3
Université de Montréal
La synthèse de prostacycline induite par le VEGF-A165 requiert l’hétérodirnérisation des récepteurs du VEGF
par
Paul-Eduard Neagoe Département de phanTiacologie
Faculté de Médecine
Mémoire présenté à la Faculté des Études Supérieures en vue de l’obtention du grade de
Maîtrees Sciences (M. Sc.) en pharmacologie /radecanf\\ Janvier 2005 7005
s
© Paul-Eduard Neagoe, 2005\J%/
uc8
Université
tJI’h
de Montréal
Direction des bibliothèques
AVIS
L’auteur a autorisé l’Université de Montréal à reproduire et diffuser, en totalité ou en partie, par quelque moyen que ce soit et sur quelque support que ce soit, et exclusivement à des fins non lucratives d’enseignement et de recherche, des copies de ce mémoire ou de cette thèse.
L’auteur et les coauteurs le cas échéant conservent la propriété du droit d’auteur et des droits moraux qui protègent ce document. Ni la thèse ou le mémoire, ni des extraits substantiels de ce document, ne doivent être imprimés ou autrement reproduits sans l’autorisation de l’auteur.
Afin de se conformer à la Loi canadienne sur la protection des renseignements personnels, quelques formulaires secondaires, coordonnées ou signatures intégrées au texte ont pu être enlevés de ce document. Bien que cela ait pu affecter la pagination, il n’y s aucun contenu manquant. NOTICE
The author of this thesis or dissertation has granted a nonexclusive license allowing Université de Montréal to reproduce and publish the document, in part or in whole, and in any format, solely for noncommercial educational and research purposes.
The author and co-authors if applicable retain copyright ownership and moral rights in this document. Neither the whole thesis or dissertation, nor substantial extracts from it, may be printed or otherwise reproduced without the author’s permission.
In compliance with the Canadian Privacy Act some supporting forms, contact information or signatures may have been removed from the document. While this may affect the document page count, it does flot represent any loss of content from the document.
Faculté des Études Supérieures
Ce mémoire intitulé:
La synthèse de prostacycline induite par le VEGF -A165 requiert l’hétérodimérisation des récepteurs du VEGF
a été évalué par un jury composé des personnes suivantes
André de Léan, Ph.D. Président-rapporteur Martin G. Sirois, Ph.D. Directeur de recherche Artur Femandes, M.D., Ph.D. Évaluateur externe
111
RÉSUMÉ
Nous avons précédemment démontré que la synthèse endothéliale du facteur d’activation plaquettaire (PAF) par le vascttÏar endotheÏiat grrni’thfactor(VEGF-A165) passe par l’activation du récepteur hornodirnérique VEGFR-2/R-2. Le VEGF-A165 est aussi capable d’induire la synthèse de prostacycline (PGI2), toutefois les récepteurs impliqués sont inconnus. Le VEGF-A165 (agoniste des récepteurs VEGFR-1, VEGFR 2 et NRP-l) et le VEGF-AI2I (agoniste des récepteurs VEGfR-1 et VEGFR-2) (l0 M) augmentent la synthèse de PGI7 de 70 et 40 fois respectivement par rapport aux valeurs témoins dans les cellules endothéliales d’aortes bovines (BAEC). Cependant, aucun agoniste des récepteurs VEGFR-l (VEGf-B et P1Gf) ou VEGFR-2 (VEGF-C) n’a induit la synthèse de PGJ2. Un traitement des BAEC avec des antisens oligomères dirigés contre l’ARNrn des récepteurs VEGFR-1 ou VEGFR-2 a diminué jusqu’à 79% la synthèse de PGb induite par le VEGF-A165. De plus, un traitement avec l’inhibiteur de VEGFR-2 a inhibé de 95% l’effet du VEGF -A165 sur la relâche de PGI7. Ensemble, ces résultats suggèrent que l’hétérodimérisation de VEGFR-l/R-2 est essentielle pour la production de PGI2 et que la NRP-1 potentialise les effets du VEGF-A165. Par la suite, nous avons démontré que l’activation des récepteurs VEGFR-2/R-2 et VEGFR l/R-2 mène à la phosphorylation de la p42144 MAPK et de la cPLA2, qui est nécessaire à la relâche d’acide arachidonique (AA). De plus, la synthèse de PGI2 passe par l’activation de la COX-1, qui convertit en cascade 1’AA en PGG2 et en PGH2. La conversion de la PGH2 en PGI2 nécessite la dénitrosylation de la PGI2 synthase qui est induite par le VEGF -A165 contrairement au VEGF-C.
ABSTRACT
We previously reported that vascular endothelial growth factor (VEGF-A165) is mediating endothelial platelet-activating factor (PAF) synthesis upon activation of hornodirneric receptors VEGFR-2/R-2. VEGF-A65 is also capable of mediating prostacyclin (PGI2) synthesis, but the VEGF receptors implicated are stili unknown. VEGF-A165 (VEGFR-1, VEGFR-2 and NRP-l agonist) and VEGf-A121 (VEGFR-l and VEGFR-2 agonist) induced a 70- and 40-fold increase in PGI2 production as compared to the basal level in bovine aortic endothelial ceils (BAEC). However, the VEGFR-l agonists (VEGF-B and PIGF) and VEGFR-2 agonist, VEGF-C, did flot increase significantly the PGI2 release. Treatment of BAEC with antisense oligonucleotides targeted against VEGFR-I or VEGFR-2 rnRNA decreased the release of PGI2 induced by VEGF-A165 up to 79%. Furtherrnore, treatrnent of EC with a specific VEGFR-2 inhibitor dirninished by 95% the PGI2 release induced by VEGF-A165. Taken together, these resuits are suggesting that PGI2 synthesis in EC requires VEGFR-1/R-2 heterodirnerization and that NRP-1 coreceptor is potentiating VEGF-A165-rnediated PGI2 release. In addition, we found that VEGFR-2/R-2 or VEGFR-l/R-2 activation lead to p42144 MAPK and cPLA2 phosphorylation and subsequent activation, which is required for arachidonic acid (AA) release. Furthermore, PGI7 synthesis requires COX-l activation, converting the AA in PGG2 and the latter in PGH2. The conversion of PGH2 in PGI2 requires the PGI2 synthase denitrosylation, which is induced by VEGF-A165 as opposed to VEGf-C.
V
TABLE DES MATIÈRES RÉSUMÉ
ABSTRACT iv
LISTE DES FIGURES vii
REMERCIEMENTS ix
1.0 INTRODUCTION
1.1 BIOLOGIE DU SYTÈME VASCULAIRE
1.1.1 L’endothéliurn et ses fonctions physiologiques 1 1.1.2 Les fonctions physiologiques de l’endothélium 2 1.1.3 Les fonctions pathologiques de l’endothélium 3 1.2 LE VASCULAR ENDOTHELIAL GROWTH FACTOR (VEGF) 4
1.2.1 Origines et fonctions générales du VEGF 4
1.2.2 Isoformes et analogues du VEGF 5
1 .2.3 Généralités sur les récepteurs du VEGF 8
1.2.4 Le VEGFR-1, récepteur de clairance 10
1.2.5 Le VEGFR-2, médiateur principal des actions du VEGF-A 12
1.2.6 La neuropiline, corécepteur du VEGFR-2 13
1.2.7 Le VEGF R-3, un récepteur des vaisseaux lymphatiques 14
1.2.8 Dimérisation des récepteurs du VEGF 17
1.3 TONUS VASCULAIRE ET PROSTACYCLINE (PGP,) 19
1.3.1 Principaux régulateurs du tonus vasculaire NO et PGI2 19 1.3.2 Mécanismes de synthèse de la prostacycline 20 1.3.3 Rôles de la prostacycline et les inducteurs de sa synthèse 23
1.3.4 La nitrosylation un phénomène nouveau et opposé à la
phosphorylation 24
1.4. Problématique et but de l’étude 25
2.0 ARTICLE 26 SUMMARY 31 INTRODUCTION 33 METHODOLOGY 35 RESULT$ 40 DISCUSSION 47 REFERENCES 54 FIGURE LEGENDS 59 FIGURES 64 3.0 DISCUSSION 74
3.1 Les récepteurs du VEGF impliqués dans la synthèse de PGI2 75 3.2 L’hétérodimérisation des récepteurs VEGFR- Y et VEGFR-2 est nécessaire
pour la synthèse de prostacycline 79
3.3 Voies de signalisation de la synthèse de PGI2 induite par le VEGF -A165... $0 3.4 Différences et similitudes entre la signalisation des récepteurs
homodirnères et hétérodimères du VEGF $2
4.0 CONCLUSION $5
vii
LISTE DES FIGURES
Figure 1: Les isoformes du VEGF 7
Figure 2: Les récepteurs du VEGF et leurs analogues respectifs 9 Figure 3: Voies de signalisation induites suite à l’activation des récepteurs du VEGF. ..16 figure 4: Voie de synthèse des prostaglandines à partir de l’acide arachidonique 22
Article
figure 1: PGL synthesis rnediated by VEGF-A isoforrns 64 figure 2: NRP-1 coreceptor potentiates PGI2 synthesis induced by VEGF-A165 65 figure 3: Effect ofVEGFR-1 and VEGFR-2 agonists on PGI2 synthesis 66 figure 4: Expression and regulation ofVEGF receptors by colTesponding agonists 67 Figure 5: Heterodimerization capacity ofVEGFR-l and VEGFR-2 subunits 6$ Figure 6: Contribution ofVEGF receptors on PGI2 synthesis induced by VEGF-A
isoforrns 69
f igure 7: Regulation ofVEGF receoptors phosphorylation by selective inhibitors 70 figure 8: CelI signaling pathway by which VEGF-AI65 promûtes PGÏ2 synthesis 71 figure 9: $-Nitrosylation ofPGI2 synthase in presence ofVEGF-A165 and VEGf-C 72 Figure 10: Proposed signaling pathway by which VEGF-A165rnediates PGI2 synthesis..73
LISTE DES ABRÉVIATIONS
AA Acide arachidonique
bFGF : BasicfibrobÏast growthfctctor BAEC : Cellules endothéliales d’aorte bovine CE : Cellules endothéliales
CMLv : Cellules musculaires lisses vasculaires
COX : Cyclooxygénase
cPLA2 Phospholipase A2 cytosolique
eNOS Synthase endothéliale du monoxyde d’azote iPLA2 Phospholipase A2 indépendante du calcium MAPK : Mitogen-activatedprotein kinase
MKK MA?kinase kinase
NO Monoxyde d’azote
NRP-1 : Neuropiline-l
PAF Facteur d’activation plaquettaire
PGI2 Prostacycline
PLA2 : Phospholipase A2
PIGF PlacentaÏ growth factor
PLC Phospholipase C
sPLA2 : Phospholipase A2 sécrétée
VEGF VascuÏar endotheÏial groii’thfiictor
ix
REMERCIEMENTS
Je tiens à remercier le Dr Martin G. Sirois, pour m’avoir accueilli dans son laboratoire et de m’avoir aidé à développer mon «assiduité scientifique» en me permettant d’effectuer un stage et la formation de maîtrise. Ses conseils, ses idées et son support financier m’ont grandement aidé dansma démarche scientifique.
