© Vincent Picher-Martel, 2019
L’implication de l’ubiquiline-2 dans l’agrégation de
TDP-43 et la pathogénèse de la sclérose latérale
amyotrophique
Thèse
Vincent Picher-Martel
Doctorat en neurobiologie
Philosophiæ doctor (Ph. D.)
Québec, Canada
L’implication de l’ubiquiline-2 dans l’agrégation de TDP-43
et la pathogénèse de la sclérose latérale amyotrophique
Thèse
Dr Vincent Picher-Martel MD MSc
Sous la direction de :
Dr Jean-Pierre Julien PhD
Dr Nicolas Dupré MD MSc
iii
Résumé
La sclérose latérale amyotrophique (SLA) est la maladie du motoneurone la plus fréquente chez les adultes. Elle se caractérise par une perte progressive des motoneurones supérieurs et inférieurs menant à une paralysie et au décès entre deux à cinq ans après le début des symptômes. Environ 10% des patients ont une forme familiale (fSLA). Jusqu’à 15% des patients atteints de la SLA peuvent également présenter une démence fronto-temporale (DFT). La DFT se présente par des troubles de comportement et un changement de personnalité. Plusieurs gènes sont identifiés dans fSLA, incluant superoxide dismutase 1 (SOD1), TAR DNA-binding protein 43 (TDP-43), Fused in sarcoma (FUS), optineurin (OPTN), Tank-binding kinase 1 (TBK1) et ubiquiline-2 (UBQLN2). Ces mutations ont mené à la compréhension de plusieurs mécanismes pathologiques. L’un des mécanismes les mieux décrit est la formation d’inclusions cytoplasmiques de TDP-43. Ces inclusions contiennent d’autres protéines telles qu’UBQLN2 et ubiquitine et représentent le marqueur neuropathologique classique de la maladie. UBQLN2 est une protéine impliquée dans le système de dégradation du protéasome (UPS) et l’autophagie. Des mutations dans le gène UBQLN2 ont été lié à l’agrégation de TDP-43 dans des cellules en culture. En revanche, nous possédons très peu de connaissances sur les mécanismes pathologiques impliquant UBQLN2.
Dans cette thèse, nous avons utilisé des neurones en culture pour surexprimer les formes native et mutante d’UBQLN2 humain et pour étudier l’effet sur la délocalisation cytoplasmique et
l’agrégation de TDP-43. La surexpression d’UBQLN2WT ou d’UBQLN2P497H entraînait une
accumulation cytoplasmique et une agrégation de TDP-43. Puisque notre groupe a déjà démontré une interaction entre la sous-unité p65 de NF-κB et TDP-43, nous avons analysé l’activation du facteur NF-κB par la surexpression d’UBQLN2 WT ou P497H. Nous avons observé une activation du facteur avec l’expression des deux formes d’UBQLN2. Cette activation était dépendante de la MAPK p38 en réponse à un stress cellulaire et une accumulation cytoplasmique d’UBQLN2/TDP-43. L’augmentation de l’activité de NF-κB causait une mort neuronale et celle-ci était réversible par le traitement des cellules avec la Withaferine A, un inhibiteur de NF-κB.
iv
Puisque nous avons déterminé qu’il y avait un effet synergique important sur l’agrégation de TDP-43 par la surexpression d’UBQLN2, nous avons décidé de générer un nouveau modèle de souris transgénique avec mutation dans UBQLN2 et dans TDP-43. Les souris ont été générées par
l’introduction du gène UBQLN2P497H sous le contrôle du promoteur du gène neurofilament lourd
(NFH). Les souris simples transgéniques UBQLN2P497H étaient ensuite croisées avec nos souris
TDP-43G348C précédemment décrites pour produire des souris double transgénique UBQLN2P497H;
TDP-43G348C. Bien que les souris simples transgéniques UBQLN2P497H développaient seulement
un trouble cognitif léger, les souris doubles transgéniques développaient les caractéristiques classiques de la SLA/DFT avec agrégation cytoplasmique de TDP-43, perte de motoneurones, dégénérescence axonale et atrophie musculaire, troubles moteurs et cognitifs et une gliose entourant les motoneurones. Nous avons ensuite utilisé ce modèle pour approfondir notre compréhension des mécanismes d’agrégation d’UBQLN2 et de TDP-43 et de leur rôle dans l’inflammation. Nous avons observé que les microglies provenant des souris doubles transgéniques étaient plus sensibles à la stimulation aux lipopolysaccharides (LPS) et que l’activité NF-κB était plus importante dans les cerveaux provenant des souris doubles transgéniques. Nos résultats ont également suggéré que l’agrégation d’UBQLN2 entraînait une séquestration d’ubiquitine, réduisant ainsi l’efficacité du protéasome et la clairance de TDP-43. D’ailleurs, l’augmentation du pool d’ubiquitine a permis d’améliorer l’efficacité du protéasome et favoriser le retrait de TDP-43 des agrégats et d’augmenter sa dégradation.
En conclusion, cette thèse démontre un rôle important de la protéine UBQLN2 dans la délocalisation de TDP-43 sous formes d’agrégats en culture cellulaire et en modèle animaux. Cela suggère qu’UBQLN2 et TDP-43 possède un rôle synergique dans la physiopathologie de la SLA et dans la neuroinflammation qui leur est associée. Notre modèle double transgénique pourra certainement être utilisé pour tester des possibilités thérapeutiques. De plus, l’interaction entre UBQLN2 et TDP-43 pourrait être ciblé pour traiter la SLA/DFT.
v
Abstract
Amyotrophic lateral sclerosis (ALS) is the most common motor neuron disease in adults. It is characterized by progressive lost of upper and lower motor neurons leading to paralysis and eventually death from 2 to 5 years after the onset of the symptoms. Approximately 10% of the patients have a family history and the remaining patients have a sporadic form of the disease. Up to 15% also have fronto-temporal dementia (FTD) with prominent behavioral and personality changes. Numerous genes are known to be mutated in the familial form of the disease. This includes mutation in superoxide dismutase 1 (SOD1), TAR DNA-binding protein 43 (TDP-43), Fused in sarcoma (FUS), optineurin (OPTN), Tank-binding kinase 1 (TBK1) and ubqiquilin-2 (UBQLN2). These mutations lead to various pathological mechanisms. One of the most defined mechanism is the formation of cytoplasmic aggregates of TDP-43. These inclusions are positive for other proteins such as UBQLN2 and ubiquitin and are the pathological hallmark of the disease. UBQLN2 plays a central role in the ubiquitin proteasome system (UPS). Mutations in UBQLN2 have been link to TDP-43 pathology in vitro. However, the mechanisms underlying TDP-43 pathology induced by UBQLN2 are still unknown.
In this thesis, we used neurons in culture and overexpressed human UBQLN2WT and human
UBQLN2P497H to study the interaction between TDP-43 and UBQLN2. We demonstrated that the
overexpression of UBQLN2 species enhanced TDP-43 cytoplasmic accumulation and aggregation. Since TDP-43 is known to interact with the p65 subunit of the NF-κB transcriptional factor, we analyzed the effect of UBQLN2 overexpression on the p65 activation. We observed an
increase in NF-κB activation in cells transfected with either UBQLN2WT or UBQLN2P497H. We
observed that the hyperactivation of NF-κB was caused by the action of the p38 MAPK in response to cellular stress and UBQLN2/TDP-43 cytoplasmic accumulation. This increase in NF-κB activity enhanced motor neuron death which was reversible by treatment with Withaferin A, a known NF-κB inhibitor.
Because we observed an important synergistic effect in TDP-43 aggregation with UBQLN2 overexpression, we decided to generate a new transgenic mouse model with mutations in both
vi
UBQLN2 and TDP-43. Mice were generated with the expression of UBQLN2P497H gene under the
control of the neurofilament heavy (NFH) promoter. The single transgenic UBQLN2P497H were
then bred with our previously described TDP-43G348C transgenic mice. Whereas the single
UBQLN2P497H transgenic mice developed only mild cognitive impairment, the double transgenic
UBQLN2P497H; TDP-43G348C mice developed typical features of ALS/FTD with important
TDP-43 cytoplasmic aggregation, motor neurons loss, axonal degeneration, muscle atrophy, as well as motor and cognitive symptoms and gliosis. We took advantage of our new generated double transgenic mice to analyze the interaction between UBQLN2 and TDP-43 and to study the effect of aggregation of both proteins on inflammatory pathways. We observed that microglia from double transgenic mice were hyperresponsive to intraperitoneal injection of lipopolysaccharide (LPS) and that NF-κB activity was increased in double transgenic mice. Our results also suggested that UBQLN2 up-regulation induced TDP-43 aggregation by the sequestering of ubiquitin proteins into aggregates and the reduction of the UPS efficacy. Thus, increasing the pool of ubiquitin promoted the UPS function with ensuing reduction of TDP-43 cytosolic accumulation.
