction des occurrences (actions préventives).
37
2.3.2.3 Intérêts de la gestion du risque
Pour le patient
La gestion du risque permet de montrer que, dans le cadre des connaissances et des techniques du moment, les risques associés à la fabrication, à la distribution et à l’usage du produit ont été correctement identifiés, et que les dispositions pour en assurer la maîtrise ont été définies et mises en œuvre permettant ainsi de mettre à la disposition des patients des médicaments présentant les caractéristiques de qualité, sécurité et efficacité optimales et reproductibles.
Pour la qualité
La gestion du risque permet d’identifier au travers une approche structurée et rigoureuse les failles et faiblesses du système impliquant des interfaces entre différents services. L’intérêts pour la qualité se développe au travers de l’analyse, de l’évaluation et en somme de la maîtrise des risques.
Pour l’amélioration continue
La gestion du risque permet non seulement de bien connaître les produits, les équipements, les procédés et processus (analyse et évaluation) mais aussi de développer la démarche d’amélioration continue (maîtrise).
38
Approche rétrospective : l’analyse du risque d’une déviation
Dans une approche rétrospective du risque qualité par conséquence de l’occurrence d’une déviations il faut considérer les deux paramètres du risque de la manière suivante :
Occurrence(s) de la non-conformité ;
la gravité des conséquences potentiels sur la qualité du produit et l’impact pour le patient (sévérité).
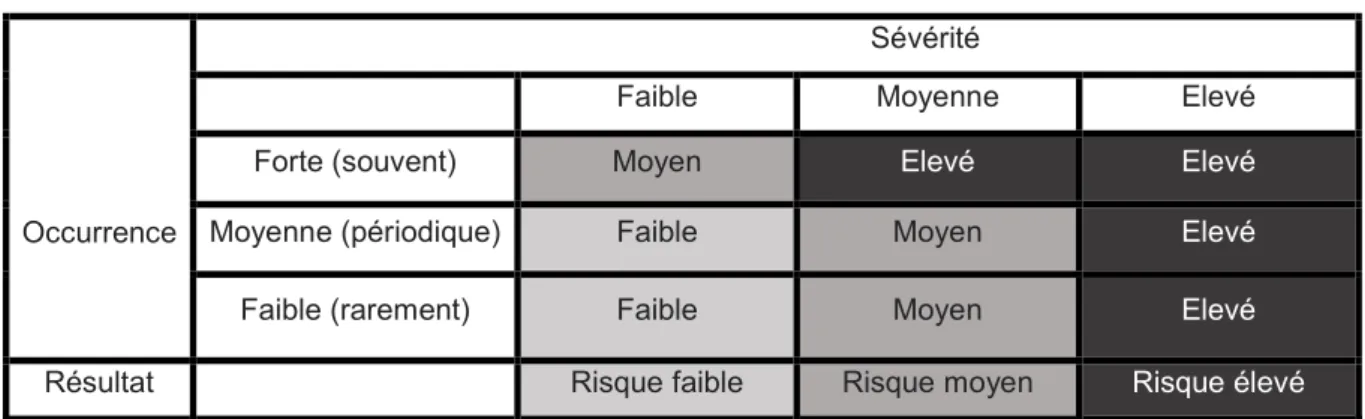
Tableau 1 : Modèle de détermination de la criticité
Occurrence
Sévérité
Faible Moyenne Elevé
Forte (souvent) Moyen Elevé Elevé
Moyenne (périodique) Faible Moyen Elevé
Faible (rarement) Faible Moyen Elevé
Résultat Risque faible Risque moyen Risque élevé
L’analyse du risque fait partie intégrante de la gestion des déviations. En effet lorsqu’un incident qualité survient l’analyse du risque intervient dès le début de l’investigation afin de déterminer la criticité de l’écart et de son mode de défaillance. Cette évaluation est nécessaire afin de mesurer l’ampleur des efforts à mettre en œuvre pour adopter une maîtrise proportionnelle à l’importance du risque.
Lors de l’analyse des solutions à mettre en œuvre pour contrer le risque (Corrections et CAPA), l’évaluation sur la réduction et / ou l’élimination potentielle du risque que les actions permettent, justifie les ressources allouées et apporte de la pertinence au choix.