Je remercie le Département de pharmacologie et la faculté des études supérieures de l’Université de Montréal pour leur support financier et la qualité des études offertes.
Je remercie mes collègues de travail pour leur soutien moral et scientifique, Judith f avier, Stéphanie Lapointe, Catherine Marchand, Katty Mallet, Pascal Bernatchez, Simon Rollin. Un merci spécial à Ricardo Maliba et Alexandre Brkovic, puisque avec eux, 1’ «atmosphère de travail » dans le laboratoire est «très décontractée ».
Je remercie Dr André de Léan, Dr Martin G. Sirois et Dr Artur Fernandes pour l’évaluation de mon mémoire de maîtrise.
Le plus important des merci est cependant adressé à mes parents, pour leur support financier, moral et leurs conseils qui se sont toujours avérés judicieux. Sans leur support, je n’aurais pu me rendre aussi loin.
1.1 BIOLOGIE DU SYTÈME VASCULAIRE
1.1.1 L’endothélium et ses fonctions physiologiques
L’endothélium, composé de cellules endothéliales (CE), tapisse l’intérieur des vaisseaux sanguins, constituant ainsi le seul point de contact avec le sang. Depuis sa découverte au milieu du XIXe siècle, l’endothélium fait l’objet d’une attention particulière, puisque son rôle est davantage de maintenir une homéostasie au niveau de l’échange de nutriments que d’être seulement une barrière physique entre le sang et les tissus. Cette homéostasie se traduit par les multiples fonctions de l’endothéljum. Tout d’abord l’endothéliurn vasculaire est une barrière de perméabilité sélective, lui permettant de filtrer les substances nocives pour l’organisme. Ensuite, c’est une barrière hémocompatible composée d’un tissu synthétique, métabolique et sécrétoire. L’endothélium joue aussi un grand rôle dans le maintien de l’équilibre des propriétés thrombogénique et anti-thrombogénique, conférant au sang sa viscosité optimale. Le tonus vasculaire est aussi un élément qui est finement régulé par les cellules endothéliales, qui peuvent moduler la vasoconstriction ou la vasodilatation en envoyant des messages appropriés aux cellules musculaires lisses. Les CE sont les premières à participer dans le processus d’angiogenèse, qui permet entre autres, la vascularisation des régions tissulaires lésées, favoi-isant ainsi la réparation tissulaire. Ces rôles combinés assurent un fonctionnement optimal du système vasculaire et contribuent ainsi à l’homéostasie.
1.1.2 Les fonctions physiologiques de l’endothélium
L’angiogenèse se définit comme le processus de formation de nouveaux vaisseaux sanguins à partir de vaisseaux préexistants, la distinguant de la vasculogenèse, qui est une formation de vaisseaux de novo, à partir des hémangioblastes [1]. Ce dernier processus, permet notamment la formation d’un plexus vasculaire primitif dans l’embryon, premier signe de développement du système cardiovasculaire [2, 3]. L’angiogenèse permet à ce plexus vasculaire de prendre de l’expansion et d’avoir une meilleure organisation avec, comme finalité, le système cardiovasculaire
[4].
L’angiogenèse peut être tant un processus physiologique, que pathologique. Durant les changements cycliques de l’endométriurn [5, 6], lors de la guérison d’une blessure profonde [7], ou en conditions ischémiques, nous sommes en présence de phénomènes physiologiques durant lesquels, l’angiogenèse joue un rôle positif.Le premier facteur proangiogénique découvert fut le bctsic fibrobÏctst Growth factor [8], qui jusqu’à aujourd’hui, est considéré comme le principal facteur angiogénique. Cependant, la découverte d’autres facteurs angiogéniques, tels le Vascittar EndotheliaÏ GrowtÏi Factor (VEGF) et les angiopoïétines, a permis de mieux comprendre le fonctionnement de l’angiogenèse. Toutefois, il a été démontré que le VEGF est le seul facteur proangiogénique ayant des activités proinfiammatoires, cette dernière observation a suscité un engouement particulier, expliquant ainsi pourquoi le VEGF est l’un des facteurs de croissance les plus étudié dans le système vasculaire.
1.1.3 Les fonctions pathologiques de l’endothélium
Les pathologies vasculaires et les tumeurs sont régulées par l’hypoxie, l’inflammation et l’angiogenèse. En effet, l’angiogenèse pathologique est souvent une réponse de l’organisme aux effets de l’hypoxie due à l’éloignement du site pathologique des vaisseaux sanguins existants. L’inflammation est un processus de défense contre une agression invasive des tissus. Elle se caractérise par un changement des propriétés de l’endothélium, qui devient plus perméable et entraîne éventuellement la fonnation d’oedème et de rougeur. Les néovaisseaux sanguins tumoraux sont caractérisés par leur désorganisation et leur nombreuses fenestrations. Ces conditions entraînent un état hypoxique dans certaines parties de la tumeur, qui, couplée à une surexpression des facteurs proangiogéniques, induit des nouveaux processus angiogéniques. De plus, la pennéabilité vasculaire élevée, un indicateur de la réponse inflammatoire, est prédominante dans ces néovaisseaux tumoraux et entraîne, également à son tour, la relâche des facteurs de croissance tumoraux et vasculaires. Les pathologies vasculaires non tumorales, comme la plaque athérosclérotique ou l’arthrite rhurnatoïde, sont aussi régulées par l’hypoxie et l’inflammation, mais possèdent des vaisseaux sanguins plus structurés et moins fenestrés. Le remodelage vasculaire fait partie du processus angiogénique induit par l’inflammation. En effet, lors de la réponse inflammatoire, les cellules du système immunitaire relâchent plusieurs facteurs angiogéniques, qui vont procéder â une réorganisation des vaisseaux sanguins existants et même en former de nouveaux. Ce phénomène, appelé le sii’itch angiogénique, est difficilement détectable au niveau temporel, ce qui explique la difficulté de traiter ces pathologies [4].
4
1.2 LE VASCULAR ENDOTHELIAL GROWTH FACTOR (VEGF)
1.2.1 Origines et fonctions générales du VEGF
Le VEGF a été découvert au début des années $0, comme une molécule de 34 à 42 kDa sous le nom de Vctsctt/ar PerrneabiÏity Factor (VPF). Cette molécule était secrétée par des cellules tumorales et induisait la pennéabilité microvasculaire chez les rats et les hamsters [9]. En 1989, Fen-ara et coil. ont purifié une molécule par des techniques de combinaison d’affinité à l’héparine, chromatographie d’affinité à l’héparine-sépharose et HPLC. Ils ont trouvé que cette molécule possédait les mêmes propriétés et la même structure que le VPF, ses effets étant spécifiques aux cellules endothéliales, ils la nommèrent donc, le VascuÏar Endothetial Grrni’th Factor (VEGF,) [10]. Au cours des années, il a été possible de démontrer la capacité du VEGF à induire plusieurs activités biologiques, notamment dans la formation du plexus vasculaire à partir d’angioblastes, dans la vasculogenèse, et dans la formation du système vasculaire mature à partir du plexus vasculaire primaire, par l’entremise de l’angiogenèse [4]. En induisant un influx de calcium, le VEGF augmente aussi la conductivité hydraulique des vaisseaux sanguins [11]. Cette induction de la penTléabilité vasculaire est nécessaire dans les premières étapes de l’angiogenèse [12]. Le VEGF est une molécule multifonctionnelle, capable de promouvoir la migration et la prolifération des CE ainsi que l’angiogenèse. Toutefois, le VEGF se distingue des autres facteurs de croissance tels le EGF et le bfGF par sa capacité d’induire l’inflammation [13, 14]. En effet, le VEGF est le seul facteur de croissance proangiogénique ayant une activité proinflammatoire, ce dernier effet étant médié par sa capacité d’induire la
synthèse du facteur d’activation plaquettaire (PAF), qui augmente la perméabilité vasculaire et contribue ainsi à l’angiogenèse pathologique [15-17].