In conclusion, this thesis demonstrates an important role of UBQLN2 species in TDP-43 mis-localization and aggregation in vitro and in vivo. It also suggests that UBQLN2 and TDP-43 possess a synergic role in neuroinflammation. Certainly, our double transgenic mice could be used to study future therapeutic avenues and these mechanisms could be targeted to treat TDP-43-associated ALS/FTD pathology.
vii
Table des matières
RÉSUMÉ ... III ABSTRACT ... V LISTES DES FIGURES ... XI LISTE DES ABRÉVIATIONS ... XII À CES NOMBREUSES SOURIS, ... XV REMERCIEMENTS ... XVI AVANT-PROPOS ...XVIII
INTRODUCTION ... 1
1.1 LES CARACTÉRISTIQUES CLINIQUES DE LA SCLÉROSE LATÉRALE AMYOTROPHIQUE ... 1
1.1.1 Généralités ... 1 1.1.2 Historique ... 1 1.1.3 Épidémiologie ... 2 1.1.4 Présentation clinique ... 3 1.1.5 La démence fronto-temporale ... 7 1.1.6 Diagnostic ... 8 1.1.7 Traitement ... 10
1.2 MÉCANISMES PATHOLOGIQUES ET MOLÉCULAIRES ... 12
1.2.1 L’excitotoxicité induite par le glutamate ... 12
1.2.2 La pathogénèse associée à SOD1 ... 14
1.2.3 Les neurofilaments ... 16
1.2.4 Le stress du réticulum endoplasmique ... 17
1.2.5 Les cellules gliales et la neuroinflammation ... 18
1.3 TARDNA-BINDING PROTEIN 43(TDP-43) ... 20
1.3.1 Métabolisme des ARNs ... 20
1.3.2 Granules de stress ... 22
1.3.3 Modifications post-traductionnelles ... 23
1.3.4 Délocalisation et agrégation de TDP-43 ... 24
1.4 LE FACTEUR NUCLÉAIRE KAPPA-B(NF-ΚB) ... 24
1.4.1 Membre de la famille NF-κB ... 25
1.4.2 Voies de signalisation ... 26
1.4.3 NF-κB dans le système nerveux ... 28
1.4.4 NF-κB dans la sclérose latérale amyotrophique ... 28
1.5 UBIQUILINE-2 ... 30
1.5.1 Structure ... 30
1.5.2 Le système ubiquitine-protéasome ... 31
1.5.3 L’autophagie et l’implication d’UBQLN2 ... 32
1.5.4 UBQLN2 dans la sclérose latérale amyotrophique ... 34
1.6 HYPOTHÈSES ET OBJECTIFS DE LA THÈSE ... 35
2 CHAPITRE 2. UBIQUILIN-2 DRIVES NF-ΚB AND CYTOPLASMIC TDP-43 IN NEURONAL CELLS ... 37
2.1 RÉSUMÉ ... 39
2.2 ABSTRACT ... 40
2.3 BACKGROUND ... 40
2.4 RESULTS ... 42
2.4.1 UBQLN2 up-regulation induces NF-κB activation ... 42
2.4.2 UBQLN2 expression in Neuro2A cells leads to cellular stress via MAP kinase pathway ... 44
viii
2.4.4 UBQLN2 aggregates are dynamic structures ... 47
2.4.5 UBQLN2 up-regulation enhances vulnerability to NF-κB-mediated neuronal death and causes an ER-stress 48 2.5 DISCUSSION ... 50
2.6 METHODS ... 54
2.6.1 Cell culture, transfection and cell treatment ... 54
2.6.2 Protein extraction and Western blot analysis ... 54
2.6.3 Luciferase assay ... 55
2.6.4 Immunofluorescence ... 56
2.6.5 Plasmids construction ... 56
2.6.6 MTS assay ... 57
2.6.7 Live cell imaging ... 57
2.6.8 Statistical analysis ... 57
2.7 REFERENCES ... 59
2.8 FIGURE LEGENDS ... 63
2.8.1 Figure 1. NF-κB activation induced by hUBQLN2 is supressed with hUBQLN2 siRNA. ... 63
2.8.2 Figure 2. hUBQLN2 expression in Neuro2A cells caused cellular stress via MAP kinase pathway. ... 64
2.8.3 Figure 3. hUBQLN2 co-localized with TDP-43 and P62 but not with NF-κB and IκB-α. ... 65
2.8.4 Figure 4. hUBQLN2 inclusions were dynamic. ... 66
2.8.5 Figure 5. UBQLN2 induced a NF-κB-mediated neuronal cell death and an ER-stress response. ... 67
2.9 ADDITIONAL FILES ... 68
2.9.1 Additional file 1. (.avi) Movement and fusion of UBQLN2 inclusions in Neuro2A. ... 68
2.9.2 Additional file 2. Lack of inclusion bodies in cell expressing TDP-43. ... 68
3 CHAPITRE 3 FROM ANIMAL MODELS TO HUMAN DISEASE: A GENETIC APPROACH FOR PERSONALIZED MEDICINE IN ALS ... 69
3.1 RÉSUMÉ ... 71
3.2 ABSTRACT ... 72
3.3 INTRODUCTION ... 73
3.4 CLINICAL MANIFESTATION AND EPIDEMIOLOGY OF AMYOTROPHIC LATERAL SCLEROSIS ... 74
3.4.1 Classical ALS ... 74 3.4.2 Bulbar ALS ... 74 3.4.3 Fronto-temporal dementia ... 75 3.4.4 Epidemiology ... 75 3.4.5 Neuropathology ... 76 3.4.6 Diagnostic criteria ... 77
3.5 MUTATION IN FAMILIAL CASES OF ALS ... 77
3.5.1 Superoxide dismutase 1 (SOD1) ... 77
3.5.1.1 Specific disease characteristics in humans... 77
3.5.1.2 Mouse models ... 78
3.5.1.3 Other models ... 80
3.5.1.4 Biomarkers ... 82
3.5.1.5 Personalized medicine ... 83
3.5.2 TAR-DNA-binding protein (TDP-43)... 84
3.5.2.1 Specific disease characteristics in humans... 84
3.5.2.2 Mouse models ... 86
3.5.2.3 Other models ... 87
3.5.2.4 Biomarkers ... 88
3.5.2.5 Personalized medicine ... 89
3.5.3 C9ORF72 ... 90
3.5.3.1 Specific disease characteristics in humans... 90
3.5.3.2 Mouse models ... 91
3.5.3.3 Other models ... 92
ix
3.5.3.5 Personalized medicine ... 94
3.5.4 Fused in sarcoma (FUS) ... 95
3.5.4.1 Specific disease characteristics in humans... 95
3.5.4.2 Mouse models ... 95 3.5.4.3 Other models ... 96 3.5.4.4 Personalized medicine ... 97 3.5.5 Other genes ... 98 3.5.5.1 Ubiquilin-2 (UBQLN2) ... 98 3.5.5.2 Optineurin (OPTN) ... 99 3.6 CONCLUSION ... 100 3.7 DECLARATIONS ... 100 3.8 ABBREVIATIONS ... 101 3.9 REFERENCES ... 103 3.10 FIGURE LEGENDS ... 125
3.10.1 Fig.1 Timeline of gene discovery and pathogenic mechanisms in ALS ... 125
3.10.2 Fig.2 Clinical findings in Amyotrophic lateral sclerosis (ALS)... 126
3.10.3 Fig.3 Gene therapy mechanism of action ... 127
3.10.4 Fig.4 Neuropathological findings in human sALS cases and animal models of ALS. ... 129
4 CHAPITRE 4 NEURONAL EXPRESSION OF UBIQUILIN-2 EXACERBATES TDP-43 PATHOLOGY IN TDP-43G348C MICE THROUGH INTERACTION WITH UBIQUITIN ... 135
4.1 RÉSUMÉ ... 137
4.2 ABSTRACT ... 138
4.3 BACKGROUND ... 138
4.4 METHODS ... 141
4.4.1 Generation of Transgenic mice ... 141
4.4.2 Quantitative Real-Time PCR ... 142
4.4.3 Accelerating rotarod ... 142
4.4.4 Step-through passive avoidance test ... 142
4.4.5 Pole test ... 143
4.4.6 Cat-walk analysis ... 143
4.4.7 Mice sacrificed, protein extraction and immunoblotting ... 143
4.4.8 Immunofluorescence and immunohistochemistry ... 144
4.4.9 H&E staining and ventral root preparation ... 145
4.4.10 Cell culture and transfection ... 146
4.4.11 Immunoprecipitation ... 146
4.4.12 Lipopolysaccharide treatment and cytokines array ... 147
4.4.13 Proteasome assay ... 147
4.4.14 Statistical analysis ... 148
4.5 RESULTS ... 148
4.5.1 Generation of UBQLN2P497H mutant mice ... 148
4.5.2 TDP-43 proteinopathy in double transgenic mice ... 149
4.5.3 Motor neuron loss, muscle atrophy and axonal degeneration in double transgenic mice ... 151
4.5.4 Motor and cognitive deficits in double transgenic mice ... 151
4.5.5 Gliosis and inflammatory profiles ... 153
4.5.6 hUBQLN2P497H increased TDP-43 cytosolic accumulation through ubiquitin sequestering ... 154
4.6 DISCUSSION ... 157
4.7 CONCLUSIONS ... 160
4.8 LIST OF ABBREVIATIONS ... 161
4.9 DECLARATIONS ... 162
4.10 TABLE 1OLIGOPRIMERS USED FOR PLASMID CONSTRUCTION AND Q-RT-PCR ... 163
4.11 REFERENCE ... 163
x
4.12.1 Fig. 1 UBQLN2P497H transgenic mouse line generation ... 167
4.12.2 Fig. 2 Cytosolic TDP-43 accumulations in double transgenic mice ... 168
4.12.3 Fig. 3 TDP-43/UBQLN2 inclusions at 8 months of age ... 169