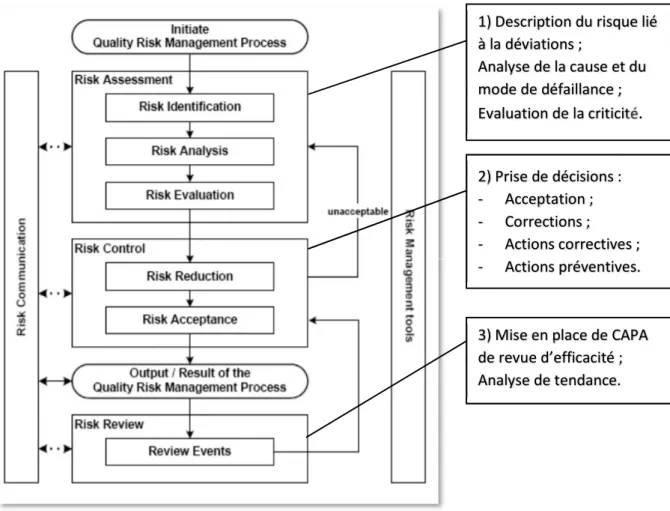
Un modèle de gestion du risque qualité est schématisé dans l’ICH Q9 sous forme d’un diagramme. Ce diagramme sur lequel est appliquée la méthodologie de gestion d’une déviation est présenté en figure 4.
39
Figure 4 : Calque de la gestion d’une déviation sur le procédé classique de gestion du risque qualité de l’ICH Q9 (6)
Chaque étape de ce diagramme peut varier d’un cas à un autre, il en va de même pour le procédé de gestion d’une déviation. Toutefois un processus robuste prendra en compte l’ensemble des étapes avec un niveau de détail adapté au risque / à la déviation considéré(e). Les prises de décision liées à des problèmes qualité peuvent être améliorées par l’utilisation de méthodes de gestion du risque qualité. Il est nécessaire de définir et d’utiliser une approche structurée permettant de prendre des décisions vraiment reliées au contexte, ce qui permettra de démontrer aux autorités compétentes ainsi qu’aux clients des garanties accrues quant à la capacité d’une entreprise à traiter les risques et les déviations.
1) Description du risque lié à la déviations ; Analyse de la cause et du mode de défaillance ; Evaluation de la criticité. 2) Prise de décisions : - Acceptation ; - Corrections ; - Actions correctives ; - Actions préventives.
3) Mise en place de CAPA de revue d’efficacité ; Analyse de tendance.
40
CHAPITRE SECOND : L’AMELIORATION CONTINUE DES
PRATIQUES
41 L’amélioration continue est un processus de mise en valeur du système de management de la qualité permettant d'améliorer les performances globales, de satisfaire aux exigences et d’être à l’écoute des clients. Cette démarche est constante, graduelle et implique tous les acteurs de l’entreprise. (13)
En effet, chaque axe d’amélioration peut amener de meilleures conditions de travail dans les ateliers de production, et participer au développement de la culture qualité des équipes opérationnelles. Cela permet une amélioration au quotidien, un changement de pratiques ou encore une nouvelle façon de penser. (14)
A travers cette dernière partie nous allons découvrir deux initiatives d’amélioration continue des pratiques liée aux deux missions de l’assurance qualité produit définies dans la première partie de cette thèse.
Ces initiatives ont été mises en place dans un contexte : celui des laboratoires UPSA et plus précisément les deux usines de production Agenaises. L’organisation et les méthodologies présentées ne sont donc pas inscrites comme références mais peuvent se présenter comme des exemples.