1.2.2 Isoformes et analogues du VEGF
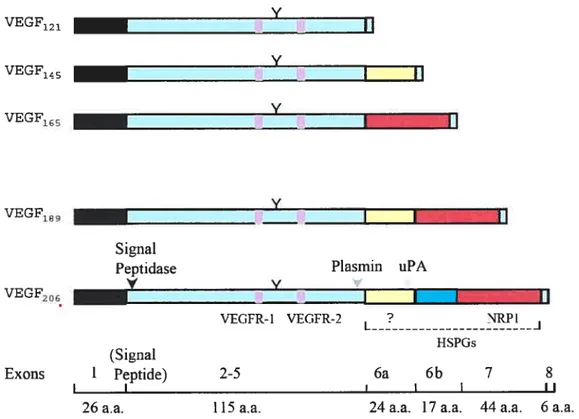
Le gène du VEGF-A est finement régulé par épissage alternatif, ce qui lui peni-let d’être secrété sous plusieurs isofonues comportant 121, 145, 165, 189, ou 206 acides aminés (120, 144, 164, 18$ et 205 chez la souris) [1$-20]. Le VEGF-A165 est l’isoforme le plus abondamment retrouvé dans la circulation sanguine, pouvant induire tous les effets biologiques associés au VEGF, ce qui en fait l’isofonue le plus étudié jusqu’à présent. Les exons 6 et 7 du gène du VEGF confèrent des propriétés différentes concernant la sécrétion et la liaison membranaire du VEGF. En effet, l’isofonue 121 du VEGF ne possède pas ces exons, ce qui en fait l’isoforme le plus soluble dans la circulation libre, ne possédant aucune affinité envers la matrice extracellulaire (MEC) contenant de l’héparan sulfate [21]. L’isoforme 165 possède le transcrit de l’exon 7, lui conférant la possibilité de liaison à la neuropiline. Les isoformes 189 et 206, qui possèdent l’exon 6 et 7, se retrouvent complètement séquestrés au niveau de la matrice extracellulaire [20] et ne peuvent être relâchés que par clivage enzymatique [22, 23]. Les autres isoforrnes du VEGF possèdent des affinités variables envers la MEC, le résultat étant un équilibre entre la forme soluble et la forn-ie liée [21]. Même s’il a été démontré que certains isoformnes sont à eux seuls capables de développer un réseau vasculaire chez la souris [24, 25], l’organisation de ce réseau est fortement influencée par le type d’isoforme présent pendant son développement. Ainsi, l’organisation adéquate de ce réseau n’est accomplie que par la présence de tous ces isoformes [26]. Le VEGF-A a
6 plusieurs analogues: Placenta! Growth Factor (P1GF), VEGF-B, VEGF-C, VEGF D et VEGF-E. Le PIGF a 53% d’homologie avec le VEGF-A165 et présente aussi les mêmes sites de glycosylation que ce dernier [27] alors que le VEGF-B a 43% d’homologie avec le VEGF-A165 et seulement 30% avec le P1GF, ne présentant pas de site de glycosylation [2$]. Le VEGF-C est identique à seulement 30% avec le domaine d’homologie du VEGF -A165, étant encodé seulement par les exons 3 et 4 du gène [29], alors que le VEGF-D présente 61% d’homologie avec le VEGF-C, mais seulement 11,8% d’homologie avec le VEGF-A165 [30]. Finalement, le VEGf-E est un homologue du virus Orf qui présente de 17 à 27% d’homologie protéique avec le VEGF -A165 [31].
VEGF121 VEGF15 VEGF165 VEGF189 Signal Peptidase V V X V II I I
Figure 1 : Les isoformes du VEGF
Le gène du VEGF produit plusieurs isoformes par épissage alternatif. Le VEGF206 est le seul à contenir la totalité des exons, et plus spécifiquement, les exons 6 et 7 en totalité, lui conférant ainsi une séquestration complète à la matrice extracellulaire. Le VEGf165 possède l’exon 7, lui permettant ainsi de se lier à la neuropilline-1, tandis que le VEGF121 manque les exons 6 et 7 étant incapable de lier le corécepteur NRP- 1. (Stringer et coli. , The spÏice variants of vascular endotheÏiaÏ growthjactor
(VEGF) andiheirreceptors, Journal ofCell Science, 2001. 114 : 853-$65) Y I II VEGF206 Exons Plasrnm uPA , Y VEGfR-1 VEGFR-2 L ? NRPI HSPGs (Signal Peptide) 2-5 6a 6b 7 8 I I I II
$
1.2.3 Généralités sur les récepteurs du VEGF
Les analogues du VEGF peuvent lier trois récepteurs différents, soit le VEGFR-1 (Fit-1), VEGFR-2 (Flk-l ou KDR) ou VEGFR-3 (Fit-4). Ces récepteurs sont de type tyrosine kinase et forment une sous-famille caractérisée par sept domaines extracellulaires ressemblant à l’immunoglobuline, une seule région transmembranaire et un double domaine tyrosine kinase (Figure 1). Les trois types de récepteurs ont été localisés à la surface des cellules endothéliales de vaisseaux sanguins [32], mais le VEGFR-3 est surtout localisé sur la surface des cellules endothéliales des vaisseaux lymphatiques [33]. Ces récepteurs, sont présents sous forme dc monomère à la surface cellulaire mais ils se dirnérisent en présence du ligand. Les différents analogues du VEGF peuvent recruter des homodimères, ce qui signifie le recrutement de deux récepteurs semblables, mais il a aussi été démontré que certains ligands peuvent recruter des hétérodimères formés de VEGFR- I et VEGFR-2 [34, 35], ou de VEGFR-2 et VEGFR-3 [36].
10
1.2.4 Le VEGFR-1, récepteur de clairance
Le VEGFR-1 (Fit-1) a été identifié et cloné en 1992 par le groupe de Williams [37]. Ce récepteur a un poids moléculaire de 180 kDa et peut lier avec haute affinité (Kd 10 pM) le VEGF-A, VEGf-B et le PÏGF. Cependant, c’est seulement en 1995, qu’il a été possible de déterminer par l’entremise d’une souris knockottt Flt-F’, que ce récepteur est essentiel à l’organisation de la vasculature embryonnaire, mais n’est pas nécessaire pour la différenciation des cellules endothéliales. Par ces faits, le knockout était létal pour l’embryon au jour 8.5 [38]. Cependant, le VEGF R-1 a été rapidement associé à un récepteur de clairance, puisque la mutation du domaine intracellulaire tyrosine kinase du récepteur n’entraîne aucune létalité ou changement morphologique chez les souris in utero aux jours 8.5 et 9.5 [39]. Cette hypothèse de récepteur de séquestration a été appuyée par la découverte d’une forme soluble du VEGFR-1, composée seulement du domaine extracellulaire du VEGFR-l, qui agirait comme un antagoniste naturel du VEGF [40]. Cependant, d’autres études ont permis de conférer des rôles importants à ce récepteur dans le développement embryonnaire, soit la régulation de la migration et l’adhésion des cellules endothéliales et l’organisation des vaisseaux sanguins [38, 41, 42].
Les études de l’activation de VEGFR-1 ont été effectuées soit sur des récepteurs transfectés dans des cellules n’exprimant pas à l’origine le VEGFR-1, soit dans des cellules primaires exprimant de façon native ce récepteur. Ces études ont permis de démontrer une faible augmentation de deux fois de l’activité tyrosine
kinase dans les cellules natives, lorsque stimulées avec le VEGF-A [43]. Il est possible que la faiblesse de l’activité tyrosine kinase native soit due en partie au manque de sites sur des séquences consensus de phosphorylation. Cependant, lorsque ces récepteurs sont surexprimés dans des cellules transfectées, l’activation du VEGFR-1 a permis d’identifier les sites d’autophosphorylation dans le domaine tyrosine kinase, soit la tyrosine 1169 (Yl 169) et Y1213. Seule la Y1169 joue un rôle dans l’initiation de la cascade enzymatique de signalisation permettant l’activation de la PLCy, un des transducteurs de signaux majeurs au noyau cellulaire [44]. Une autre étude a permis d’identifier les sites Y1213 et Yl242 comme sites majeurs d’autophosphorylation ainsi que deux autres sites mineurs, Y1327 et Y1333. Les sites Y1213 et Y1333 ont démontré une capacité de lier PLCy alors que les domaines Grb2 et SH2 ont de l’affinité pour Y1213 [45]. Le domaine SH2, faisant partie de la phospholipase Cy (PLOy), possède une activité enzymatique, alors que le domaine Grb2 est un domaine adaptateur, qui est un transducteur de signal en régulant les interactions protéine-protéine. Il a aussi été démontré que la sous-unité p$5 de la PI3K peut se lier à la Yl213 et ainsi être activée [46]. Le VEGfR-l peut donc transmettre des signaux anti-apoptotiques, donc favoriser la survie cellulaire en activant la PI3K. En activant le PLC’y, VEGFR-l active par le fait même la PKC et la p42144 MAPK. Les premières activités biologiques induites par l’activation du VEGFR-1 ont été l’induction de la migration de monocytes par un effet chemotactique [47J et la production du facteur de croissance du tissu conjonctif [4$]. De plus, il a récemment été démontré dans notre laboratoire que le P1Gf et le VEGF-B peuvent induire la transiocation de la P-sélectine endothéliale et l’adhésion
12
des neutrophiles à l’endothélium, en activant le VEGF R-1 [49]. Par conséquent, il n’est donc pas clair dans quelles circonstances le VEGFR-1 agit comme un récepteur de clairance ou comme un récepteur tyrosine kinase, en activant la voie de
signalisation de la PLCy et subséquemment la PKC, connue pour jouer un rôle important dans l’angiogenèse [50, 51].
