Étude des interactions protéiques entre les formes
d’épissage du gène UGT1A
Mémoire
Pierre Collin
Maîtrise en pharmacie avec mémoire
Maître ès sciences (M. Sc.)
Québec, Canada
Résumé
L’épissage alternatif en 3’ du gène UGT1A entraîne la production d’enzymes actives, les isoformes 1 (i1), et de protéines tronquées, les isoformes 2 (i2), qui ne possèdent pas de domaine transmembranaire (TMD) et d’activité de glucuronidation, mais plutôt des propriétés modulatrices sur l’activité enzymatique des i1 via des interactions protéiques. Nous croyons que les interactions i1-i2 impliquent plusieurs domaines d’interactions et qu’ils sont différents de ceux impliqués dans l’homo-oligomérisation des i1. Des expériences de co-immunoprécipitation démontrent que les isoformes i1 dépourvues du signal peptide +/- le TMD empêchent l’homo-oligomérisation sans affecter l’interaction i1-i2. De plus, la présence de complexes de hauts poids moléculaires observée par immunobuvardages en conditions non-réductrices démontre l’implication potentielle de ponts disulfures dans la formation des complexes i1-i2, et ce via plusieurs résidus cystéines. En somme, les résultats obtenus supportent que l’interaction i1-i2 implique plusieurs domaines protéiques et qu’ils diffèrent de ceux impliqués dans les complexes i1-i1.
Abstract
Alternative splicing of UDP-glucuronosyltranferase UGT1A gene results in the production of enzyme, isoforms i1 and i2. Unlike the active i1 proteins, i2 are truncated proteins which lack the transmembrane domain and glucuronic acid transferase activity, but have an inhibitory effect on UGT1A activity likely through the formation of hetero-oligomers with i1. We believe that i1-i2 interaction involves binding of more than one domain. Our results showed that i1, in the presence or absence of the transmembrane domain, without the signal peptide did not self-interact but instead interacted with i2. In addition, high molecular weight complexes were observed by immunoblotting under non-reducing conditions. It demonstrates the involvement of disulfide bonds in the formation of i1-i2 complexes. In summary, these results support that i1-i2 interactions involve multiple protein domains and they differ from those involved in homo-oligomerization of i1.
Table des matières
Résumé --- iii
Abstract --- v
Table des matières --- vii
Liste des tableaux --- ix
Liste des figures --- xi
Liste des abréviations --- xiii
Remerciements --- xvii
Avant-Propos --- xix
Introduction --- 1
1. Le processus d’épissage alternatif --- 3
1.1. Mécanisme de l’épissage alternatif --- 5
1.2. Régulation de l’épissage alternatif --- 8
2. La glucuronidation --- 9
2.1. Les UDP-glucuronosyltransférases --- 13
2.1.1. Nomenclature et classification des différentes familles UGT --- 13
2.2.1. La famille UGT1A --- 15
3. Oligomérisation des UGT --- 20
3.1. Interaction protéine-protéine --- 24
Problématique, hypothèse et objectifs --- 29
Résultats --- 31
Résumé --- 33
Discussion --- 61
Conclusion --- 66
Liste des tableaux
Tableau 1. Les domaines protéiques des UGT et leurs implications dans
Liste des figures
Figure 1. Processus d'épissage alternatif ... 4