4.12.4 Fig. 4 Motor neuron loss, axonal degeneration and muscle atrophy ... 170
4.12.5 Fig. 5 Motor and cognitive impairment in double transgenic mice ... 171
4.12.6 Fig. 6 Gliosis and susceptibility to NF-κB-driven inflammation ... 172
4.12.7 Fig. 7 Reversal of UBQLN2-driven UPS dysfunction and TDP-43 aggregation by upregulation of ubiquitin 173 4.12.8 Suppl Fig. 1 ... 175
4.12.9 Suppl Fig. 2 ... 176
4.12.10 Suppl Fig. 3 ... 177
DISCUSSION GÉNÉRALE ET CONCLUSION ... 178
5.1 L’IMPLICATION D’UBIQUILINE-2 DANS L’ACCUMULATION DE TDP-43 ... 179
5.2 GAIN DE FONCTIONS TOXIQUES ET PERTE DE FONCTIONS NORMALES DE TDP-43 ... 183
5.3 UBIQUILINE-2 DANS L’ACTIVATION DU FACTEUR NF-ΚB, L’INFLAMMATION ET LA MORT NEURONALE ... 186
5.4 LES SOURIS UBQLN2P497H;TDP-43G348C COMME MODÈLE MURIN DE SLA ... 190
5.5 CONCLUSION ET PERSPECTIVES FUTURES ... 194
xi
Listes des figures
FIGURE 1SPECTRE DE PHÉNOTYPES DE LA SCLÉROSE LATÉRALE AMYOTROPHIQUE ET DES MALADIES APPARENTÉES (1) ... 6
FIGURE 2LES CRITÈRES D’EL ESCORIAL RÉVISÉS POUR LE DIAGNOSTIC DE LA SLA(WWW.UPTODATE.COM) ... 9
FIGURE 3L’EXCITOTOXICITÉ INDUITE PAR LE GLUTAMATE ET LA SENSIBILITÉ DES MOTONEURONES AUX INFLUX DE CALCIUM (70) ... 13
FIGURE 4PRODUCTION D’HYDROXY-RADICAUX PAR LES FORMES MUTANTES DE SOD1(79) ... 15
FIGURE 5LE MAL-REPLIEMENT DE SOD1 NATIF OU MUTÉ ENTRAÎNE SON AGRÉGATION (96) ... 16
FIGURE 6CASCADE D’ÉVÈNEMENTS DANS LE STRESS DU RÉTICULUM ENDOPLASMIQUE (111) ... 18
FIGURE 7REPRÉSENTATION SCHÉMATIQUE DES DOMAINES DE TDP-43 HUMAIN ET DE SES MUTATIONS (143) ... 21
FIGURE 8FONCTIONS NORMALES DE TDP-43 DANS LA GESTION DES ARNS ET LA FORMATION DE GRANULES DE STRESS (142) ... 21
FIGURE 9VOIE D’ACTIVATION DU FACTEUR NF-ΚB PAR LA STIMULATION DE TNF-Α (177) ... 26
FIGURE 10VOIES D’ACTIVATION ATYPIQUE, CANONICALE ET NON-CANONICALE DE NF-ΚB(187) ... 27
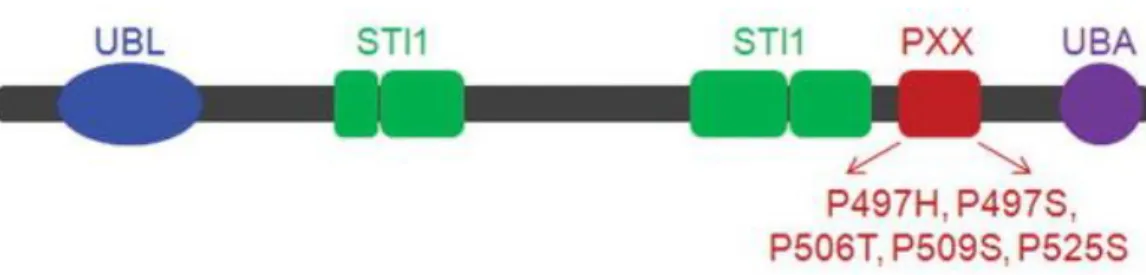
FIGURE 11REPRÉSENTATION SCHÉMATIQUE DES DOMAINES D’UBQLN2(213) ... 30
FIGURE 12LA CONJUGAISON DES UBIQUITINES ET LE SYSTÈME UBIQUITINE-PROTÉASOME (222) ... 32
FIGURE 13LA MICROAUTOPHAGIE, LA MACROAUTOPHAGIE ET L’AUTOPHAGIE MÉDIÉE PAR DES CHAPERONES (223) ... 33
FIGURE 14MÉCANISME PROPOSÉ D’AGRÉGATION D’UBQLN2 ET DE TDP-43 ... 182
FIGURE 15CO-LOCALISATION DE TDP-43 ET TIA1 DANS LES MOTONEURONES DE SOURIS DOUBLE TRANSGÉNIQUE À L’ÂGE DE 5 MOIS. ... 185
FIGURE 16MÉCANISME PROPOSÉ D’ACTIVATION DE NF-ΚB ET D’INDUCTION DE LA MORT NEURONALE PAR UBQLN2 ... 190
xii
Liste des abréviations
SLA sclérose latérale amyotrophique TDP-43 TAR-DNA binding protein 43 SOD1 superoxyde dismutase 1
FUS Fused in Sarcoma UBQLN2 Ubiquiline-2
FDA Food and drug Association OPTN optineurin
VCP valosin-containing protein TBK1 TANK-binding kinase 1 MNS motoneurone supérieur MNI motoneurone inféfieur
ALSFRS-R ALS functional rating scale revised DFT démence fronto-temporale
EMG électromyographie
EAAT-2 excitatory amino acid transporter 2
AMPA alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid GluR2 glutamate receptor 2
NF neurofilaments
RE réticulum endoplasmique UPR unfolded response protein PERK PKR-like ER kinase
IRE1 inositol-requiring transmembrane Kinase ATF6 activating transcription factor 6
XBP1 X-Box-binding protein 1
eIF2α eukaryotic initiation factor 2 alpha GFAP glial fibrillary acidic protein iPSCs induced pluripotent stem cells IL-6 interleukine-6
xiii NF-κB Nuclear factor kappa-B
TLR toll-like receptor
hnRNPs heterogeneous nuclear ribonucleoproteins NTD domaine N-terminal
CTD domaine C-terminal
NLS sequence localisation nucléaire NLS sequence d’exportation nucléaire GS granules de stress
TIA-1 T-cell restricted intracellular antigen-1 PABP poly (A)-binding protein
G3BP1 Ras Gap SH3 domain binding protein 1 UPS ubiquitin-proteasome system
IκB inhibiteur kappa-B
IKK kinases serine-thréonine IκB NEMO NF-κB essential modulator TRAF-2 TNF associate factor-2 NIK NF-κB inducing kinase SYK spleen tyrosine kinase CNTF ciliary neurotrophic factor NGF nerve growth factor
BDNF brain-derived neurotrophic factor WA Withaferine A
UBL N-terminal ubiquitin-like UBA C-terminal ubiquitin-associated STI1 stress-induced protein1-like
Ubxd8 ubiquitin regulatory X domain-containing protein 8 AMC autophagie médiée par des chaperonnes
LC3 microtubule-associated protein 1A/1B-light chain 3 APG9A autophagy-related protein 9A
xiv MAPK mitogen-activated protein kinase
AAV adeno-associated virus siRNA petit ARN interferent
xv
xvi
Remerciements
J’aimerais remercier plusieurs personnes, sans qui cette thèse n’aurait pas été réalisée. D’abord, j’aimerais remercier Dr Jean-Pierre Julien pour l’accueil dans son laboratoire au cours des 6 dernières années. J’ai particulièrement apprécié sa confiance et son ouverture à mener mes projets selon l’orientation que je voulais y apporter. Jean-Pierre m’a toujours supporté et encouragé à poursuivre en recherche dans son laboratoire pendant mes études en médecine. Il m’a laissé énormément d’autonomie, me permettant de concilier travail au laboratoire et journées de clinique bien remplies. Il a certainement été un mentor qui m’a fait grandir professionnellement.
Je remercie Dr Nicolas Dupré pour son support comme co-directeur. Nicolas m’a donné d’excellents conseils sur la conciliation recherche/clinique et sur mes possibilités de carrière en recherche comme neurologue. J’espère compléter de nombreux autres projets avec lui dans les prochaines années.
Je remercie mon amoureuse et maman de Colby, Marie-Hélène Paradis, pour sa patience au cours de ces années. Je n’ai pas été le plus présent des conjoints, mais tu ne me l’as jamais reproché. Tu peux espérer que je travaille moins maintenant que ma thèse est terminée, mais malheureusement, je ne crois pas cela va survenir. Je t’aime Guylaine.
Je ne peux assez remercier mon voisin de bureau, collègue et ami, Louis-Charles Béland. Sans lui, ma thèse aurait été beaucoup plus courte, mais n’aurait jamais été aussi plaisante. Nos nombreuses soirées à « ne pas » boire dans nos bureaux, à s’obstiner sur ton absence de connaissances en biologie et à fêter dans les congrès ont certainement marqué mes études supérieures.
xvii
Je remercie également mes parents pour leur éducation, leur support et pour leurs essaies à comprendre mes travaux de recherche. Ce n’est pas évident, je ne suis pas sûr que je comprenne mes résultats non plus.
J’aimerais également remercier mes matantes préférées Christine Bareil et Geneviève Soucy pour leur travail dévoué dans le laboratoire. Je les remercie pour le maintien de ma lignée de souris et pour leur amitié aux cours des 6 dernières années. Un merci à Mélanie pour m’avoir fait quelques commentaires sur ma thèse. J’aimerais remercier tous les autres membres des laboratoires de Jean-Pierre et Jasna pour leur amitié/aide dans les dernières années; Sai, Kallol, Jean-Pierre, Karine, Sunny, Banshi, Poojah, Sylvia, Daniel et Romina.