42
1
CONTEXTE INDUSTRIEL
1.1 LES LABORATOIRES UPSA (15)
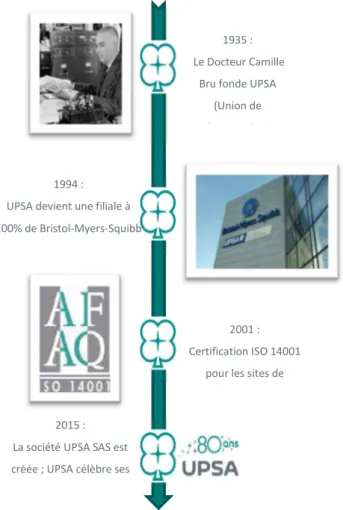
Historiquement spécialistes de la lutte contre la douleur et de l’automédication, les laboratoires UPSA (Union de Pharmacologie Scientifique Appliquée) ont été créés à Agen en 1935 par le Docteur Camille Bru.
Intégrée dans le groupe Bristol-Myers Squibb depuis 1994, UPSA est aujourd’hui à la fois une société et une marque. Depuis huit décennies, UPSA a développé une forte expertise dans la douleur, l’état grippal, la vitalité, la digestion et la qualité du sommeil. A travers sa gamme d’automédication, UPSA développe et met à disposition des patients des médicaments, compléments alimentaires et dispositifs médicaux pour soigner les maux du quotidien.
1935 : Le Docteur Camille
Bru fonde UPSA (Union de pharmacologie
1994 : UPSA devient une filiale à 100% de Bristol-Myers-Squibb
2001 : Certification ISO 14001
pour les sites de
2015 : La société UPSA SAS est créée ; UPSA célèbre ses
80 ans.
43 UPSA dispose de trois localisations en France. Le siège sociale se trouve à Rueil-Malmaison, la production se fait à Agen, les produits sont distribués à partir des sites d’Agen et de Fontenay-sous-Bois.
A l’échelle internationale, UPSA est présent dans 60 pays, principalement en Europe, en Afrique et en Asie. Annonçant une croissance à deux chiffres à l’international, UPSA s’est donnée comme objectif d’atteindre 60 % du chiffre d’affaires à l’export d’ici 2021. (16) Dédié pour l’essentiel au traitement de la douleur, UPSA est une référence dans la production de l’effervescence et produit également une large gamme de médication familiale destinée à lutter contre les maux du quotidiens. Une multitude de formes pharmaceutiques sont produites ; des formes sèches (comprimés, gélules et sachets), les formes liquides (sirops), des formes pâteuses (gels) ou encore des suppositoires ; ce qui apporte une grande diversité aux types d’atelier de production que l’on peut rencontrer, que ce soit pour la fabrication ou le conditionnement.
Trois localisations en France
① Siège social, Rueil-Malmaison (92) ② Production et distribution, Agen (47) ③ Distribution, Fontenay-sous-Bois (94)
44
Les sites de production d’Agen
La production des médicaments se fait sur deux sites de production : Guyenne et Gascogne, cependant il existe trois business units (BU), une sur Guyenne et les deux autres sur Gascogne. Une business unit est une unité organisationnelle au sein d’une entreprise définie autour d’un domaine d’activité. Une BU est dirigée de façon autonome avec des objectifs et des ressources propres (17). Accordé à cette séparation organisationnelle en trois unités de production, le service AQP d’UPSA est également divisé de la sorte. La haute direction est commune entre les trois BU, il en va de même pour les procédures réglementaires. Lorsqu’un projet global est initié, une attention particulière doit être apporté à l’applicabilité dans chacune des BU afin que les pratiques soient harmonisées.
Une stratégie pour la performance industrielle
Depuis Juin 2016 le groupe Bristol Myers Squibb redonne son autonomie à UPSA ; la création d'une entité dédiée aux activités UPSA au sein du groupe BMS en France permet de développer la marque à travers une stratégie propre et pleinement adaptée à la spécificité des produits de la marque. La création de l'entité UPSA SAS favorise ainsi la mise en œuvre d'une stratégie commerciale dédiée, adaptée aux marchés de la médication généraliste et grand public régis par des modes opératoires et des principes de fonctionnement propres. L'enjeu est également de renforcer les synergies des fonctions d’assurance qualité, de production, commerciales et de distribution pour faciliter la fluidité des circuits de décision et l'efficacité des process.
Ciblé aux sites de productions Agenais, la stratégie industrielle mise en place par la direction est claire :
Maintenir le haut niveau d’exigence qualité, sécurité et productivité. Maintenir la performance économique des sites à long terme.