Figure 2. Épissage de type U2 du pré-ARNm par le spliceosome ... 7
Figure 3. Évènements d'épissage alternatif ... 8
Figure 4. Principales enzymes responsables du métabolisme de phase II ... 11
Figure 5. Schématisation de la réaction de glucuronidation ... 12
Figure 6. Arbre phylogénétique des principaux membres de la superfamille UGT .... 14
Figure 7. Évènements d'épissage en 3’ au locus UGT1A ... 16
Figure 8. Structures primaires et tertiaires des isoformes i1 et i2 ... 18
Liste des abréviations
ABC « ATP-binding cassettes » ADN Acide désoxyribonucléique ARN Acide ribonucléique
ARNm ARN messager
BS Site de branchement co-IP Co-immunoprécipitation COOH Groupement carboxyl
CPT-11 7-éthyl-10-[4-(1-piperidino)-1-piperidino]-carbonyloxy camptothécine CYP P450 Cytochrome P450
EA Épissage alternatif
ESE « Exonic splicing enhancer » ou activateur exonique de l’épissage ESS « Exonic splicing silencer » ou inhibiteur exonique de l’épissage
FRET « Fluorescence resonance energy transfert » ou transfert d’énergie par résonnance de fluorescence
GST Glutathione S-transférase hnRNP « heterogenous nuclear RNP » i1 et i2
ISE Isoforme 1 et isoforme 2 « Intronic splicing enhancer » ou activateur intronique de l’épissage ISS « Intronic splicing silencer » ou inhibiteur intronique de l’épissage
kDa KiloDalton MRA Microrégion A NAT N-acétyltransférase NH2 nt Groupement amine Nucléotide
OATP « organic anion transporting polypeptide »
OH Groupement hydroxyl
PCR « Polymerase chain reaction » ou réaction en chaîne de la polymérase
PPT Polypyrimidine
Pré-ARNm ARN pré-messager
PS Peptide signal
SDS-PAGE « Sodium dodecyl sulfate polyacrylamide gel electrophoresis » ou électrophorèse sur gel de polyacrylamide en présence de dodésylsulfate de sodium
SH Groupement thiol
siARN « small interfering RNA » ou petit ARN interférent SN-38 7-éthyl-10-hydroxycamptothécine
SNP « Single Nucleotide Polymorphism » ou polymorphisme d’un seul nucléotide
snRNP « Small ribonuclear protein » ou petite ribonucléoprotéine nucléaire SR « serine/arginine-rich protein» ou protéine riche en sérine/arginine SULT Sulfotransférases
TMD « Transmembrane domain » ou domaine transmembranaires UDP Uridine diphosphate
UDPGA « Uridine diphosphate glucuronic acid » ou acide uridine diphospho glucuronique
Imagination is more important than knowledge.-
Remerciements
Je tiens tout d’abord à remercier les différents évaluateurs de mon mémoire, le Dr Frédéric Picard et le Dr Éric Biron. Je tiens également à remercier ma directrice de recherche, Chantal Guillemette. Merci de m’avoir accueillie dans ton laboratoire en 2010 pour mon premier stage de recherche dans le domaine de la santé, j’y aie découvert une passion et une équipe incroyable. Je tiens aussi à te remercier de ton dévouement pour le laboratoire et de m’avoir permis de participer à un congrès international à Boston et ce même si je ne présentais pas de résultats. Ce fût une belle expérience et surtout mon premier voyage aux États-Unis. Je voudrais aussi remercier Éric Lévesque, même si je n’ai pas eu l’occasion de travailler avec toi, j’admire ta passion et surtout ton enthousiasme.