Finalement, je remercie Dre Jasna Kriz d’accepter que je poursuivre en recherche dans son équipe au cours des prochaines années.
xviii
Avant-Propos
Dans le chapitre 2, l’article « Ubiquilin-2 drives NF-κB and cytoplasmic TDP-43 in neuronal cells » a été publié dans le journal Molecular Brain le 31 octobre 2015 (doi: 10.1186/s13041-015-0162-6.). Il n’y a eu aucune modification entre la version publiée et la version intégrée dans cette thèse. Je suis l’auteur principal de cet article.
Auteurs : Vincent Picher-Martel, Kallol Dutta, Daniel Phaneuf, Gen Sobue and Jean-Pierre Julien Contribution des auteurs : Vincent Picher-Martel a réalisé 95% des expériences, a analysé les résultats, fait les figures et écrit l’article. Kallol Dutta a fait quelques western blots. Daniel Phaneuf
a construit les plasmides pCMV-UBQLN2WT et pCMV-UBQLN2P497H. Gen Sobue a gentiment
fourni le plasmide DsRed-TDP-43. Jean-Pierre Julien a conçu, supervisé le projet et corrigé l’article.
Dans le chapitre 3, l’article « From animal models to human disease: A genetic approach for personalized medicine in ALS » a été publié dans le journal Acta Neuropathologica Communications le 11 juillet 2016 (doi: 10.1186/s40478-016-0340-5.). Il n’y a eu aucune modification entre la version publiée et la version intégrée dans cette thèse. Je suis l’auteur principal de cet article.
Auteurs : Vincent Picher-Martel, Paul N. Valdmanis, Peter V. Gould, Jean-Pierre Julien and Nicolas Dupré
Contribution des auteurs : Vincent Picher-Martel a fait la revue de la littérature, fait les figures et écrit l’article. Paul N Valdmanis a corrigé l’article. Peter V Gould a fournie quelques images d’immunohistochimie pour la figure 3. Jean-Pierre Julien a corrigé l’article. Nicolas Dupré a corrigé l’article et supervisé l’écriture.
Dans le chapitre 4, l’article « Neuronal expression of UBQLN2P497H exacerbates TDP-43
xix
Neurobiology le 30 octobre 2018. Il n’y a eu aucune modification entre la version publiée et la version intégrée dans cette thèse. Je suis l’auteur principal de cet article.
Auteurs : Vincent Picher-Martel, Laurence Renaud, Christine Bareil and Jean-Pierre Julien
Contributions des auteurs : Vincent Picher-Martel a construit le plasmide utilisé pour générer les souris, fait la caractérisation des souris, fait plus de 95% des expériences de l’article, analysé les données, fait les figures et écrit l’article. Laurence Renaud a fait les analyses des cytokines et aidé pour la culture cellulaire. Christine Bareil a génotypé et maintenue la lignée de souris. Jean-Pierre Julien a supervisé le projet et corrigé l’article.
1
Introduction
1.1 Les caractéristiques cliniques de la sclérose latérale amyotrophique
1.1.1 Généralités
La sclérose latérale amyotrophique (SLA), ou la maladie de Lou Gehrig, se caractérise par une perte progressive des motoneurones supérieurs et inférieurs menant à une faiblesse progressive et à une paralysie. Les patients décèdent généralement à l’intérieur de 2 à 5 ans après le début des symptômes. Il n’existe pas de traitement efficace modifiant réellement le cours de la maladie. Nous connaissons plusieurs gènes dans la forme familiale de la maladie incluant superoxyde dismutase 1 (SOD1), TAR-DNA binding protein 43 (TDP-43), C9ORF72, Fused in Sarcoma (FUS) et ubiquiline-2 (UBQLN2). Grâce à l’identification de ses gènes, plusieurs mécanismes pathologiques sont connus, mais le lien exact entre ces mécanismes demeure imprécis.
1.1.2 Historique
La première description de la sclérose latérale amyotrophique est largement attribuée au Dr Jean-Martin Charcot en 1869. D’ailleurs, la SLA est parfois appelée la maladie de Charcot dans certains pays, dont la France. En revanche, il n’a pas été le premier à décrire des cas pouvant être attribué à cette maladie, mais il a été le premier à bien décrire les trouvailles pathologiques de la maladie (2). En 1853, l’anatomiste français Jean Cruveilhier a décrit une femme souffrant d’une atrophie musculaire progressive avec une paralysie bulbaire. Après le décès de la patiente, Dr Cruveilhier a principalement décrit une atrophie des racines antérieures de la moelle épinière (Cruveilhier J et al., 1853). En 1862, l’anatomiste anglais Jacob Augustus Lockhart Clarke a décrit un cas d’un homme de 43 ans avec paralysie, atrophie musculaire, spasticité et fasciculations (Radcliffe CB et al., 1862). À l’autopsie, Dr. Clarke a décrit une perte des cellules dans la corne ventrale avec une pâleur diffuse de la substance blanche de la moelle. Dr. Clarke fut en fait le premier à décrire une pathologie centrale pouvant être reliée à la SLA. En 1865, Jean-Martin Charcot a décrit un patient similaire avec sclérose des colonnes latérales de la moelle épinière, atrophie des racines ventrales, mais avec une substance grise corticale normale (Charcot J-M et al., 1865). Ces trouvailles lui ont permis de former l’hypothèse que les neurones moteurs de la corne antérieure était la source de
2
l’atrophie musculaire progressive. Charcot a également décrit en détails les étapes de la maladie en considérant une variation importante entre les modes de présentation et d’évolution.
Aux États-Unis, la maladie est aussi appelée la maladie de Lou Gehrig en l’honneur du célèbre joueur de baseball des Yankees de New-York décédé de la maladie en 1941 à l’âge de 37 ans. Les connaissances physiopathologiques de la SLA n’ont pas évolué de manière importante jusqu’à la découverte d’une mutation dans le gène SOD1 en 1993 (3). Cette découverte a permis de changer considérablement la recherche sur la SLA en permettant le développement de modèles animaux avec mutation dans le gène SOD1. L’un des deux seuls traitements acceptés pour traiter la maladie, le riluzole, a été indroduit au début des années 90 après avoir été testé dans les modèles SOD1. Le deuxième traitement, l’edaravone, a été accepté par la Food and Drug Association (FDA) américaine en 2017. Pour l’instant, il n’est pas disponible au Canada.
1.1.3 Épidémiologie
L’incidence de la SLA est d’environ 1 à 2,6 nouveaux cas / 100 000 habitants par année. Une revue systématique avec méta-analyse récente regroupant plus de 13 000 patients dans 45 études a réussi à bien démontrer l’incidence de la SLA par continent (4). En Amérique du nord et en Europe, l’incidence annuelle est aux alentours de 1,81 nouveaux cas/100 000 habitants alors qu’elle est d’environ 0,83 nouveau cas/ 100 000 habitants en Asie. La prévalence de la maladie est aux alentours de 6 personnes par 100 000 habitants (5). Au Canada, on estime qu’environ 3000 personnes vivent actuellement avec la maladie et que 2 à 3 personnes décèdent de la maladie à chaque jour (6). L’âge moyen au début des symptômes est de 61,8 ± 3,8 ans et de 64.4 ± 2.9 ans au diagnostic (7).
Plusieurs facteurs de risques pour développer la SLA peuvent être énumérés. En revanche, très peu d’entre eux sont basés sur des évidences solides et ayant une causalité établie. Le facteur génétique est certainement le mieux décrit avec environ 5 à 10% des patients qui sont atteints d’une forme familiale. À ce jour, 26 gènes ont été reliés à la forme familiale de la maladie. Parmi les plus
3
communs on peut nommer par exemple; C9ORF72, TDP-43, SOD1, FUS, optineurin (OPTN), UBQLN2 et valosin-containing protein (VCP), TANK binding kinase 1 (TBK1) (8). Le tabagisme a été relié à un risque augmenté de développer la SLA dans des études prospectives et rétrospectives (9-13). Cependant, certaines études, incluant une méta-analyse, n’ont démontré aucune association avec le tabagisme (14, 15).
La SLA a certainement commencé à attirer l’attention en 1941 lors du décès de Lou Gehrig. Son décès a mené à plusieurs études sur le rôle du sport et de l’activité physique dans le développement de la maladie. Une incidence augmenté de SLA a été observée chez les joueurs de soccer jusqu’à 15 fois l’incidence normale lors de carrière professionnelle prolongée (16). L’explication pathologique de ce phénomène n’est pas connue, mais pourrait être secondaire aux traumatismes crâniens répétés (17, 18). Une méta-analyse publié en 2016 a confirmé une augmentation significative de SLA chez les joueurs de soccer et de football professionnel, mais les évidences sur l’activité physique en général demeure non-concluantes (19). Certains auteurs considèrent les chocs électriques ou l’exposition aux champs magnétiques comme étant un facteur de risque. En revanche, une revue a démontré que parmi treize études de cas ou de cohortes, seulement trois études avaient des résultats à la limite du significatif (20).
L’exposition à des métaux lourds ou à des toxines environnementales seraient relié au développement de la maladie. Depuis les années 70, de nombreuses études ont mentionné la relation entre la SLA et l’exposition au plomb (10, 21-23), mais d’autres études ont échoué à démontrer une relation (24-26). Cette relation a été étudiée avec une revue systématique et méta-analyse. Cette étude regroupait neuf études cas-contrôles et a démontré un risque relatif de 1,81 (1.39 à 2,36) de développer la SLA avec une exposition occupationnelle au plomb. Le risque attribuable au plomb seulement était estimé à 5%.