1.2.5 Le VEGFR-2, médiateur principal des actions du VEGf-A
Le VEGfR-2 fut découvert en 1992 et fut nommé KDR (Kinase Dornain Region) [52]. Son poids moléculaire est de 200-230 kDa et son affinité pour le VEGF-A est moindre que celle du VEGFR-1, se situant entre 75 et 125 pM [53]. Le VEGFR-2 a plusieurs ligands faisant tous partie de la grande famille du VEGF. Ainsi, le VEGF-A, VEGF-E et les formes matures du VEGF-C et VEGF-D peuvent lier et activer le récepteur 2 du VEGF [54-57]. Ce récepteur est exprimé dans les cellules endothéliales du système sanguin et lymphatique [36]. Son rôle au niveau embryonnaire se situe surtout au niveau de la vasculogenèse, en stimulant la migration et la prolifération des cellules endothéliales, et subséquemment la maturation vasculaire [58, 59]. Le VEGFR-2 joue un rôle prédominant dans la médiation des processus physiologiques et pathologiques induits par le VEGF. Ainsi, suite à la phosphorylation du VEGFR-2 par son ligand, plusieurs tyrosines dans le domaine kinase intracellulaire ont été identifiées comme sites d’autophosphorylation menant à des cascades enzymatiques diverses. Ainsi, la Y1175 est responsable du recrutement et de l’activation de la PLCy, menant à l’activation de la p42/44 MAPK [60], ainsi que le recrutement de la protéine adaptatrice Sck qui aide au couplage du VEGFR-2 avec la p42/44 MAPK [61],
menant à une augmentation de la synthèse d’ADN et subséquemment, de la prolifération cellulaire. L’autre tyrosine impliquée dans la signalisation de PLCy est la Y95 1, qui active cette dernière par l’intermédiaire de VRAP (VEGfR-associated protein), qui participe aussi au recrutement de la PI3K [62]. Récemment, la Y1214 a été déterminée comme responsable de l’activation de la Cdc42, qui active ultérieurement la SAPK2 (Stress-activated Protein Kinase)/p38 MAPK, deux protéines essentielles au remodelage de l’actine dans les fibres de stress. Ces cascades enzymatiques mènent à l’activation de la MAPKAPK-2 (MAPK ctctivated
protein kinase-2,) et Hsp27 (Heat shock protein 27,), déclanchant ainsi un
remodelage de l’actine et ainsi la motilité cellulaire [63]. Cette voie de signalisation mène à la polymérisation de l’actine et le rassemblement d’adhésions focales, convergeant vers la fonuation de fibres de stress et la contraction des cellules endothéliales qui est nécessaire pour induire la migration et la prolifération cellulaire. Deux autres tyrosines, la Y1054 et Y1059 participent à l’autophosphorylation suivant l’activation du récepteur, mais elles ne sont associées à aucune molécule adaptatrice [43]. Le VEGF R-2 peut donc moduler plusieurs activités biologiques comme la relâche de NO et de prostacycline via l’activation de c-Src [64] et de la PI3K [65], la translocation de la P-sélectine, l’adhésion des neutrophiles [49] et la synthèse de PAF [66].
1.2.6 La neuropiline, corécepteur du VEGfR-2
L’activité du VEGFR-2 peut être modulée par les protéoglycans semblables à l’héparine associés à la membrane cellulaire, qui peuvent agir comme corécepteurs et lier le VEGF-A, augmentant ainsi sa capacité de liaison au récepteur [67].
14
Cependant, le principal corécepteur du VEGFR-2 est la neuropiline (NRP). Il existe deux types de NRP, soit NRP-1 et NRP-2 [68], les deux pouvant être des récepteurs pour les sémaphorines, des molécules guidant les axones par l’effet de répulsion en liant la collapsine [69]. Dans leurs fonctions de corécepteurs pour le VEGFR-2, elles sont impliquées dans le développement vasculaire (NRP-1) et le développement lymphatique (NRP-2) [70, 71]. Le principal corécepteur est la NRP-l, que nous retrouvons exprimé sur les cellules endothéliales et tumorales. La NRP-1 a été classée comme un corécepteur dû à son manque d’un domaine tyrosine kinase intracellulaire, lui conférant ainsi l’incapacité d’activer des voies de signalisation intracellulaires. Cependant, ce corécepteur aide à stabiliser la liaison entre le VEGF A65 et le VEGFR-2, ainsi, il augmente l’efficacité de transduction du signal [72]. Une seule étude a été capable de démontrer jusqu’à maintenant la capacité directe de la NRP-l d’activer des voies de signalisation menant à la migration cellulaire [73].
1.2.7 Le VEGFR-3, un récepteur des vaisseaux lymphatiques
Le VEGFR-3 est un récepteur de 195 kDa retrouvé surtout dans les vaisseaux lymphatiques, il a une grande affinité pour le VEGF-C et le VEGF-D [54, 57]. 11 participe au remodelage vasculaire dans l’embryon, puisque son absence entraîne une accumulation de fluide et une défaillance cardiaque [74], ceci étant dû soit au manque du récepteur, soit au surplus de disponibilité du VEGF-C et VEGF-D pour le VEGFR-2 [75]. Ce récepteur participe activement au maintient et à la croissance des vaisseaux lymphatiques, la lymphangiogenèse, ce qui en fait une
cible de choix pour la suppression des métastases tumorales passant par les nodules lymphatiques [76-78].
Le VEGFR-3 est retrouvé principalement sur les cellules endothéliales vasculaires et capillaires, mais dans une proportion moindre que sur les CE lymphatiques [79]. La plupart des études sur les voies de signalisation induites par le VEGF R-3, ont été effectuées sur les vaisseaux lymphatiques. Ainsi, l’activation du VEGFR-3 dans les CE lymphatiques induit leur prolifération, migration et survie [801. Le VEGF-C a été un des ligands les plus étudiés du VEGFR-3, possédant aussi la capacité de se lier et activer le VEGfR-2. En effet, le propeptide dirnérique du VEGF-C passe par un processus de maturation peptidique, qui clive la molécule, la rendant ainsi apte à lier le VEGFR-2 et augmentant son affinité pour VEGFR-3 [56]. Les voies de signalisation induites par l’activation du VEGfR-3 dans les CE lymphatiques ressemblent à celles induites par l’activation du VEGFR-2 dans les CE des vaisseaux sanguins. Le VEGFR-3 active la voie de la PKC et subséquemment la p42744 MAPK, ainsi que la voie de la PI3K avec l’activation subséquente de Akt/PKB, ces deux voies étant impliquées dans la transmission de signaux mitogéniques et anti-apoptotiques [80, 81].
Figure 3 : Voies de signalisation induites suite à l’activation des récepteurs du VEGF Les icepteurs dimériques du VEGF, sont activés suite à la liaison d’un agoniste faisant partie des analogues du VEGF-A. Suite à l’activation des dimèms, l’autophosphorylation des résidus spécifiques tymsine kinase entraînent l’association avec les VEGFR-associated protein (VRAP), PLCy et Sck, menant à l’activation de diverses voies de signalisation. Le dimèi du VEGFR-2 a été principalement associé aux activités biologiques induites par le VEGF-A, notamment la survie, la migration, la perméabilité et la prolifération cellulaire. Cependant le VEGFR-1 peut aussi induire certaines activités biologiques mais à plus faible échelle. (Hoeben et coll., Vascular Endotheliat Growth Factor andAngiogenesis, Phaimacological Reviews, 2004. 56 549-580)
Q
VEGFR-INEGFR-1 or VEGFR-2NEGFR-2 or VEGFR-INEGFR-2 Ceil membrane III H III HI p1p2P(Y951) P(Y996) P(Y1054)
______ P(Y1O59)f ______ P(Y1175) P(Y1214) DAG
()
()
+ Ca2 ER SURVIVAL PERMEABIUTY Focal adhesion turnover Ca2/
PCI2 MIGRATION Gene Transcription PROLIFERATION1.2.8 Dimérisation des récepteurs du VEGF
Même si l’activation du VEGFR-2 semble suffisante pour induire les voies de signalisation, et subséquemment les activités biologiques du VEGF-A165, il demeure néanmoins que des études récentes suggèrent que le VEGFR-l puisse réguler les activités biologiques du VEGFR-2. Premièrement, en transfectant des CE avec des récepteurs chimériques, contenant seulement les domaines transmembranaires et cytoplasmiques du VEGFR-l et du VEGF R-2, une récente étude a démontré que le domaine cytoplasmique du VEGFR-l peut avoir un rôle d’inhibition, contrecarrant l’augmentation de la croissance cellulaire induite par l’activation du VEGFR-2. Ainsi, en stimulant avec le ligand de la portion extracellulaire des chimères, seulement la transduction du signal engendrée par la portion cytoplasmique du récepteur a contribué à inhiber la migration cellulaire [82]. D’après cette étude les effets de l’activation du VEGFR-l sur la croissance cellulaire sont donc à l’opposé de l’activation du VEGFR-2 dans les CE. Deuxièmement, l’hétérodirnérisation de VEGFR-1 avec VEGFR-2 peut être induite en absence de ligand et elle augmente en présence d’homodimères de VEGF-A ou d’hétérodimères de VEGF-A/P1GF [83]. L’activation de VEGF R-l induit un cross-taÏk intrarnoléculaire qui augmente l’efficacité de la cascade signalétique induite par le VEGFR-2, mais aucune preuve de transphosphorylation entre les récepteurs hétérodimériques du VEGF n’a été démontrée. Toutefois, l’activation des hétérodimères de VEGFR-1 et VEGFR-2 (VEGFR-l/R-2) induit une plus forte phosphorylation de la PLCy et une plus grande migration des CE que celles induites
18
par les cellules exprimant uniquement le VEGFR-2, alors que les CE exprimant uniquement le VEGFR-l n’ont démontré aucun effet [35].