Merci aux différents membres du laboratoire que j’ai pu côtoyer au courant des trois dernières années. Premièrement, je tiens à remercier Mario. Merci de toujours avoir eux quelques minutes pour répondre à mes questions. Ton dévouement pour les étudiants est admirable. Nos différentes discussions sur la science m’ont permis de développer mon esprit tant critique que scientifique. Merci pour tes milles et un conseil pour les différentes expériences, on aimerait tous avoir un Mario dans notre laboratoire. J’espère que l’on va avoir l’occasion d’aller prendre un petit Varlet ensemble! Merci à Lyne pour ta gentillesse et ta bonne humeur. J’aimerais aussi te remercier pour ton aide dans le labo et tes conseils. J’ai bien aimé pouvoir te côtoyer depuis ton retour de congé de maternité et n’oublie pas! Avril n’est pas un beau mois pour aller dans le sud! Finalement, je te souhaite que tes filles finissent par se lever tard un jour…! Merci à Mélanie pour ton aide durant ma maîtrise et ton rire contagieux. Je tiens aussi à te remercier pour le congrès à Boston, cela a été un très belle expérience, bonne chance dans ta future carrière! Christine, je tiens à te remercier de m’avoir sauvé la vie avec Vincent durant cette fameuse soirée de la clé. Cette histoire va rester mon anecdote préféré. Sans oublier la soirée de la Saint-Jean! En espérant que de telles situations ne se reproduisent plus! Je te souhaite beaucoup de bonheur à Montréal et j’espère que l’on va se revoir à l’occasion. Merci à Étienne, mon maitre de stage! Je tiens à te remercier pour le temps que tu as consacré pour mes différentes questions et surtout d’avoir toujours pris quelques minutes pour me répondre et ce même si tu étais occupé! J’ai beaucoup appris et apprécié de travailler avec toi, j’espère ravoir cette occasion. J’admire ta
passion pour ton travail, tu vas faire un excellent chercheur et un excellent professeur, j’en suis sure. Je vais garder de très bons souvenirs de nos soirées bien arrosés. Je ne croyais pas être capable de trouver une personne avec un foie aussi tenace, enfin! Je te souhaite beaucoup de succès! Vincent, merci pour ton aide, ta bonne humeur, ton enthousiasme et ta disponibilité. J’ai bien aimé avoir eu la chance de travailler avec toi et j’espère un jour ravoir cette chance. J’oubliais, je suis sure que tu vas faire un papa incroyable!!! J’espère que tu vas venir me visiter à Montréal et qu’on va ravoir la chance d’aller jouer une petite partie de tennis! Finalement, j’aimerais te remercier toi, Étienne et Mario, pour premièrement rire de mes jokes plates, mais aussi pour les différentes discussions scientifiques, cela à été très formateur et cela ma aussi montré que j’aimais la science! Merci Johanie pour ton aide, surtout dans les derniers mois pour la rédaction de mon mémoire, sans toi, j’aurai perdu des heures. Bonne chance pour ta future carrière! Isabelle L., je n’ai pas eu l’occasion de travailler avec toi dans le labo, mais merci pour ta bonne humeur et ta gentillesse, bonne chance pour ton post-doc! Isabelle G., merci pour ta bonne humeur, bonne chance dans ta futur carrière. Sylvia, merci pour ton aide en anglais, surtout pour mes demandes de bourse! Bonne chance pour ton doctorat! Merci aussi à nos trois Français! Je n’ai eu que quelques mois pour vous connaitre, mais c’est assez pour vous dire que vous êtes des gens incroyables, bonne chance et à la prochaine! Merci à Patrick et Véronique de toujours avoir votre porte ouverte pour nos questions. Merci également à toute l’équipe de Olivier Barbier, vous êtes tous incroyable. J’espère avoir la chance de travailler avec des collaborateurs comme vous dans le futur.
Finalement, je voudrais remercier Lucie. Merci d’avoir été patiente durant ces deux années à distance, on va enfin être ensemble! Merci de m’avoir écouté, supporté et de toujours être la pour moi. Tu es ce qui m’est arrivé le mieux dans ma vie, tu es ma source de bonheur. Merci aussi à ma mère, sans qui je ne serais pas rendu là aujourd’hui et ce même si nous avons passé des situations et des moments difficiles. C’est en parti grâce à toi que je suis rendu là et que j’ai pu me rendre aussi loin. Merci.
Avant-Propos
Le présent mémoire intitulé « Étude des interactions protéiques entre les formes d’épissage du gène UGT1A », présenté à la faculté des études supérieures de l’Université Laval pour l’obtention du grade de Maître ès Sciences (M.Sc) est rédigé sous la forme d’insertion d’un article.