1.1.4 Présentation clinique
La SLA se caractérise par une perte progressive des motoneurones supérieurs (MNS) et inférieurs (MNI). Les patients atteints de la maladie se présentent avec une faiblesse progressive évoluant généralement depuis quelques semaines. Dans la forme classique, soit environ 70% des cas, les
4
faiblesses débutent à une main ou à une jambe. Ces faiblesses vont généralement progresser pour atteindre le membre controlatéral et ensuite atteindre les autres régions du corps. Lorsque les symptômes affectent d’abord les mains, les patients ont généralement une lenteur, une rigidité et une certaine incoordination dans leur mouvement. Ils expriment une difficulté croissante à remplir leurs tâches quotidiennes ou encore à manipuler de petits objets. Lorsque les symptômes affectent les jambes, les patients se plaignent de difficultés à tourner, de difficultés à se lever d’une chaise ou à monter des marches, de jambes lourdes ou d’un trouble de l’équilibre avec chutes.
Lors de l’évaluation clinique par un médecin, il est possible de mettre en évidence des signes d’atteintes des motoneurones supérieurs et inférieurs. La spasticité et l’augmentation des réflexes ostéotendineux ou la préservation des réflexes avec un muscle faible et atrophié caractérise l’atteinte du MNS. Il est également possible d’observer un signe de Babinski, soit une élévation pathologique des orteils lors de la stimulation de la plante du pied, ou encore un signe de Hoffman caractérisé par une réponse en flexion de l’index lors du frottement de l’ongle du majeur. L’atteinte du MNI est plutôt mise en évidence par une atrophie musculaire, une faiblesse importante, une baisse des réflexes et des fasciculations. Les fasciculations sont des petits frétillements des muscles dénervés visibles à l’œil nue et classique de la maladie. Environ 30% des patients se présentent plutôt avec des symptômes d’origine bulbaire. Les signes et symptômes d’une atteinte du MNS au niveau bulbaire incluent une lenteur de la parole, un réflexe palmo-mentonier positif ou un réflexe « jaw jerk ». L’atteinte du MNI s’observe par la présence d’une faiblesse au niveau du visage associée à une dysarthrie, une baisse de l’élévation du palais, une atrophie de la langue et des fasciculations de la langue.
L’évolution naturelle de la maladie est très variable d’un patient à l’autre. Les experts se plaisent à affirmer qu’il existe autant d’évolution différente que de patients atteints de la maladie. En général, les patients décèdent de la maladie 2 à 5 ans après le début des symptômes. Cependant, une évolution courte sur moins de 6 mois ou encore une évolution sur plus de 6 ans sont fréquemment observés. Plusieurs facteurs peuvent influencer l’espérance de vie des patients. D’abord, l’âge est un facteur pronostic clé de la maladie avec une espérance de vie inversement relié à l’âge au début des symptômes (27). La présence de symptômes bulbaires au moment du
5
diagnostic est également un facteur indépendant de mauvais pronostic comparativement aux présentations périphériques (28). La progression des déficits musculaires et de la fonction respiratoire lors des six premiers mois est également un élément clé de mauvais pronostic (28). De manière évidente, un début de symptômes avec atteinte respiratoire est un mauvais facteur pronostic. Les formes familiales n’ont généralement pas une espérance de vie plus courte que les
formes sporadiques. Par contre, certaines mutations, tel que la mutation SOD1A4V, présentent une
évolution rapide et un décès dans la première année (29). Certaines études ont évalué l’utilité de mesure clinique pour quantifier le pronostic des patients. Une étude rétrospective utilisant la capacité vitale forcée, l’échelle du conseil de recherche médicale (MRC), l’échelle bulbaire et le score ALSFRS a démontré que ces échelles étaient toutes des facteurs indépendants de pronostic
(30, 31). L’état nutritionnel évalué avec un index de masse corporel sous 18,5 kg/m2 est également
un facteur de mauvais pronostic (32).
La SLA peut être incluse dans une catégorie de maladies touchant les motoneurones. Cette catégorie inclut également la sclérose latérale primaire et l’atrophie musculaire progressive. Ces trois maladies forment un spectre de maladie avec l’atrophie musculaire progressive à un extrémité (avec une atteinte des MNI seulement) et la sclérose latérale primaire à l’autre extrémité (avec une atteinte des MNS seulement). Il n’est pas clair si ces trois maladies sont en fait des variantes d’une seule maladie ou encore trois maladies à part entière (1) (Fig. 1). Quand les symptômes débutent au niveau du bulbe, il est également possible d’utiliser les termes parésie bulbaire progressive, quand on observe une prédominance d’atteinte du motoneurone inférieur, ou parésie pseudobulbaire lors d’atteinte du motoneurone supérieur. Ces variantes sont souvent difficiles à distinguer cliniquement et rendent le diagnostic de la maladie complexe. Il peut être particulièrement important de différencier ces formes d’atteinte du motoneurone pour l’inclusion dans les essais cliniques et pour évaluer le pronostic des patients (33). En effet, les patients avec la sclérose latérale primaire ont une espérance de vie supérieure par rapport aux patients avec la forme classique de la SLA.
6
Plusieurs patients se plaignent également de symptômes non-moteurs. Une revue systématique a récemment mise en évidence la prévalence de ces symptômes (34). Les symptômes
neuropsychiatriques incluant des symptômes dépressifs, de l’apathie, de l’irritabilité touche de 6 à 29% des patients selon les études (35-38). La dépression majeure touche environ 6-13% des patients alors qu’elle touche environ 5-6% de la population générale (39). La fatigue sévère affecte jusqu’à 30-40% des patients (36). La labilité émotionnelle qui est également connu comme l’affect pseudobulbaire se caractérisant par des rires ou des pleures incontrôlés est présente chez environ 50% des patients (34). Les douleurs musculaires sont très prévalentes touchant environ 20-80% des patients (40, 41). Finalement, la sialorrhée importante est certainement l’un des problèmes non-moteurs majeurs des patients atteints de la SLA. Elle peut toucher environ 20 à 50% des personnes atteintes de la maladie (33, 42).
Figure 1 Spectre de phénotypes de la sclérose latérale amyotrophique et des maladies apparentées (1)
7 1.1.5 La démence fronto-temporale
Bien que l’on ait longtemps cru que les personnes atteintes de la maladie gardaient toutes leurs capacités cognitives, il est maintenant clair qu’il y a une forte association entre la SLA et la démence fronto-temporale (DFT). Les premières évidences reliant la DFT et la SLA remontent au début des années 90’ avec la présentation d’une série de 70 cas avec maladie du motoneurone et troubles cognitifs (43). Il a été démontré que les cas de SLA ou SLA-DFT avaient des trouvailles neuropathologiques similaires incluant des inclusions d’ubiquitine (44). Ces inclusions sont ensuite devenues le critère pathologique du continuum SLA-DFT. En 2006, la découverte de la présence de TDP-43 comme la composante majeure des inclusions cytoplasmiques dans la SLA et dans la DFT a confirmé la relation entre les deux pathologies (45). Plusieurs mutations ont été mise en évidence dans des familles avec SLA-DFT, incluant des mutations dans le gène C9ORF72, TDP-43, UBQLN2 et SQSTM1. La prévalence de la DFT parmi les patients atteints de la SLA varie selon les études. Il est suggéré qu’environ 50% des patients ont certains troubles cognitifs, mais que seulement 10 à 15% des patients remplissent les critères de la démence (46, 47). De manière similaire, environ 15% des patients avec DFT développent une SLA.
La démence fronto-temporale peut être divisée en trois sous-catégories ; la DFT comportementale, l’aphasie primaire progressive sémantique et l’aphasie primaire progressive non-fluente. Toutes ces formes ont une pathologie d’inclusion de TDP-43, de TAU ou d’autres protéines. La démence fronto-temporale comportementale (DFTc) est la plus commune chez les patients atteints de SLA. Il y a seulement quelques cas rapportés d’aphasie primaire progressive avec maladie du motoneurone (48, 49). La DFTc se caractérise cliniquement par des changements de comportement et de personnalité. Les patients démontrent une importante désinhibition avec des comportements sociaux inappropriés. Ceux-ci peuvent inclure par exemple le fait de toucher ou embrasser des étrangers ou encore d’uriner ou de flatuler en public. Ils démontrent généralement une apathie ou une perte de l’empathie. Certains patients souffrent d’hyperoralité avec un goût élevé pour certains aliments ou ont des comportements de rage alimentaire. Certains essaient de mettre une quantité excessive de nourriture dans leur bouche ou de manger des objets non comestibles. Finalement, les comportements obsessifs et stéréotypés sont fréquents. Avec l’évolution de la maladie, les gens peuvent présenter des troubles de mémoires, mais ceux-ci ne sont pas au premier plan.