C’est dans ce contexte que de nombreux plans d’amélioration continue sont mis évidence et que des projets d’optimisations voient le jour.
45
1.2 LE PROCESSUS DE REVUE DES DOSSIERS ETABLI
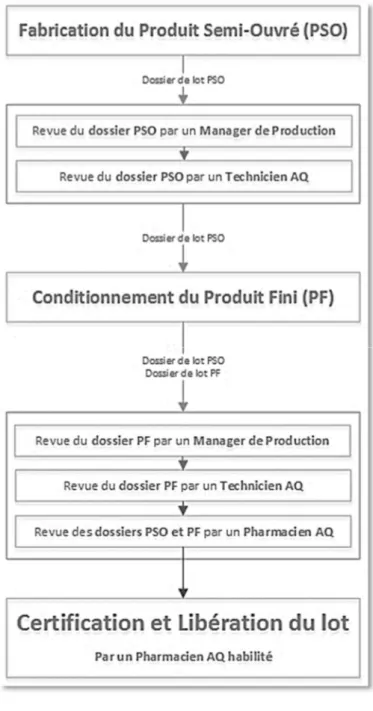
Le circuit de revue
Un dossier de lot de production est un document préétabli relatif aux opérations et aux conditions générales de fabrication et conditionnement spécifiques à chaque lot ; il est numéroté, agrée, daté et visé et rassemble tous les éléments nécessaires à la libération du produit final (Cf. § I.1.1.3.). Il est spécifique à une formulation, une taille de lot, un processus industriel.
Le dossier de lot suit le chemin logique de fabrication et de conditionnement du produit. Avant d’être transmis au pharmacien libérateur une étape préalable de vérification (revue) des dossiers a lieu.
La revue des dossiers de lots est une activité essentielle pour s’assurer de la conformité des produits pharmaceutique à leur dossier d’AMM. Cette revue s’assure également que le produit se conforme aux directives et procédures en vigueur et à d’autres textes législatifs et règlementaires comme ceux émis par la DGCCRF, et que le dossier de lot est renseigné dans le respect des règles documentaires des BPF (complété en temps réel, conformément aux instructions de conditionnement, avec un crayon bleu indélébile, etc.) et du data integrity (la notion de data integrity sera développé plus loin dans ce document).
Toutes les étapes de la production sont analysées afin de vérifier que les paramètres de chaque procédé ont été contrôlés et ont respectés les normes approuvées.
Comme vu en première partie de cette thèse il existe deux dossiers de lot : Le dossier de fabrication ou dossier du produit semi-ouvré (PSO) Le dossier de conditionnement ou dossier du produit fini (PF).
46 A l’issue de la fabrication et du conditionnement, il y a une première vérification par les conducteurs de procédé de fabrication et les conducteurs de ligne de conditionnement. Ceux-ci s’assurent qu’ils ont bien réalisé toutes les opérations et qu’ils ont bien renseigné toutes les informations nécessaires.
Le dossier du PSO une fois sorti de l’atelier est tout d’abord revu par un manager de production. Il peut s’agir d’un responsable de production ou d’un animateur d’équipe (AE).
47 Il est ensuite revu par un technicien AQ (TAQ) qui réalisera la libération informatique du PSO, dans le progiciel de gestion intégré, pour permettre le conditionnement (cette libération du PSO n’engage pas la possibilité de mise sur le marché car il ne s’agit pas d’une référence enregistrée à la vente, et la libération effectuée n’est pas celle appliquée au PF).
Après le conditionnement, manager de production et technicien AQ vont chacun leur tour revoir le dossier PF. Il est à noter que chaque revue de dossier (de fabrication et de conditionnement) est suivie d’un engagement de conformité signé de la part du vérificateur. Une fois que le dossier PF a été revu, les deux dossiers PSO et PF sont transmis au pharmacien AQ pour la revue finale en vue de la certification. Après avoir vérifié la conformité des deux dossiers, s’être assuré que le lot a été analysé conforme par le laboratoire de contrôle qualité, et avoir pris en compte l’absence de déviations, de change control ou tout autre évènement non clôturé, le pharmacien habilité peut alors certifier et libérer le lot.