1.3 TONUS VASCULAIRE ET PROSTACYCLINE (PGI2)
1.3.1 Principaux régulateurs du tonus vasculaire NO et PCI2
Le tonus vasculaire est déterminé par l’état contractile des cellules musculaires lisses vasculaires (CMLv) de la paroi du vaisseau sanguin. Cet état est influencé par l’activité des substances vasoconstrictrices, qui augmentent la concentration calcique, ou vasodilatatrices, qui diminuent cette concentration de calcium. Parmi les substances vasoconstrictrices nous retrouvons la vasopressine, l’angiotensine II et l’endothéline, tandis que l’acétylcholine, la prostacycline et le NO sont des molécules vasodilatatrices. Le NO est relâché par les CE, lorsque les forces de cisaillement d’un tonus presseur augmentent. Le NO exerce son effet vasodilatateur de manière paracrine sur les CMLv par l’entremise de l’activation de la guanylate cyclase (GC) engendrant ainsi une augmentation de GMPc. Ce dernier permet la déphosphorylation de la chaîne légère de rnyosine, entraînant une relaxation des CMLv [84, 85].
La prostacycline est aussi relâchée par les cellules endothéliales en réponse à différents facteurs, agissant de façon paracrine. La PGI2 se lie à son récepteur (IP), faisant partie de la famille des récepteurs couplés à des protéines G, situé sur la surface des CMLv. Son activation entraîne une augmentation de la production de l’AMPc et une diminution du calcium libre intracellulaire, provoquant ainsi la relaxation des CMLv [$6, $7]. La prostacycline agit de façon intracrine, en induisant l’apoptose cellulaire via l’activation des récepteurs nucléaires de la famille des
20 PPAR (Peroxisorne Proflferator-Activated Receptors). Cependant les actions des prostaglandines sur les PPAR ne sont pas encore totalement élucidées [881.
1.3.2 Mécanismes de synthèse de la prostacycline
La prostacycline est dérivée de l’acide arachidonique (AA), un produit lipidique synthétisé à partir du clivage des glycérophospholipides membranaires par des phopsholipases A2. Les PLA2 ont été classées en plusieurs groupes, soit les
PLA7 cytosoliques (cPLA) [89], les PLA2 secrétées (sPLA2) [90] et les PLA2
indépendantes du calcium (iPLA2) [91]. Ces enzymes sont très spécifiques dans leur clivage membranaire, catalysant l’hydrolyse de la position sn-2 des phospholipides membranaires afin de relâcher l’AA. Ce dernier est ensuite converti en PGH2 par des cyclooxygénases. Trois isoformes des COX ont été caractérisés jusqu’à présent, la
forme constitutive, COX-l, la forme inductible, COX-2 et un troisième isoforme, la
COX-3, qui est une variante provenant de l’épissage alternatif du gène de la COX
1. La COX-l joue un rôle dans l’homéostasie vasculaire, l’isoforme COX-2 est
induit par des stimuli inflammatoires, soit des endotoxines bactériennes ou des cytokines, comme le VEGF [92] et la COX-3, qui a été trouvée dans le cerveau canin, semble être impliquée dans la régulation de la douleur et de la fièvre [93-95]. La COX-1 est surtout employée par les CE dans la synthèse des prostaglandines à court terme, allant jusqu’à quelques minutes. Ses produits sont principalement la PGb et la PGE2, la dernière étant produite dans une moins grande quantité à court ten-ne dans les CE [96]. Inversement, la COX-2 est impliquée dans la régulation du processus inflammatoire à long terme, et dépendamment du type cellulaire et des agonistes induisant sa synthèse et son activité, les produits finaux sont la PGI2 ou la
PGE7 [97, 98]. Appelées aussi PGH7 synthases, les COX sont des enzymes à activité oxydoréductase. Elles catalysent l’oxydation dc l’AA en PGG2, pour ensuite le réduire sous la fonrie PGH, le précurseur de toutes le prostaglandines et thrornboxanes [99]. Les différentes PG synthases et isornérases transforment le PGH7 en prostaglandines primaires (PGI2, PGD-,, PGE2 et PGF2). La PGI2 a un temps de demi-vie très court (t1/7 = 2-3 minutes), étant rapidement transformé en
6-keto PGF1, son métabolite plus stable (t117 = 30 minutes). La thromboxane A2 est
synthétisée par la thrornboxane A synthase dans les plaquettes seulement et agit de manière contraire à la PGI2 endothéliale en provoquant une vasoconstriction et favorisant l’agrégation plaquéttaire [100]. Un débalacement de l’équilibre PG1/TxA, vers une surproduction de la TxA2 peut avoir des conséquences néfastes sur la vasculature, pouvant déclancher des thromboses et des vasospasmes. [100,
= Arachidonc acid ‘ COX I ,r K ndomuhacin ‘
/
-‘‘ COOH Q ‘_-// 00H PCG2 COX-1orCO 2 COQH cocu C00H PGI synthae -TXA sytrase o P2 TH HO/
PGç C00H K Z / “-cooli OH OH / Qj-/ G-Reto-PGF i Z “— ‘“C00H/
K 0H 0H OH PGE --N C00H 0H (9u, lltc-PGF2) o OH PGD2 Figure 4 Voie de synthèse des prostaglandines à partir de l’acide arachidonique La voie classique de synthèse des prostaglandines part de la libération de l’acide amchidomque (AA) par les phospholipases A2 (PLA2). La sPLA2 et la cPLA2 sont les principaux isoformes retrouvés dans les CE. La transformation de l’acide amchidomque (AA) en PGG2 par l’action de la COX-l ou COX-2. Ces deux enzymes catalysent ensuite la formation de la PGH2, le puicurseur des pmstaglandines et des thromboxanes. La PGI2 est synthétisée par la PGI2 synthase â partir de la PGH2, mais cette forme est instable et rapidement transformée dans son métabolite inactif, la 6-keto-PGF1 . La PGI2 synthase est l’enzyme prédominaiite par rapport aux autres svnthases dans les cellules endothéliales, ce qui signifie que la synthèse des autres prostaglandines est absente ou marginale (Vane et coll. Cyciooxi.’genases 1 and 2, Annu Rev Pharmacol Toxic, 1998. 38: 97-1201.3.3 Rôles de la prostacyclïne et les inducteurs de sa synthèse
La PGI est majoritairement produite et secrétée par les cellules endothéliales [102], mais peut aussi être trouvée dans les cellules musculaires lisses [103] et les cardiomyocytes [104]. Sa durée de vie est très courte lorsque secrétée, puisqu’elle est rapidement transformée en son métabolite stable, le 6-keto PGFI(L. Lorsque
relâchée par les CE, la PGI2 se lie sur son récepteur IP situé sur la surface des cellules musculaires lisses et, par l’entremise de l’AMPc, l’interaction entre les filaments d’actine et de rnyosine est inhibée, ce qui permet la relaxation des CMLv, et son effet vasodilatateur [105]. La prostacycline est aussi un inhibiteur de l’agrégation plaquettaire, via leur récepteur IP, mais les voies de signalisation menant à cette inhibition ne sont pas encore claires [106]. La synthèse de PGI2 dans les cellules endothéliales peut être induite par divers agonistes, tels la bradykinine, l’ADP, l’ATP, l’acide arachidonique exogène, l’acétylcholine, la vasopressine, l’angiotensine et le VEGF-A165. Le VEGF-A165 est donc un vasodilatateur indirect et ce rôle dans l’inflammation et l’angiogenèse s’ajoute à ceux mentionnés auparavant. La relâche de PGI2 induite par le VEGE est induite, en partie via l’activation de la p42144 MAPK et l’activation subséquente de la cPLA2 [96]. Puisque l’activation des MAPK a été attribuée largement à la signalisation induite par le VEGFR-2, des études ont suggéré ce récepteur comme le seul responsable de la relâche de PGI2 [64].
24 1.3.4 La nitrosylation : un phénomène nouveau et opposé à la
phosphorylation
La phosphorylation et la nitrosylation sont deux mécanismes régulant les protéines dans la cellule. Le premier mécanisme nous a permis de comprendre les voies de signalisation cellulaire, alors que le deuxième a été moins exploré. Cependant, depuis quelques années, la découverte des mécanismes de régulation du NO a pen1is d’approfondir nos connaissances sur la nitrosylation, dont l’action a été spécifiquement étudiée sur deux enzymes impliquées dans la synthèse de facteurs vasodilatateurs, soit la eNOS et la PGI2 synthase [107]. Ces études ont permis de distinguer la nitrosylation de la phosphorylation en deux points (I) la nitrosylation est effectuée sur des résidus spécifiques de cystéine ou tyrosine alors que la phosphorylation est souvent retrouvée sur des résidus tyrosine, sérine ou thréonine et (2) la nitrosylation inhibe l’activité de ces deux enzymes, contrairement à l’effet le plus rencontré de la phosphorylation, l’activation protéique [108, 109]. La eNOS est une enzyme limitante dans la production de NO à partir de la L-arginine et donc, la production de NO en grande quantité amènerait une boucle de régulation négative, puisque le NO et ses dérivés oxydés participeraient à l’inactivation de la eNOS et ainsi diminuerait la production de NO. Dans le cas de la PGI2 synthase, la nitrosylation est effectuée sur les résidus tyrosines, mais la raison de cette nitrosylation de l’enzyme limitante de la production de PGI2 est encore inconnue.