8 1.1.6 Diagnostic
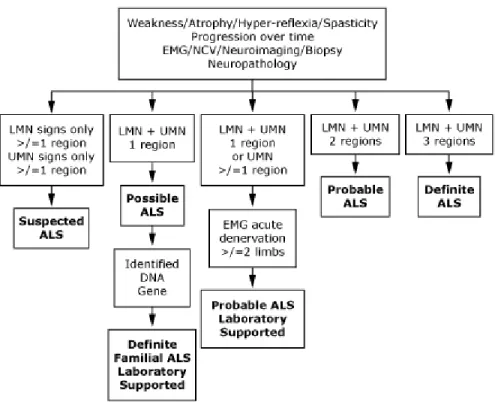
Le diagnostic de la SLA prend régulièrement plusieurs mois avant d’être considéré vu l’évolution progressive de la maladie. Une revue systématique a suggéré un délais moyen de 12,6 ± 2,6 mois entre le début des symptômes et le diagnostic (7). Plusieurs critères peuvent être utilisés pour réaliser le diagnostic avec une présentation clinique correspondante. Les critères d’El Escorial sont certainement les plus utilisés et ont été révisé en 2000 par la fédération mondiale de recherche du les maladies du motoneurones (50). Selon ces critères, le diagnostic de la maladie requiert : 1) une évidence de dégénérescence du motoneurone inférieur par l’évaluation clinique, électrophysiologique ou neuropathologique, 2) une évidence de dégénérescence du motoneurone supérieur par évaluation clinique et 3) une progression des symptômes et des signes d’une région à une autre. De plus, il est nécessaire d’éliminer des diagnostics alternatifs pouvant mimer la maladie par électrophysiologie, par un examen neuropathologique ou par imagerie neurologique. Le diagnostic peut ensuite être classé en différents niveaux de certitude selon le nombre de régions atteintes (bulbaire, cervical, thoracique ou lombosacrée) (Fig. 2). La SLA cliniquement définie nécessite obligatoirement une atteinte du motoneurone supérieur et inférieur dans la région bulbaire et à deux niveaux dans la moelle ou à trois niveaux dans la moelle sans atteinte bulbaire. La SLA cliniquement probable nécessite obligatoirement une atteinte du motoneurone inférieur et supérieur dans deux régions différentes. La SLA probable supporté par laboratoire nécessite une atteinte MNI et MNS dans une région ou une atteinte MNS dans une région ou plus avec dénervation dans plus de deux membres à l’électromyographie EMG. La SLA possible nécessite une atteinte du MNS et MNI dans une région. L’ajout d’une mutation génétique connue amène le diagnostic de SLA familial supporté par laboratoire. La présence d’une atteinte du MNS ou du MNI dans une seule région rend le diagnostic de la SLA suspecté.
9
Figure 2 Les critères d’El Escorial révisés pour le diagnostic de la SLA (www.uptodate.com)
L’une des plus grandes lacunes des critères d’El Escorial est la lenteur à laquelle ils nous permettent de faire le diagnostic de la maladie. En effet, plusieurs patients développent une atteinte du MNI ou du MNS tard au cours de la maladie, ce qui nous empêche de réaliser un diagnostic rapide. La sensibilité des critères est donc très basse en pratique clinique. Par exemple, seulement 56% des patients avec SLA clinique rencontrait les critères de SLA définie ou probable et jusqu’à 10% décédaient sans atteindre une certitude plus élevée que la SLA « possible » (51). En revanche, ces critères demeurent particulièrement pertinents pour la recherche. En 2008, un consensus d’expert a suggéré l’utilisation de critères électromyographiques dans les critères d’Awaji (52). Ces critères incluaient la présence de potentiels d’unité motrice instable (dégénérescence chronique), de potentiel de fasciculations et des fibrillations. Les critères d’Awaji ajouté à l’algorithme d’El Escorial démontraient une sensibilité de 95% pour un diagnostic défini comparativement à 18% pour les critères d’El Escorial et 53% pour les critères d’El Escorial en utilisant ses propres critères à l’EMG. Ces résultats étaient particulièrement intéressants pour les présentations bulbaires. En utilisant les critères d’Awaji, le temps moyen entre les symptômes et
10
le diagnostic semble réduire de 15,2 mois à 9,0 mois (53). Une étude prospective multicentrique récente a révélé une sensibilité des critères d’Awaji de 57% contre 45% pour les critères d’El Escorial avec une spécificité de 99% dans les deux cas (54).
1.1.7 Traitement
Comme mentionné ci-haut, il n’existe aucun traitement modifiant réellement l’évolution de la maladie. La majorité des patients reçoivent du riluzole, un inhibiteur glutamatergique qui diminue la toxicité glutamatergique induite par les astrocytes sur les motoneurones (voir section 1.2). Quatre essais cliniques ont évalué l’effet du riluzole sur la survie et la survie des patients sans trachéostomie. Le premier essai clinique publié en 1994 était un essai clinique à double aveugle, contrôlée avec un placebo sur 155 patients. Après 12 mois, les auteurs ont observé une survie de 58 % dans le groupe placebo et 74% dans le groupe riluzole (p=0,014) (55). Une deuxième étude multicentrique à double insu, contrôlée avec un placebo étudiant différentes doses de riluzole (50, 100 et 200 mg par jour) a été publié par le même groupe en 1996 (56). Cette étude contenait 959 patients avec SLA probable ou définie. Après 18 mois de suivi, 50,4% des patients dans le groupe placebo et 56,8% des patients dans le groupe riluzole (risque ajusté 0.65, p=0,002) étaient vivants et sans trachéostomie. La survie semblait prolongée dans le groupe 200 mg, mais les effets secondaires étaient plus marqués. Une troisième étude, publiée par le même groupe, a étudié l’effet sur la survie et la sécurité chez les patients en phase avancée de la maladie ou âgé de plus de 75 ans (57). L’étude contenait 168 patients et avait un suivi de 18 mois. Les auteurs n’ont observé aucun avantage sur la survie des patients en phase avancée de la maladie, mais la molécule était bien tolérée. Une dernière étude japonaise n’a démontré aucun avantage sur la survie, mais contenait seulement 195 patients, avait plusieurs objectifs primaires différents et les données sur la survie sans trachéostomie n’étaient pas disponibles. Une revue Cochrane publiée en 2012 a réalisé une méta-analyse avec revue systématique sur l’utilité du riluzole en SLA. Cette revue incluait les trois premières études dans son analyse et excluait la dernière étude japonaise par manque de données sur la survie des patients. La méta-analyse suggère un gain de 9% dans la probabilité de survie à 12 mois (placebo 49%, riluzole 58%) et une médiane passant de 11,8 à 14,8 mois. Donc, le riluzole augmente la survie des patients de 2 à 3 mois. Un suivi de la fonction hépatique demeure nécessaire pendant le traitement.
11
Plus récemment, la FDA américaine a approuvé l’utilisation de l’edaravone dans le traitement pour la SLA. L’edaravone est une molécule antioxydante approuvée pour le traitement de l’AVC ischémique au Japon depuis 2001. Elle a démontré une efficacité à améliorer les capacités motrices
et à diminuer la mortalité des motoneurones chez les souris SOD1G93A (58). Cependant, aucun effet
sur la survie n’a été observé. Quatre essais cliniques et une analyse post-hoc a mené à l’acceptation de l’edaravone par la FDA. Contrairement aux études sur le riluzole, ces essais avaient comme objectif primaire de démontrer un effet sur le score ALS Functional Rating Scale (ALSFRS-R), un score évaluant l’autonomie et les symptômes des patients. Avec un score maximum de 48 points, l’ALSFRS-R évalue la parole, la salivation, la dysphagie, l’écriture, l’hygiène, la capacité à marcher et à monter des marches et la dyspnée.
Une première étude de phase 2 étudiant deux doses (30 et 60 mg) sans groupe contrôle a démontré une perte de seulement 2,3 ± 3,6 points sur le score ALSFRS-R durant les 6 mois de l’étude. Les patients avaient précédemment une perte 4,7 ± 2,1 points dans les 6 mois avant le traitement (59). La première étude de phase 3 à double aveugle, contrôlée avec un placebo sur 206 patients n’a démontré aucun avantage de l’edaravone sur l’ALSFRS-R à 6 mois (60). Les auteurs ont observé une baisse de 6,35 ± 0,84 points dans le groupe placebo et 5,70 ± 0,85 points dans le groupe edaravone (p=0,411). Le même groupe a ensuite réalisé une étude d’extension de 6 mois à double aveugle et contrôlée par un placebo avec les mêmes patients (61). Cette étude n’a également démontré aucun avantage sur le score ALSFRS-R. De plus, les patients recevant l’edaravone avaient plus d’effet secondaire sérieux (52,1% contre 28,9%). Les auteurs ont ensuite réalisé une analyse post-hoc afin de cibler les patients pouvant bénéficier de l’edaravone (62). Cette analyse post-hoc a démontré que les patients en début de maladie, soit à l’intérieur de 2 ans du début des symptômes, avec une bonne fonction respiratoire et au moins deux points pour chaque item du ALSFRS-R pourraient potentiellement bénéficier de l’edaravone. Ainsi, une dernière étude à double aveugle, contrôlée avec un placebo, incluant seulement ces patients, a été réalisée (63). L’étude incluait 137 patients et a démontré une réduction de 2,49 points sur l’échelle ALSFRS-R (p=0,0013) sans augmentation des effets secondaires. Les auteurs suggèrent que les patients traités avec l’edaravone ont une réduction de 33% de perte de points à 6 mois. Il n’y avait aucune
12
différence sur la survie des patients. Ainsi, l’edaravone pourrait être bénéfique chez une fraction des patients avec évolution lente et en début de maladie, mais d’autres études doivent être réalisées pour confirmer ces résultats.
1.2 Mécanismes pathologiques et moléculaires
Notre compréhension de la majorité des mécanismes pathologiques cellulaires provient des causes génétiques de la maladie. Des mutations dans plusieurs gènes différents peuvent contribuer aux mêmes mécanismes pathologiques. Parmi ces mécanismes, on peut inclure une toxicité induite par le glutamate, le stress oxydatif, une mauvaise régulation des ARNs, la formation d’agrégats intracytoplasmiques, la sécrétion de cytokines inflammatoires, la réduction du transport axonal et la réduction dans l’efficacité des mécanismes de clairance cellulaire. La pathogénèse de la SLA est certainement multifactorielle et constitue une interaction complexe entre tous ces mécanismes.