L’article présenté dans ce mémoire intitulé « Protein-protein interactions between the bilirubin-conjugating UDP-glucuronosyltransferase UGT1A1 and its shorter isoform 2 regulatory partner derived from alternative splicing » dont je suis second auteur a été publié dans la revue « Biochemical Journal » en novembre 2012. J’ai participé à ce projet en effectuant des expérimentations sous la supervision de l’étudiante au doctorat Mélanie Rouleau, d’abord en participant au clonage de protéines tronquées. Ces constructions étaient essentielles afin de confirmer l’implication de plusieurs domaines d’interaction entre les isoformes 1 et 2 des UGT1A et l’implication possible des ponts disulfures dans ces interactions. Nous avons choisi l’isoenzyme UGT1A1, car cette UGT est l’une des plus étudiées et est impliquée notamment dans l’homéostasie de la bilirubine. Pour ce faire, nous avons créé des vecteurs d’expression des protéines tronquées à l’aide de réactions en chaîne par polymérase (PCR) et de « Overlap extension PCR » (OE-PCR). Par la suite, j’ai participé au clonage de ces vecteurs ainsi qu’à l’analyse de séquençage. Toujours sous la supervision de Mélanie Rouleau, j’ai procédé à la co-transfection transitoire des vecteurs exprimant les protéines tronquées dans des cellules humaines dépourvues d’UGT (HEK293). Nous avons finalement effectué des co-immunoprécipitations (co-IP) en utilisant les lysats des différentes cellules transfectées et ensuite procédé à des immunobuvardages de type Western pour confirmer l’expression des protéines UGT1A1.
J’ai donc participé à l’exécution d’expériences dans cette étude, notamment à la construction des vecteurs, à la transfection, aux co-immunoprécipitations et aux immunobuvardages. Avec les Drs Guillemette et Bellemare, l’auteur principal, Mélanie Rouleau, a planifié et supervisé les expériences de laboratoire, elle a participé à l’exécution des clonages et des co-IP, a effectué les gels non-réducteurs ainsi que les expériences de localisation protéique. Elle a également effectué l’analyse des résultats et la rédaction de
l’ébauche du manuscrit. Ma directrice de recherche, Chantal Guillemette, a conceptualisé l’étude, planifié les expériences avec l’aide de Mélanie Rouleau et de Judith Bellemare ainsi que l’analyse des résultats, la rédaction et la révision du manuscrit. Mario Harvey, assistant de recherche, a participé à l’interprétation des résultats. L’article qui est inséré dans ce mémoire est identique à la version qui a été publiée. Ma participation dans la réalisation des expériences contenues dans cet article scientifique est estimée à 35 %.
Ce mémoire comprend une introduction où je présente une revue de la littérature sur le sujet de l’article ainsi que l’hypothèse et les objectifs de la recherche. Suivent ensuite le résumé en français de l’article et le manuscrit original rédigé en anglais. Ce dernier contient les sections suivantes : résumé, introduction, matériel et méthodes, résultats, discussion, bibliographie, tableaux et figures. Cette section est suivie d’une discussion générale portant sur l’ensemble des résultats et propose des perspectives pour la suite du projet, et une conclusion. Finalement, une liste des références citées dans l’introduction et dans la discussion termine ce mémoire.
Introduction
Les organismes vivants sont constamment exposés à une variété de molécules exogènes provenant principalement de l’environnement et de l’alimentation et doivent être en mesure de les éliminer. Lors de la prise de médicaments, l’organisme doit aussi être capable de prendre en charge ces molécules afin de les métaboliser pour éviter leurs effets toxiques. Toutefois, les voies métaboliques liées à la clairance des drogues sont caractérisées par une grande variabilité principalement liée à l’âge, au sexe, à l’origine ethnique et à l’environnement (Ma and Lu 2011). De plus, la génétique peut fortement influencer l’élimination de molécules. En effet, plusieurs évidences dans la littérature indiquent que la réponse suite à la prise d’un médicament peut varier selon la composition génomique d’un individu (Lerer and Segman 2006; Eby 2012; Levesque et al. 2013; Stockmann et al. 2013). Par exemple, la présence du variant UGT1A1*28 du gène UGT1A1 est associée à une diminution du taux d’élimination du métabolite actif de l’Irinotecan (CPT-11), le SN-38. Cet agent anticancéreux est utilisé en première instance dans le traitement du cancer du côlon (Guillemette 2003; Chen et al. 2012). Toutefois, une accumulation de ce dernier peut engendrer des effets secondaires graves (Gupta et al. 1994; Ando et al. 2000; Iyer et al. 2002). À cause de l’étroite fenêtre thérapeutique de plusieurs médicaments, cette variabilité ne doit pas être prise à la légère, car elle peut occasionner des effets secondaires dus à une forte accumulation du médicament en circulation ou une non-réponse pour cause de sous-dosage (Spear et al. 2001; Lesko and Schmidt 2012). Dans ce contexte, il est important d’étudier les différentes voies métaboliques et les polymorphismes génétiques pouvant affecter des gènes impliqués dans ces processus.