1.4. Problématique et but de l’étude
Au cours des dernières années nous avons démontré que le VEGF -A165 augmente la peniiéabilité vasculaire, la transiocation de la P-sélectine endothéliale et l’adhésion des neutrophiles suite à la synthèse du facteur d’activation plaquettaire (PAF) [17, 49]. Nous avons déterminé que les homodimères du récepteur VEGFR-2 induisent un effet maximal de ces activités biologiques. De plus, il a été démontré que le VEGF -A165 est capable d’induire la synthèse de PGI2 dans les cellules endothéliales. Cependant, les récepteurs du VEGF et les voies de signalisation subséquentes participant à la synthèse de PGI2 ne sont pas encore claires. Conséquemment, nous avons voulu investiguer le rôle des récepteurs VEGFR-l, VEGfR-2 et NRP-l dans la synthèse de PGI2. Pour ce faire, le modèle des cellules endothéliales d’aorte bovine (BAEC) a été choisi pour plusieurs raisons: la présence des récepteurs du VEGF, la grande concentration de PGI2 synthétisée et le modèle de cellules endothéliales artérielles. En utilisant des approches pharmacologiques, comme l’activation des récepteurs du VEGF par des agonistes spécifiques et l’utilisation d’inhibiteurs, nous avons voulu déterminer la contribution des récepteurs du VEGF dans la synthèse de PGI. Nous avons par la suite confirmé cette contribution par un traitement par antisens afin de bloquer la synthèse protéique. Nous avons aussi voulu étudier les différentes voies de signalisation menant à la synthèse de la PGI2, ceci dans le but ultérieur de pouvoir déterminer la contribution de la PGI2 dans les processus inflammatoires et angiogéniques induits par le VEGF-A165. Nous avons utilisé des techniques d’irnrnunobuvardage de type Western, ainsi que le dosage du métabolite stable de la PGb, la 6-ketp PGFIŒ par ELISA.
2.0 ARTICLE
Identification des auteurs
1. Identification
Paul-Eduard Neagoe
Département de pharmacologie, Faculté de médecine
2. Description de l’article
Titre : VEGF-A165-induced prostacyclin syiithesis requires the heterodimerization ofVEGFR-l and VEGfR-2
Liste des auteurs: Neagoe Paul-Eduard, Caroline Lemieux, Martin G. Sirois Soumission à la revue: «Journal of Biological Chemistry», Octobre 2004
3. Déclaration de tous les coauteurs autres que l’étudiant
A titre de coauteur de l’article idenifié ci-dessus, je suis d’accord pour que
Paul-Eduard Neagoe inclue cet article dans son mémoire de maîtrise qui a pour titre La synthèse de prostacycline induite par le VEGF-A1 requiert I’hétérodimérisation des récepteurs du VEGF
Caroline Lemieux
2
I
iÔ/û11
Coauteur Date
M artin G. Sirois
2$
Q
cc:
Objet : From the JBC te: Manuscript M4:12017
06/01/05 08:20
M4 :12017
Dear Dr. Sirois:
Your manuscript entitled “Vascular endothelial growtli factor
(VEGF)-A165-induced prostacyclin synthesis requires the activation of VEGF receptor -1 and -2 heterodimer lias been accepted for publication and is tentatively scheduled for an April issue. Your paper will first lie pubuished online on the day it was accepted as a JBC Papers in Press and can lie seen at www.jbc.org . This date is considered the formai date of
publication. Ail structures/coordinates referenced in an accepted
manuscript must lie deposited and the data released BEFORE final publication of the manuscript. If the data lias flot been released, please contact PD3 for its immediate release.
Now that your manuscript lias been accepted for publication, the redactory office at our printer, CADMUS Professionai Communications, requires source files including a separate Word file for text and graphic TIFF or EPS files for the figures. Please go to this web site to upload your source files directly to the redactory office: http://rapidsubmission.cadmus.com/jbc Publication of the print version of your manuscript will be delayed until we receive these materials. If you have any questions, please contact
Cadrnus at: Thank you in advance for your quick
response.
You will receive page proofs in approximately six weeks after submission of the source files; please return promptly to expedite publication. You will lie bulled after publication for page charges, reprints you order,
half-tone, electron micrographs and authors alterations as applicable. The current charge for lialf-tone figures is $25.00 each plus $120.00 if you order special paper. Color Figures, if any, will be charged at e rate of $300.00 per Figure.
JEC publishes a colored illustration relevant to an article in the saine issue. The cover illustration should lie scientifically interesting and visually attractive and sliould NOT lie e complex, data laden figure. The illustration submitted need not lie a figure in the paper but should lie closely related to the subject of the paper. To sulimit an illustration for consideration please send a high quality color PRINT fwe cannot accept electronic figures) . The print must lie at least 7 inches wide by 6 inches
tau to lie considered. The figure can lie larger but must be in the same proportion as 7 inches wide by 6 inclies tau or it will not lie considered.
Include also an explanatory caption cf 50-60 words as well as the
manuscript nurnber. Send te: Elyse Pierson, Department cf Biological
Sciences, 371 Serra Mali, Stanford University, Stanford CA, 94305-5020
(650—725 4819)
Sincerely yours,
Richard W. Hanson
Associate Editor
30
Vascular endothelial growth factor
(VEGF)-A165-induced prostacyclin synthesis requires the
activation of VEGF receptor -1 and -2 heterodimer
Paul-Eduard Neagoe, Caroline Lemieux and Martin G. Sirois*
Research Center, Montreal Heart Institute, Department of Pharmacology, Université de Montréal, Montreal (QC), Canada
Dr. Sirois is recipient of a scholarship from the Canadian Institutes of Health Research (CIHR), and this work was supported by grants from CIHR (MOP-4391 9),
Heart and Stroke Foundation of Quebec and Birks Farnily foundation to Dr. Sirois.
Running Titie: VEGF-rnediated PGI2 synthesis
Key words: PGb, VEGF analogs, VEGF receptors, neuropilin-1, phospholipase A2
*Conespofldence shouÏd be addressed to:
Martin G. Sirois, PhD
Research Center
Montreal Heart Institute 5000, Belanger Street
Montreal (QC), Canada, HIT 1C8 Phone: (514) 376-3330 (ext: 3583) fax: (514) 376-1355
SUMMARY
We previously reported that vascular endothelial growth factor (VEGf)-A165
inflarnrnatory effect is rnediated by acute platelet-activating factor (PAF) synthesis from endothelial celis (ECs) upon the activation of VEGF receptor-2 (VEGFR-2) and its coreceptor, neuropilin-l (NRP-1). In addition, VEGF-A165 prornotes the release of othei- endothelial mediators including nitnc oxide (NO) and prostacyclin (PGL). However, it is unknown whether VEGF-A165 is mediating PGI2 synthesis through VEGF receptor-1 (VEGFR-1) and/or VEGF reccptor-2 (VEGFR-2) activation, and whether the coreceptor neuropilin- I (NRP- 1) potentiates VEGF-A165 activity.
In this study, PGI2 synthesis in bovine aortic endothelial ceils (BAEC) was assessed by quantifying its stable metabolite (6-keto-prostaglandin F1; 6-keto-PGF1) by ELISA. Treatment ofBAEC with VEGF analogs. VEGF-A165 (VEGF1, VEGF R-2 and NRP-l agonist) and VEGF-A1R-21 (VEGFR-I and VEGfR-R-2 agonist) (up to I0 M) increased PGI2 synthesis by 70- and 40-fold, within I 5 minutes. Treatment with VEGFR-1 (P1GF, and VEGF-B) or VEGFR-2 (VEGF-C) agonist did not increase PGb synthesis. Combination ofVEGFR-l and VEGF R-2 agonists did not increase PGI2 release. Pretreatment with a VEGFR-2 inhibitor, abrogated PGI2 release rnediated by VEGF-A16 and VEGF-A121, and pretreatrnent ofBAEC with antisense oligomers (AS) targeting VEGFR-1 or VEGFR-2 mRNA reduced PGI2 synthesis mediated by VEGF-A165 and VEGF-A121 up to 79%. In summary, our data demonstrate that the activation of VEGFR-l and VEGFR-2 heterodirner (VEGFR l/R-2) is essential for PGI2 synthesis mediated by VEGF-A165 and VEGF-A1-,1,
32 which carmot be reproduced by parallel activation of VEGfR-1 and VEGFR-2 homodimers with corresponding agonists. In addition, the binding of VEGF-A1 to NRP-l potentiates its capacity to prornote PGI2 synthesis.
INTRODUCTION
Vascular endotheliai growth factor (VEGF-A) is known as an inflarnrnatory cytokine participating in the wound healing, tissue regeneration and physiological angiogenesis, but also for its capacity to prornote pathologicai angiogenesis in turnor growth, atherosclerosis and proliferative retinopathies (1,2). There are five different VEGF-A isoforrns, of 206, 189, 165, 145 and 121 arnino acids and aiso several VEGF analogs: placental growth factor (P1Gf-1 and -2), VEGf-B, VEGf-C, VEGF-D, and a viral homoiog, VEGf-E (2). The actions ofVEGF family members are mediated by the activation of selective tyrosine kinase receptors including
VEGFR-l (Fit-1) and VEGFR-2 (flk-1/KDR), which are alrnost exclusively
expressed on endothelial ceils (ECs), and VEGFR-3 (f It-4), which is mainly Iirnited to the lympliatic endothelium (2). VEGF-A binds to VEGFR-1 and VEGfR-2; P1GF-1, PÏGf-2 and VEGF-B bind to VEGFR-1; VEGF-C and D bind to VEGFR-2 and R-3: whereas VEGf-E interacts only with VEGFR-2 (2,3). Recent studies also reported that neuropilin- I (NRP- Î), a transmembrane receptor, acts as a coreceptor by enhancing the binding of VEGF-A165 to VEGFR-2 and potentiates various VEGF-A165 biologicai activities (3-6). Such selectivity is attributable to the presence ofVEGf-A exon 7 in VEGF-A165, a domain that is lacking in VEGF-A121, VEGf-C , VEGf-D and P1Gf-1 (4,7).