1.2.1 L’excitotoxicité induite par le glutamate
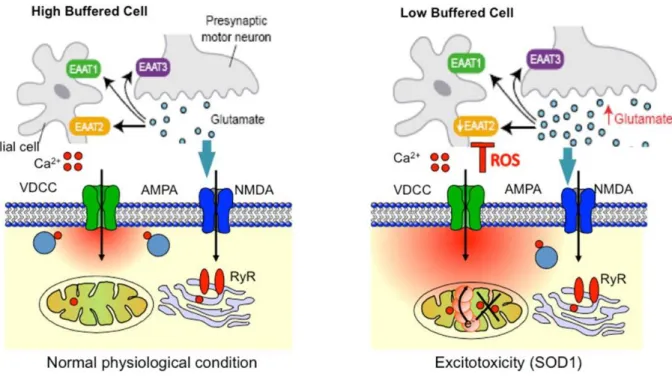
La toxicité induite par le glutamate est un mécanisme important tant dans la forme familiale que sporadique (64). Les évidences de son implication proviennent d’études animales et humaines. Une baisse de l’expression et de l’activité du transporteur excitatoire d’acide aminé 2 (EAAT-2 ou GLT1) a été observée dans le cortex moteur et la moelle épinière de patient SLA par mauvais épissage de son ARNm (65). Le transporteur EAAT-2 sert normalement à la réabsorption post-synaptique de glutamate dans les neurones et les cellules gliales afin de contrôler le niveau extra-cellulaire de glutamate (66). Son absence augmente ainsi le niveau de glutamate et créé une excitotoxicité. L’inhibition du transporteur EAAT-2 dans les souris SOD1 cause un mort des motoneurones (67) et accélère l’évolution de la maladie (68). D’un autre côté, la surexpression du transporteur par l’entremise d’antibiotiques de la classe des β-lactames semble protecteur chez des souris SOD1 (69) (Fig. 3).
13
Figure 3 L’excitotoxicité induite par le glutamate et la sensibilité des motoneurones aux influx de calcium (70)
Du côté post-synaptique, les récepteurs alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) sont impliqués dans la toxicité. Il est bien décrit que les motoneurones sont sensibles aux influx de calcium (71-73). La perméabilité au calcium des récepteurs AMPA est dépendante de la présence de la sous-unité GluR2 et son absence augmente l’influx de calcium (74). Une souris
double transgénique SOD1G93A; GluR2-/- a présenté une accélération importante de la perte de
motoneurones et une survie plus courte (75). Des évidences suggèrent que les motoneurones humains sont plus sensibles à l’excitotoxicité induite par le glutamate car ils possèdent moins de sous-unité GluR2 que les autres neurones (76). De plus, les motoneurones des patients atteints de la maladie ont une capacité de tampon du calcium réduit par rapport aux autres motoneurones (75). Ceci pourrait en partie expliquer la susceptibilité des motoneurones dans la SLA. Le riluzole agit sur ce mécanisme pathologique en bloquant la sécrétion de glutamate, en diminuant la cascade d’activation des récepteurs glutamatergique et en stabilisant la forme inactivée des canaux sodiques (77).
14 1.2.2 La pathogénèse associée à SOD1
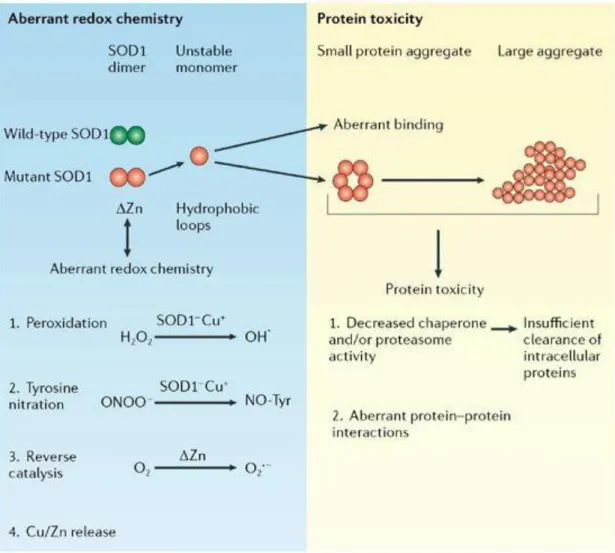
Le premier gène identifié dans la forme familiale de la maladie fut le gène Cu/Zn superoxyde dismutase 1 en 1993 (3). À ce jour, plus de 170 mutations dans le gène SOD1 ont été reconnues. Les mécanismes pathologiques associés à SOD1 demeurent les plus étudiés et les mieux décrits et la majorité de nos connaissances sur la SLA nous proviennent en fait d’études sur SOD1. SOD1 est une métalloprotéine catalysant la transformation des anions superoxydes en dioxygène et peroxyde d’hydrogène, éliminant ainsi les radicaux libres. La forme native de SOD1 est un homodimère où chacun des monomères lie un zinc et un cuivre pour créer une protéine mature et stable (78).
La première hypothèse reliant les mutations dans le gène SOD1 et la mort des motoneurones est une altération de son activité enzymatique. Une altération dans sa structure favoriserait la liaison de substrats anormaux avec le cuivre (79). SOD1 mutant peut par exemple lier des peroxynitrites ou des peroxydes d’hydrogènes et ainsi favoriser la nitration de ses résidus tyrosine et a production d’hydroxy-radicaux (80, 81) (Fig. 4). La protéine SOD1 mutée peut également catalyser des anions superoxydes par une baisse de la liaison au zinc (82). Les évidences qu’une mutation dans le gène SOD1 induirait une réaction oxydative sont controversées. Le dommage oxydatif a été démontré
dans les souris SOD1G93A et les patients sSLA (83, 84), mais n’a pas été observé dans les souris
15
Figure 4 Production d’hydroxy-radicaux par les formes mutantes de SOD1 (79)
Plusieurs mutations, à l’exception de celles ayant un impact sur la liaison au zinc ou au cuivre, n’ont aucun impact sur son activité dismutase (86-88). Ainsi, une autre hypothèse pour expliquer la toxicité de SOD1 consiste en la formation d’agrégats toxiques et insolubles de SOD1 causée par une instabilité conformationnelle de la protéine mutée. Ces agrégats sont trouvés dans les motoneurones et les astrocytes des patients avec la forme familiale et sporadiques (89). Les agrégats apparaissent au début des symptômes et augmentent avec la progression de la maladie (90, 91). Certaines mutations ont davantage tendance à former des agrégats en modifiant l’interface dimérique, en jouant sur les boucles électrostatiques ou sur la liaison aux métaux (92). Ces changements structurels induisent un mauvais repliement de la protéine (misfolded SOD1) (Fig.
16
protection des neurones malades, plusieurs évidences démontrent qu’il existe une distribution de cellules à cellules de la protéine mal-repliée. En effet, des neurones en culture exposés à des agrégats de SOD1 muté présentent une agrégation de SOD1 muté endogène persistant jusqu’à plus de 30 jours (93). Une exposition à des agrégats de SOD1 peut également induire une agrégation de la forme native de SOD1 (94). Il n’est pas encore clair quelle est l’implication in vivo de la distribution de cellules à cellules de SOD1. Cependant, l’injection de SOD1 mal-replié dans le nerf sciatique induit une progression de la pathologie de manière caudo-rostral (95).
Figure 5 Le mal-repliement de SOD1 natif ou muté entraîne son agrégation (96)
1.2.3 Les neurofilaments
Les neurofilaments (NF) font partie des filaments intermédiaires, l’une des trois classes de filaments composant le cytosquelette. Les neurofilaments peuvent être divisés en quatre sous-unités : lourd (heavy, H), moyen (medium, M), léger (light, L) et l’alpha-internexine qui est remplacé par la périphérine dans le système nerveux périphérique (97). Une accumulation anormale de neurofilament et de périphérine est observée dans les sphéroïdes et les inclusions hyalines des motoneurones dans la SLA (98). Des mutations dans le gène NFH sont reconnues dans un faible nombre de patients SLA et ces mutations peuvent mener à l’hyperphosphorylation des neurofilaments. L’un des premiers indicateurs de maladie dans les souris SOD1 est l’accumulation de neurofilaments dans les motoneurones (99-101). Les neurofilaments sont nécessaires aux transports à travers les axones et leur accumulation cytoplasmique entraîne des dérangements dans ce transport (102). Ceci peut donc entraîner des défauts métaboliques comme
17
l’absence de mitochondrie aux synapses (103) ou un dysfonctionnement du transport des granules de ribonucléoprotéines (97).