Cette certification/libération se réalise dans un LIMS (Laboratory Information Management System), grâce à un accès sécurisé et propre au pharmacien habilité. Le LIMS implémente de façon automatique le registre de certification et transmet l’autorisation de déplacement du lot vers un stock destiné à la vente dans le progiciel de gestion intégré.
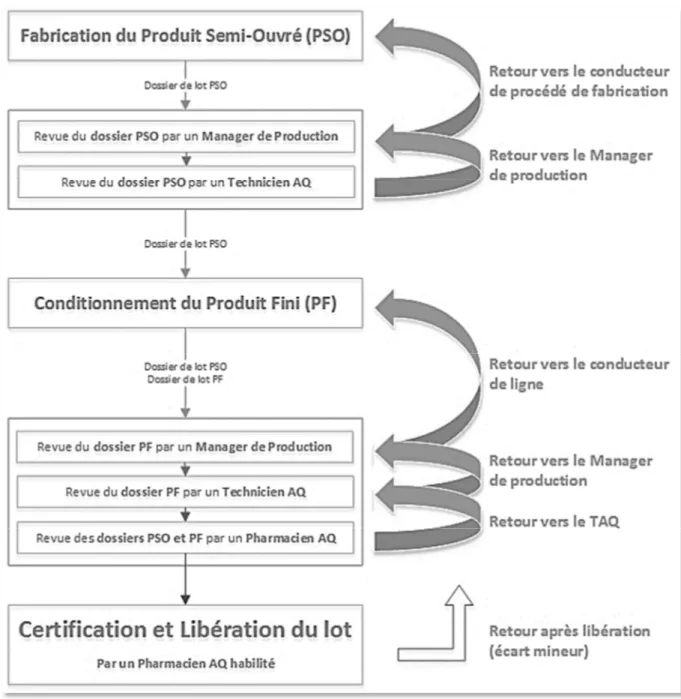
Les retours de dossiers
Lors de la revue d’un dossier de lot par l’assurance qualité produit, des questions peuvent se poser, des écarts peuvent être mis en évidence. Le dossier est alors renvoyé vers le personnel de production afin que des réponses soient apportées aux questions, et que les écarts soient justifiés ou corrigés.
48
Figure 8 : Application des retours au flux de revu des dossiers
Sur le logigramme présenté en figure 8, nous pouvons voir le schéma d’application des retours au flux de relecture des dossiers. De manière générale un dossier remonte le flux jusqu’à ce que le retour soit traité par la personne experte/compétente et/ou par la personne ayant réalisé les contrôles, les manipulations, etc.
49 Les retours sont dans la majorité des cas lié à des écarts, il en existe deux types :
Les écarts liés à la production/au produit : non-conformité de l’attribut d’un produit, non-conformité d’un paramètre du procédé, opération manquante ou non conforme, etc.
Les écarts documentaires : information manquante, erronée ou non compréhensible.
Ces écarts peuvent être classés en trois catégories de criticité :
Les écarts mineurs : ces écarts n’affectent pas la compréhension et sont sans impact sur la qualité du produit, le retour pourra être traité après libération du lot.
Exemples : donnée critique erronée/illisible/absente avec moyen de contrôle ailleurs dans le dossier ; saisie d’une donnée mineure erronée/illisible/absente sans impact ; etc.
Les écarts majeurs : ces écarts peuvent affecter la compréhension mais n’ont pas d’impact sur la qualité du produit. Toutefois, le retour devra être traité au plus vite car il est bloquant pour la libération du lot.
Exemples : test effectué mais non documenté ; donnée critique erronée/illisible/absente sans moyen de contrôle ailleurs dans le dossier ; utilisation de documentation non officielle ; etc.
Les écarts critiques : ces écarts ont un impact sur la qualité du produit, ils sont donc bloquant pour la libération. Les écarts critiques sont considérés comme des déviations et nécessitent une investigation.
Exemples : Manque test ; mauvais arrondi avec impact sur la conformité du produit ; modification des normes hors circuit d’approbation ; action réalisés/vérifiée par la même personne ; annexe manquante/erronée/incomplète ; etc.