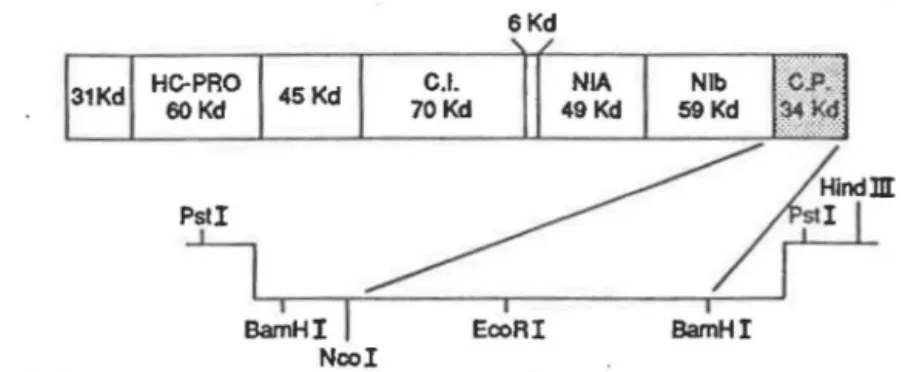
Expression de gènes du virus de la mosaïque du navet dans E. Coli
Texte intégral
Figure
Documents relatifs
Cette quatrième série d’essais de co-expression révélait aussi la présence d’un complexe formé entre les protéines Snu13p et Pih1p (Figure 64, piste 129) et entre les
A partir de l’indice de consommation (IC), du tx moyen de protéines consommables par l’homme dans les rations animales, et la teneur moyenne en protéines des produits animaux, il
Etude de l’organisation des gènes des protéines du lait au sein du noyau et attachement de ces gènes à la matrice nucléaire.. Sciences du
Cette hypothèse peut s’appliquer aux HAd40 et 41 qui comportent également deux fibres: une courte et une longue, la fibre longue peut interagir avec le CAR alors que le récepteur
Statistical analysis consisted of separate mixed-design analyses of variance (ANOVA) for the depen- dent variables (cumulative fixation duration and mean fixa- tion duration) and
The sec- ond part of the book, also in two chapters, ad- dresses Bentham's censorial jurisprudence: first, his considerations on legal reform and, second, his ideas on
The constant challenge of immigrant integration, especially those of a Muslim background manifests in a conflicting discourse of multiculturalism hesitating between
Dans les stades tardifs de l’infection virale où la protéine VP16 du MDV-1 est détectée par l’AcMo I13, l’absence de phosphorylation des protéines de tégument avant