1.2.4 Le stress du réticulum endoplasmique
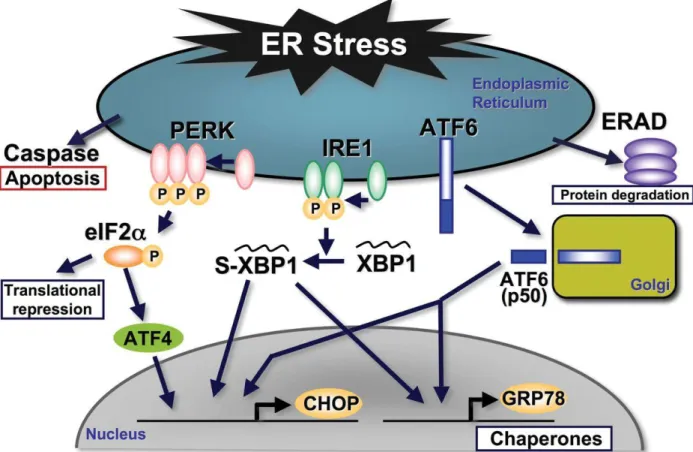
Le réticulum endoplasmique (RE) est le premier compartiment cellulaire où les protéines sont synthétisées et repliées. Plusieurs situations de stress peuvent entraîner une dysfonction de l’organelle et entraîner ce qu’on appelle le stress du RE (ER-stress) (104). Le stress du RE entraîne l’activation de la réponse de repliement des protéines (Unfolded response protein UPR) qui rétabli l’homéostasie du RE en améliorant les mécanismes de contrôles (105). Cependant, l’exposition chronique au stress du RE peut entraîner des dommages irréversibles et l’apoptose. L’UPR peut être activée par trois senseurs de stress : la PKR-like ER kinase (PERK), l’inositol-requiring transmembrane Kinase (IRE1) et l’activating transcription factor 6 (ATF6). L’activation de IRE1 entraîne la production du facteur de transcription X-Box-binding protein 1 (XBP1) qui contrôle l’expression de gènes impliqués dans le repliement et le contrôle des protéines du RE et de la dégradation du RE (ERAD) (106, 107). La phosphorylation de PERK induit la phosphorylation d’eukaryotic initiation factor 2 alpha (eIF2α) qui réduit le mal-repliement et induit l’activation du facteur ATF4. Ce dernier favorise l’autophagie et l’apoptose (105) (Fig. 6). En résumé, l’UPR intègre de l’information à propos de l’intensité du stress et favorise une adaptation au stress ou une mort cellulaire programmée. Les différents facteurs de l’UPR (PERK, ATF6, IRE1, eIF2α) sont activés dans la moelle épinière de patients SLA (108). Ainsi, plusieurs modèles animaux ont suggéré l’implication de stress du RE dans la pathogénèse SLA avec mutations dans les gènes FUS, TDP-43, SOD1 et ataxine-2 (109). De manière intéressante, deux médicaments inhibant le stress du RE, le salubrinal et le guanabenz, ont permis de diminuer la neurotoxicité dans un modèle de C. elegans et un modèle de zebrafish avec mutation TDP-43 (110).
18
Figure 6 Cascade d’évènements dans le stress du réticulum endoplasmique (111)
1.2.5 Les cellules gliales et la neuroinflammation
Depuis quelques années, les cellules gliales sont certainement reconnues comme des joueurs importants dans la SLA et dans la mort neuronale. Des études dans les souris SOD1 ont suggéré une implication des astrocytes et des microglies entourant les motoneurones dans le début et dans l’évolution de la maladie. En effet, une gliose importante est retrouvée dans les tissues de patients et de souris transgénique SOD1 et TDP-43. Les cellules gliales peuvent avoir une fonction protectrice ou neurotoxique (112). Ce concept d’implication des cellules gliales dans la mortalité neuronale est souvent référé à la toxicité cellulaire non-autonome.
Les astrocytes répondent à un stress environnant en adoptant un phénotype réactif et en développant des longs processus épais remplis de glial fibrillary acidic protein (GFAP). Les astrocytes provenant de cas sporadiques exposés à des neurones en culture cause une mort des
19
motoneurones par un mécanisme dépendant de SOD1 (113). Des évidences plus récentes ont démontré que des cellules souches pluripotentes induites (iPSCs), provenant de patients SOD1 et différenciés en astrocytes, peuvent induire une mort des motoneurones dans des souris non-transgénique (114). Au contraire, des iPSCs, différenciés en astrocytes provenant de personnes saines, semblent neuroprotectrice lorsque transférés à des souris SOD1 (115). Les astrocytes activés expriment une réduction de l’expression du transporteur EAAT-2, ce qui cause une excitotoxicité induite par le glutamate (cf. section 1.2.1). Les astrocytes induisent également une toxicité par la sécrétion de cytokines et des facteurs neurotoxiques tel que des chimiokines et l’IL-6 (11l’IL-6) (117). Ces cytokines pro-inflammatoires activent la voie de signalisation Bid et NF-κB qui participent à la mort neuronale (118). Cependant, une autre étude a démontré que l’activation du facteur NF-κB dans les astrocytes n’était pas reliée à la mortalité des neurones (119).
La microglie est considérée comme la première ligne de défense du SNC contre les infections. Les cellules microgliales peuvent échantillonner l’environnement, présenter des antigènes et possèdent des récepteurs CD14 et Toll-like receptor (TLR) impliqués dans la réponse immunitaire innée. Selon les cytokines environnantes, les cellules microgliales peuvent acquérir un phénotype neurotoxique ou neuroprotecteur. La présence des cytokines TGFβ, IL-4 et IL-10 favorisent un phénotype neuroprotecteur alors que les cytokines TNF-α, IL-1β et IL-6 favorisent un état neurotoxique (120). Les cellules microgliales activées développent une forme amiboïde avec élargissement de leur corps cellulaire. Les évidences suggèrent que les microglies ont probablement un rôle dichotomique selon le stade de la maladie. Une étude récente très intéressante a utilisé un système d’imagerie d’activation microgliale in vivo pour vérifier l’activité inflammatoire ou anti-inflammatoire en temps réel (121). Chez les souris SOD1, au début de la maladie, les microglies exprimaient un profil anti-inflammatoire avec haut niveau de sécrétion d’IL-10, une cytokine anti-inflammatoire. Il est intéressant de noter que les souris traitées avec un bloqueur des récepteurs IL-10 avaient un début de maladie beaucoup plus rapide, suggérant un rôle protecteur des microglies en début de maladie. Le rôle des microglies semble varier selon la présence ou l’absence d’une pathologie SOD1. En effet, la suppression de SOD1 dans des
microglies de souris (122) ou le transfert de microglies SOD1WT dans des souris SOD1G93A (123)
20
associée à TDP-43 n’est pas bien connu. Notre laboratoire a démontré que les cellules microgliales qui expriment davantage de TDP-43 produisent plus de cytokines pro-inflammatoires et de médiateurs neurotoxiques après stimulation avec LPS (124).
1.3 TAR DNA-binding protein 43 (TDP-43)
TAR DNA-binding protein 43 (TDP-43) est encodé par le gène TARDBP ou ALS10. TDP-43 a été identifié pour la première fois comme liant le motif d’ADN riche en pyrimidine TAR sur le virus de l’immunodéficience humaine type 1 (VIH) (125). Plusieurs études subséquentes ont suggéré que TDP-43 était principalement impliqué dans la régularisation des ARNm en jouant sur l’épissage, la translation, le transport et la dégradation (126-128). TDP-43 a été découvert comme étant présent dans les agrégats ubiquitinés dans la SLA et la DFT en 2006 (45, 129) et des mutations dans le gène ont été découvertes deux ans plus tard (130). La protéinopathie associée à TDP-43 est présente non seulement dans la SLA et la DFT, mais également dans la maladie d’Alzheimer (131), dans la maladie de Parkinson (132) et dans la maladie d’Huntington (133). La localisation cytoplasmique et l’agrégation de TDP-43 demeure, à ce jour, le principal élément diagnostic neuropathologique de la SLA. Les sections suivantes serviront à démontrer son rôle pathologique dans la maladie.
1.3.1 Métabolisme des ARNs
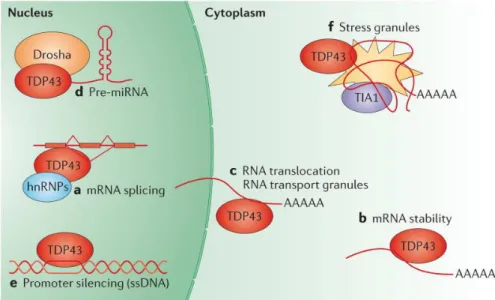
TDP-43 est un membre de la famille des ribonucléoprotéines hétérogènes liant l’ARN (hnRNPs) (134). Il est composé d’un domaine N-terminal (NTD), de deux motifs de reconnaissance d’ARN (RRM1-2) et d’un domaine C-terminal riche en glycine (CTD) (Fig. 7). Il est impliqué dans la stabilité des ARNm, dans leur transport vers le cytoplasme et dans le contrôle du niveau d’expression de certains ARNm en liant leur promoteur. De plus, il lie les domaines riches en UG à la fin des introns ou les régions 3’ non-traduit de plus de 6000 ARNs incluant son propre ARN afin de réguler l’épissage alternatif (135-140). Des évidences récentes suggèrent que TDP-43 a un rôle important dans la répression des exons cryptiques non-conservés (141). Ces exons, lorsqu’introduit dans l’ARNm, peuvent créer des codons stop prématurés ou des codons faux-sens. TDP-43 est également impliqué dans la régularisation des microARNs. Il se localise dans les fibres
21
périchromatines, là où les miRNA sont synthétisés, et lie le complexe de synthèse Drosha (142) (Fig. 8).
Figure 7 Représentation schématique des domaines de TDP-43 humain et de ses mutations (143)
Figure 8 Fonctions normales de TDP-43 dans la gestion des ARNs et la formation de granules de stress (142)
La délocalisation de TDP-43 du noyau vers le cytoplasme dans la SLA suggère certainement une perte des fonctions normales de TDP-43 à l’intérieur du noyau. Une mutation dans TDP-43 semble