Stimulation of ECs witb VEGF-A16 can prornote prostacyclin (PGI2) synthesis which is a potent vasodilator and an inhibitor of platelet aggregation (8-10). Consequently, imbalance in PGb production can be involved in the pathophysiology
34
of rnany thrornbotic and cardiovascular disorders. The induction of PGI2 can be mediated upon the activation of different phospholipase A2 enzymes that catalyse the cleavage of arachidonic acid (AA) from membrane glycerophospholipids. Subsequently, AA is converted in PGH2 by the action oftwo cyclooxygenase (COX) isofonTis, either the constitutive foi-m, COX-1, or the inducible form, COX-2. The newly forrned PGH2 is then transforrned into PGI2 by the action of the PGI2 synthase (Il-14). However, it is unknown whether the members of the VEGF superfamily are mediating PGI2 synthesis either through VEGFR-1 and/or VEGFR-2 activation, and if NRP-1 is contributing to potentiate VEGF-A165-rnediated PGI2 synthesis.
During last years we have shown that VEGf-A165 increases vascular penneability, endothelial P-selectin transiocation and neutrophil adhesion upon the synthesis of platelet-activating factor (PAF) by ECs (5,15). We subsequently investigated the contribution of VEGF receptors and assessed that ail these biological activities are mediated through the activation of VEGFR-2, and that these effects are potentiated by the presence of NRP-1 (4,5,16). Consequently, by using VEGF analogs and by regulating VEGF receptor activity either with selective inhibitors or by antisense
treatment, we investigated the contribution of VEGF members and their
METHODOLOGY Ccli Culture
Endothelial celis were harvested from bovine aol-tas (BAEC), cultured in Dulbecco’s modified eagle medium (DMEM; Life Technologies, Inc., Burlington, ON, Canada) containing 5% fetal bovine serum (FBS; Medicorp, Inc., Montreal, QC, Canada) and
antibiotics (Sigma, St-Louis, MO), BAEC were characterized as described
previously (16) and used between passages 3 to 5.
EndotheÏial PGI Snthesis
BAEC were seeded in 6-well plates and cultured up to 3 days post-confluence. Culture medjum was rernoved and celis were rinsed with HBSS (Hank’s balanced sait solution) / HEPES (10 mM; pH 7.4) (Sigma). Celis were stirnulated in HBSS/HEPES plus CaC12 (5 mM) with phosphate buffered saline (PBS) solution or VEGF analogs, VEGF-A165 (PeproTech Inc., Rocky Hill, NJ), VEGF-A121, PIGF, VEGF-B, VEGF-C (R & D Systems, Minneapolis, MN) at various concentrations
(10 - lOE8 M) and up to 30 minutes. In another set of experiments, BAEC were
pretreated with selective inhibitors of: VEGFR-l and VEGFR-2 (VTK), VEGFR-2 (SU1498) p38 MAPK (SB203580), MEK (PD98059), cytosolic phospholipase A2; cPLA2 (AACOCf3), secreted phospholipase A2; sPLA (scalaradial) (Calbiochem, La Jolla, CA), cyclooxygenase-1 and -2; COX-l and -2 (indornethacin) or PGJ2 synthase (tranylcypromine) (Sigma) 1 5 minutes prior to stimulation with VEGF-A isofonns. Upon stimulation, the supematant was collected and PGI2 synthesis was
n
j
assessed by quantifying its stable metabolite (6-keto PGF1) accordingly to rnanufacturer’s instructions (Cayrnan Chernicals, Ann Arbor, MI).
Antisense OligonztcÏeotide Therapy
We also used an antisense oligonucleotide therapy approach to discrirninate the contribution of VEGFR-1 and VEGFR-2 on PGI2 synthesis mediated by VEGF-A isoforms. BAEC were treated with antisense oligonucleotide sequences cornplementary to bovine VEGFR-1 or VEGFR-2 rnRNA (GenBank Accession Numbers X94263 and 9429$). Antisense oligonucleotide phosphorothioate backbone sequences targeting bovine VEGFR-l rnRNA (AS-RI: 5’-CAA AGA TGG ACT CGG GAG-3’), and VEGFR-2 rnRNA (AS-R2: 5’-GCT GCT CTG ATT GTT GGG-3’), or a scrarnbled phosphorothioate sequence (AS-Scr: 5’-TGC TGG CAT GTG CGT TGT-3’) (AIphaDNA, Montreal, QC, Canada) were used. The antisense oligorners were chosen based on their capacity to selectively abrogate the protein expression of the genes targeted as previously descnbed (1 6). Briefly, BAEC were seeded at 5 X lO cells/well in 6-weIl plates, in DMEM, 5% FBS, and antibiotics with or without oligomers (5 X 1 O M/daiÏy) up to 3 days post confluence. Culture medium was removed, ceils were rinsed, stirnulated in HBSS/HEPES + CaC1 (5 mM) with PBS or VEGF-A isoforrns, and PGI2 synthesis
Preparation of GST- VEGF-A Exon 7fusion Protein
In order to evaluatc the possible potentiating effect of NRP-1 on VEGF-A165-induced PGI2 synthesis, we produced a glutathione-S-transferase (GST) fusion protein encoding exon 7 of human VEGF-A165 (GST-Ex7) (The construct of GST
fusion protein-Exon 7 was generously provided by Dr. Shay Soker, Wake Forest University, Winston-Salern, NC). Escherichia coli (DH5a) were transformed with pGEX-2TK or p2TK-exon 7 vectors to produce GST and GST-Exon 7 proteins. The recombinant proteins were purified from bacterial lysates using glutathione and heparin affinity chrornatographies, as described previously (5,6).
Western Blot Analyses of VEGF Receptors Expression and fhosphorvÏcttion
BAEC were cultured up to 3 days postconfluence, ceils were rinsed, incubated on ice, and stimulated in HBSS/KEPES + CaCI2 (5 mM) plus I mg/rnl bovine serum
albumin. In sorne experiments, BAEC were pretreated with PBS or VEGF receptor inhibitors (SU149$ or VTK) 15 minutes prior to the addition of VEGf-A16 or VEGF-A121, the ceils were kept on ice for an additional 15 minutes. Then, ceils were stimulated for 7.5 minutes at 37°C, and brought back on ice. In another set of experiments, BAEC were stimulated as above with VEGF analogs only. Upon stimulation, the media was removed, ceils were washed, and lysates prepared. Western blot analyses were performed as described previously (4,5,16). Primary antibodies used were mouse monoclonal anti-human VEGfR-1 (clone FIt-11; Sigma), polyclonal rabbit anti-mouse VEGFR-2 and goat anti-hurnan NRP-l IgG antibodies (Santa Cruz Biotechnology Inc., Santa Crnz, CA). Membranes were
3$
stripped using Re-Blot Plus Strong stripping solution (Chernicon International, Ternecula, CA) for 20 minutes, and reprobed with a mouse monoclonal anti phosphotyrosine IgG (clone 4G10; 1:4000 dilution; Upstate Biotechnology Inc.,
Lake Placid, NY) to determine VEGFR-l and VEGFR-2 phosphorylation.
Kaleidoscope molecular weight rnarkers (Bio-Rad, Mississauga, ON, Canada) were used as molecular mass standards for SDS-PAGE immunoblotting experiments. Immunoreactive bands for were visualized by enhanced cherniluminescence (ECL), digitized using a 2-dirnensional gel scanner and quantified using Quantity One sofiware (Bio Rad).
In another set of experiments we assessed by Western blot analyses the expression
and activation of selective enzymes involved in the ceil signaling pathway leading to
PGI2 synthesis. Studies were performed as described above. Primary antibodies used for immunoprecipitations and Western blot analyses were the followings: rabbit polyclonal anti-hurnan phospho-p42/44 MAPK and anti-hurnan phospho cPLA2 (CelI Signaling Technology Inc., Beverly, MA), mouse monoclonal anti-ovine COX-l and mouse monoclonal anti-human COX-2 (Cayrnan Chernicals) IgGs. Then the membranes were stripped and reprobed with rabbit anti-rat p42144 MAPK (Ceil Signaling), mouse monoclonal anti-human cPLA2 (Santa Cruz).à
Western BfotAnalvsjs ofPGI Svnthase S-nitrosyÏation, cvclooxvgenase-1 and -2 BAEC were cultured up to 3 days postconfluence and treated prior to stimulation as described above. Celis were incubated on ice for 30 minutes with either VEGF-A165
or VEGF-C (10 M), then stirnulated for 5 to 15 minutes at 37°C, and brought back on ice. Upon stimulation, the media was removed, ceils washed, and lysates prepared. Immunoprecipitation of celi lysate was performed with a rabbit polyclonal anti-bovine PGI7 synthase IgG (Cayman Chemicals). Samples were separated on a 10% SDS-PAGE, and Western blot analyses perforrned as described
previously (4,5,16). A mouse monoclonal anti-nitrotyrosine IgG (Cayrnan
Chemicals) was used to assess S-nitrosylation level of PGI2 synthase. Then, membranes were stripped and reprobed with rabbit polyclonal anti-bovine PGI2 synthase IgG as described above.