Une des principales voies du métabolisme des médicaments est la glucuronidation, une réaction catalytique rendue possible grâce à des enzymes appelées UDP-glucuronosyltransférases (UGT). L’action des UGT représente plus de 35 % du métabolisme de phase II des médicaments, ce qui démontre l’importance de ces enzymes dans la santé humaine (Evans and Relling 1999). Les différentes protéines de la superfamille des UGT se séparent principalement en deux familles distinctes (UGT1 et 2), basées principalement sur leur homologie de séquences (Mackenzie et al. 2005; Guillemette
et al. 2010). De nombreux polymorphismes de ces gènes sont connus pour influencer la capacité de glucuronidation in vivo et in vitro de certaines molécules: le cas le plus connu et le plus étudié est un polymorphisme dans la boîte TATA du promoteur du gène UGT1A1 appelé UGT1A1*28 (Bosma et al. 1995; Monaghan et al. 1996). Ce variant possède sept répétitions TA au lieu de six comme chez le sauvage et provoque une diminution de plus de 30 % des niveaux d’expression du gène UGT1A1, soit une diminution de 58 % en protéine lorsque l’individu est homozygote (Guillemette 2003; Girard et al. 2005; Strassburg et al. 2008). Ce variant est également connu pour être associé avec un syndrome léger d’hyperbilirubinémie nommé le syndrome de Gilbert (Bosma et al. 1995; Bancroft et al. 1998). Toutefois, les polymorphismes n’expliquent pas à eux seuls la variabilité.
Notre laboratoire a mis en évidence l’implication de nouveaux variants d’épissage au locus UGT1A qui pourraient être impliqués dans cette variabilité. En effet, le laboratoire a identifié un nouvel exon 5 alternatif (5b) entre l’exon 4 et l’exon 5 classique (5a), qui se retrouve dans une nouvelle classe de transcrits UGT1A, soit les UGT1A_v2/v3 (Girard et al. 2007; Levesque et al. 2007). Ces transcrits mènent à la synthèse de formes tronquées, les isoformes i2 (i2), alors que les enzymes classiques (isoformes 1 (i1)) dérivent du transcrit UGT1A_v1 (exon 5a). Des études plus approfondies des i2 ont indiqué que ces isoformes ne possèdent pas d’activité enzymatique, mais qu’elles agissent plutôt comme des modulateurs négatifs de l’activité de glucuronidation (Levesque et al. 2007; Bellemare et al. 2010a; Bellemare et al. 2010b; Bellemare et al. 2010c). Plusieurs expériences ont permis de réléver que cette inhibition de la glucuronidation est en partie expliquée par l’oligomérisation des protéines; toutefois, il est à noter que le(s) domaine(s) d’interaction n’a(ont) pas encore été identifié(s) (Bellemare et al. 2010b).
Dans ce contexte, l’objectif principal de nos travaux était de déterminer si les domaines d’interactions connus pour être impliqués dans l'oligomérisation des UGT participent à l'interaction entre les formes d’épissage i1-i2. Le second objectif était de déterminer s’il y a formation de ponts disulfures entre les isoformes i1 et i2 suggérant des liaisons covalentes entre ces protéines ainsi qu’une implication individuelle de certains résidus cystéines dans ces interactions. Pour ce faire, nous avons développé des vecteurs
d’expression d’UGT1A1 contenant des délétions de domaines précis et des mutations de cystéines. Des expériences de transfection de ces vecteurs dans une lignée cellulaire n’exprimant pas les UGT (les HEK293), des électrophorèses sur gel de polyacrylamide en présence de dodécylsulfate de sodium (SDS-PAGE) en condition non réductrice et de co-immunoprécipitations (co-IP) ont ensuite permis de répondre à ces interrogations.