StatisticalAnalvsis
Data are presented as mean ± s.e. mean. Statistical comparisons were made by
analysis of variance, followed by a Bonferroni’s t-test for multiple comparisons. Differences wcre considered significant when p <0.05.
40
RESULTS
Effect of VEGF Anctlogs and Corresponding Receptors 011Pivstacvcïin Synthesis Prostacyclin (PGI-,) synthesis in postconfluent BAEC was quantified by rneasunng its stable metabolite 6-keto PGFIŒ by ELISA. First, we performed a time- (5 to 30 minutes) and concentration- (10’’ - iO8 M) dependent assay to assess how VEGF
A165 mediates PGI2 synthesis. In control PBS-treated ceils, the production of 6-keto
PGF1 did flot change in function of tirne, at 15 minutes post-treatrnent the concentration of 6-keto PGF1 was 0.657 ± 0.056 ng/106 celis. Treatrnent with
VEGF-A165 (1 0 M) induced a rapid and transient PGI2 synthesis, within 5 minutes,
we observed a 4$-fold increase, which reached a plateau within 10 minutes (70-fold increase). In addition, VEGF-A165 (l0i
- i- M) increased PGI2 synthesis by 5-,
30- and 70-fold, respectively. Interestingly, at the highest concentration (10 M), VEGF-A165 was less efficient (33-fold increase) to mediate PGI2 synthesis (Figure
lA and B).
To determine which VEGF receptors are involved in PGI2 synthesis, we used
selective VEGF analogs. Treatment with VEGF-A121 isoforrn (10’’- l0 M), which
like VEGF-A165 binds to VEGFR-1 and VEGFR-2 but flot to NRP-l coreceptor induced as well a significant but reduced PGI2 synthesis (40-fold increase; at 1OE9 M) as cornpared to VEGF-A165 (Figure 1C). These latter data suggest that NRP-1 coreceptor might contribute to potentiate VEGF-A165 capacity to prornote PGI2 synthesis. To support this hypothesis, we treated BAEC with PIIS, VEGF-A165 and VEGf-A121 and confirmed by Western blot analysis the capacity of VEGF-AI(5 as
opposed to VEGF-A11 or PBS-treated celis to prornote VEGFR-2/NRP-1 complex formation (Figure 2A). Then, BAEC were pretreated witli a GST fusion protein containing exon 7 of human VEGF-A165 (GST-Ex7) to block the interaction of VEGF-A165 with NRP-l. Exon 7 encodes a dornain not present in VEGF-A-,1 that is responsible for the binding ofVEGF-A165 to NRP-l(3,5). Pretreatment ofBAEC with GST-Exon7 (up to 1O M) 15 minutes prior to stimulation with VEGF-A165 (1O M) reduced PGI2 synthesis to the level induced by VEGF-AI2L (Figure 2B). Pretreatment with GST (up to iO’ M), without the exon 7 insert, did flot alter VEGF-A165-induced PGI2 synthesis and neither GST-Ex7 nor GST (up to i07 M) altered significantly the basal level of PGI2 synthesis or VEGF-A121-induced PGI2 synthesis (Figure 28).
Since VEGF-A165 and VEGF-A121 are both capable to activate VEGFR-1 and VEGFR-2, we then treated BAEC with selective analogs for VEGFR-1 (PIGF and VEGF-B) and VEGFR-2 (VEGF-C) to verify the contribution of each receptor under their homodimeric conformations on PGI2 synthesis. Treatment with PIGF, VEGF
B or VEGF-C (101l - 1OE8 M) for 15 minutes did not prornote the release of PGI2
(Figure 3A-C). In order to assess if a parallel activation ofVEGFR-1 and VEGFR-2 homodimei-s may induce PGL release, BAEC were treated with the combination of VEGFR-l analogs (PÏGF or VEGF-B) with VEGFR-2 analog (VEGF-C) at 1OE M and for 15 minutes. Such combination did not increase PGI2 synthesis (Figure 3D).
42 Phosphorylation ofVEGF Receptors by C’orresponding VEGF AnaÏogs
Since VEGF-A165 and VEGf-A121 were the only VEGF analogs capable to mediate endothelial PGI synthesis, we perforrned Western blot analyses to confirm the expression of VEGFR-l and VEGFR-2, and the capacity of VEGF analogs to activate them. BAEC were treated for 7.5 minutes with VEGF analogs which is the suitab!e tirne to detect the phosphorylation of VEGF receptors as previously described (16). Ce!! lysates were irnrnunoprecipitated either with anti-VEGFR-1 or
anti-VEGf R-2 IgGs. VEGFR-1 and VEGFR-2 protein expression in BAEC was
detected by irnrnunob!otting, which is in agreement with previous reports (Figure 4A and B; upper bands) (17). The membranes were stripped and the detection of
VEGFR-1 and VEGF R-2 phosphory!ation was performed by reprobing the
membranes with anti-phosphotyrosine IgG. Treatment with VEGF-A165 and VEGF
A121 (10-o M) increased the phosphory!ation ofVEGFR-1 by 10.6- and 7.3-fold as
compared to PBS-treated ce!!s, whereas equivalent treatment with P1GF or VEGF-B did not increase VEGF R-! phosphorylation (Figure 4A; lower panel). Although, we could not detect the phosphorylation of VEGFR-1-rnediated by P1GF or VEGF-B, they were neverthe!ess flot deprived ofbiological activities since they were capable to promote endothelial P-selectin translocation (data flot shown, and as previously described) (5). Secondly, we assessed the capacity of VEGF-A165, VEGf-A121 and VEGF-C (1 O M) to mediate VEGFR-2 phosphorylation. Such treatment increased by 57.$-. 24.7- and 5.8-fold the phosphorylation of VEGFR-2 as compared to PBS treated ce!ls (Figure 43; lower pane!).
PGI Synthesis Requires VEGFR- 1 and VEGFR-2 Heterodiinerization
Our data dernonstrate that the activation of VEGFR-I or VEGFR-2 hornodimers alone or in parallel with selective VEGfR-1 or VEGFR-2 analogs did flot promote PGI2 synthesis. Consequently, we speculated that VEGF-A isoforrns induce PGI2 synthesis through the activation of VEGFR-1/R-2 heterodimer. By Western blot analysis, we observed in PBS-treated ceils that VEGFR-1 and VEGFR-2 subunits can constitutively be present under heterodimeric (VEGFR-1/R-2) state, and that a treatment either with VEGf-A165 or VEGf-C did flot modulate VEGFR-I/R-2 dirnenzation (Figure 5). SecondÏy, in order to dernonstrate that PGI2 synthesis rnediated by VEGF-A165 and VEGF-A121 is driven through the activation of VEGFR-1/R-2 heterodimer, BAEC wei-e treated with selective antisense oligomers targeting VEGFR-1 or VEGFR-2 rnRNA. We showed previously that such approach at the concentration used (5 x 1 0 M / daily) abrogated selectively by over 90% the protein expression of VEGFR-I or VEGFR-2 and the biological activities investigated by $0 to 100% (16). Treatment with selective antisense oligorners targeting VEGFR-1 (AS-RI) or VEGFR-2 (AS-R2) rnRNA reduced by 79 and 71% the synthesis ofPGI mediated by VEGf-A165, and by 73 and 62% the synthesis of PGb rnediated by VEGF-A171 respectively (Figure 6A). As negative control, BAEC were treated with a scrarnbled oligorner sequence, which did not decrease significantly the level of PGI synthesis mediated by VEGf-A165 and VEGF-A121. In addition, treatrnent of BAEC with antisense or scrarnbled oligomers did not affect the basal level of PGI2 synthesis in PBS-treated ceils (Figure 6A).
44 To support the latter study, we used as well a pharmacological approach. Pretreatment of ECs with u selective VEGFR-1 and VEGFR-2 inhibitor (VTK; I 0 M; 1C50 = 2.0 and 0.1 x 1OE6 M respectively) (18), abrogated by 100 and 90% the
synthesis of PGII mediated by VEGf-A165 and VEGF-A121 respectively. Similarly, the blockade of VEGFR-2 activity with SU 1498 (selective VEGfR-2 inhibitor 5x1OE6 M; 1C50 = 7 x 1OE M) (5,19) was sufficient as weIl to abrogate by 96 and
95% the synthesis of PGI rnediated by VEGF-A165 and VEGF-A121, respectively (Figure 6B).
In order to verify the selectivity of VEGF receptor inhibitors, we assessed their colTesponding inhibitory effect on VEGfR-1 and VEGFR-2 phosphorylation mediated by VEGF-A165. Pretreatment of BAEC with SU1498 (5 x 1OE6 M), 15 minutes prior to stimulation with VEGF-A165 (10 M; 7.5 minutes) did not affect the phosphorylation of VEGfR-1 but prevented the phosphorylation of VEGF R-2. Pretreatment with VTK prevented by 100 and 83%. the phosphorylation ofVEGFR 1 and VEGFR-2 mediated by VEGF-A165 (figure 7).
Ccli SignaÏing Pathwavs bv Which VEGf-4,65 Induces PGI.2 Svnthesis
In previous studies we have shown that VEGF-A165 induces PAF synthesis upon the activation of VEGfR-2/R-2/NRP-1 complex, and requires the activation ofboth p38 and p42/44 MAPKs, and subsequent activation of secreted phospholipase A2 type V (sPLA2-V) (20,21). Since VEGf- A165 induces PGI2 synthesis upon VEGFR-1/R-2 activation, which is also potentiated by NRP-1 coexpression, we wanted to assess