1. Le processus d’épissage alternatif
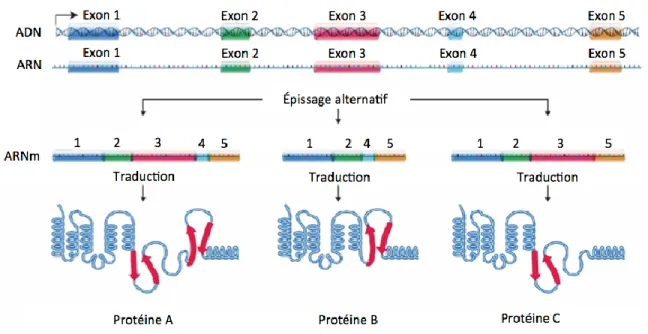
L’épissage alternatif de l’ARN pré-messager (pré-ARNm) a été initialement décrit il y a une trentaine d’années lorsqu’il a été démontré que des immunoglobulines μ différentes sont codées par un seul gène (Alt et al. 1980; Early et al. 1980). Par la suite, une multitude de cas furent découverts comme l’encodage de deux hormones par le gène « calcitonine/calcitonine-related polypeptide » dans lequel les différents transcrits sont exprimés de façon tissus-spécifique (Rosenfeld et al. 1982). De façon générale, la conséquence de l’épissage alternatif est l’inclusion ou l’exclusion de séquences exoniques; en d’autres mots, l’épissage alternatif permet un agencement alternatif des exons que l’on retrouve sur les pré-ARNm, ce qui permet la formation de différents cadres de lecture. Ce processus permet donc d’obtenir plusieurs ARN messagers (ARNm) matures à l’aide d’un seul gène, augmentant ainsi de façon considérable le nombre de protéines (Figure 1) (Adams 2008).
Figure 1. Processus d'épissage alternatif
L’épissage alternatif consiste à diversifier l’agencement entre les différents exons d’un gène, ce qui permet d’augmenter la diversité protéique en générant plusieurs protéines avec de nouvelles fonctions à partir d’un seul et même gène. Figure adaptée de Adams 2008.
Alors que le processus d’épissage alternatif (EA) chez l’Homme était méconnu, le nombre total de gènes dans le génome humain avait été estimé à 100 000, ce qui devait ainsi générer 100 000 protéines. Toutefois, le séquençage du génome humain dans les années 2000 a permis de démontrer qu’il existe plutôt environ 20 000 gènes (Zahra Abdellah 2004). La découverte de l’EA a permis d’expliquer comment il est possible d’obtenir cette grande quantité de protéines avec un nombre plus restreint de gènes. De plus, des expériences de séquençage à haut débit non-spécifiques ont permis de démontrer que plus de 95 % des ARNm sont issus de l’EA (Pan et al. 2008; Wang et al. 2008). Chez d’autres espèces, ce processus peut avoir une place encore plus grande dans la diversité de l’expression : un exemple spectaculaire est le gène Dscam de la Drosophila melanogaster qui peut générer 38 016 ARNm différents (Schmucker et al. 2000). Toutefois, il est important de noter qu’il existe aussi d’autres processus que l’épissage alternatif qui contribuent à la diversité des protéines comme l’utilisation de promoteurs alternatifs et la polyadénylation alternative (Ritter et al. 1992; Gong et al. 2001; Lutz and Moreira 2011; Moreira 2011; Pal et al. 2012).
1.1. Mécanisme de l’épissage alternatif
L’excision des introns et la jonction des exons sont rendues possible grâce à de courtes séquences hautement conservées présentes chez les pré-ARNm permettant de définir les limites physiques d’un intron, soit des sites d’épissage en 5’ et en 3’ de la jonction de l’intron et un site de branchement (BS) en 3’ (YNYURAY) (Reed 2000). Le site BS est généralement localisé à 18-40 nucléotides (nt) en aval du site d’épissage en 3’, et on retrouve une séquence de polypyrimidine (PPT) (Y10-12) entre le site BS et le 3’
(Ruskin and Green 1985; Reed 2000). Les introns du pré-ARNm sont retirés à l’aide de deux réactions de transestérification (Moore and Sharp 1993). La première réaction implique une attaque nucléophile d’un groupement 2’-OH d’un arginine du site BS sur le site d’épissage en 5’ de la jonction de l’intron. Le résultat de cette attaque permet la ligation du site d’épissage en 5’ avec le BS formant ainsi une structure en forme de lasso. Ensuite, une deuxième réaction de transestérification est rendue possible par l’attaque d’un groupement 3’-OH de l’exon en 5’ sur le site d’épissage en 5’ amenant ainsi la ligation des deux exons avoisinant l’intron. La structure en lasso produite par l’élimination de l’intron est par la suite dégradée par la cellule (Moore and Sharp 1993).
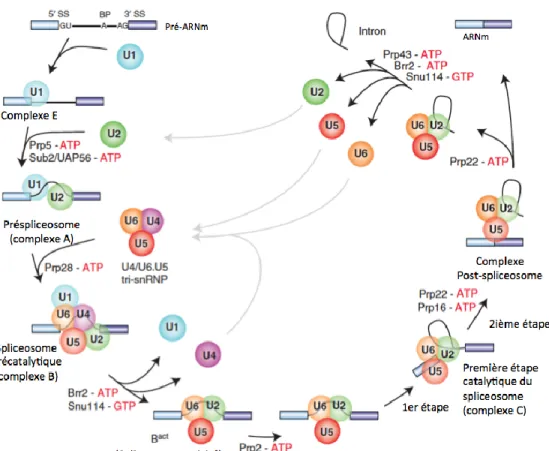
Pour permettre de telles réactions chimiques, la cellule doit faire appel à des facteurs agissant en trans qui interagissent avec le pré-ARNm pour former un complexe composé de particules ribonucléiques, le spliceosome (Kramer 1996). Ce dernier est composé de petites ribonucléoprotéines nucléaires, les snRNP, (U1, U2, U5 ET U4/U6) et de plusieurs non-snRNP. Il est à noter que l’assemblage de ces particules et d’autres facteurs d’épissage en spliceosome se déroule dans un ordre précis, et qu’il existe deux mécanismes différents dépendamment de la longueur de l’intron (Patel and Steitz 2003). Tout d’abord, lorsqu’un intron n’excède pas 200-250 nt, l’assemblage du spliceosome s’effectue directement sur l’intron et le processus est appelé « épissage de type U2 » (Figure 2) (Patel and Steitz 2003; Fox-Walsh et al. 2005; Wahl et al. 2009; Warzecha and Carstens 2012). Toutefois, la plupart des pré-ARNm de l’homme contiennent des introns variant de quelques centaines jusqu’à des milliers de nucléotides, ce qui est fortement
différent des exons qui eux ont une longueur moyenne de 120 nt (Deutsch and Long 1999; Ast 2004). Les séquences des sites d’épissage en 5’ et en 3’ et celles des points de ramification étant dégénérées, il peut y avoir de ces séquences qui apparaissent dans de longs introns. Dépendant de l’avidité des protéines en trans pour ces séquences, celles-ci peuvent mener à la présence d’exons ectopiques pouvant compromettre la traduction de la protéine active. Pour éviter ce problème, la cellule a développé une stratégie pour identifier efficacement les exons sans provoquer d’erreur : la définition de l’exon (Berget 1995). Ce processus consiste principalement en la formation de complexes d’épissage au sein même d’un exon et non pas uniquement à ses extrémités 5’ et 3'. Tout comme dans le processus d’épissage standard, le U1 snRNP s’accroche en aval de l’exon et facilite l’association de U2AF avec le PPT en amont, ce qui amène le recrutement de U2 snRNP au BS en amont de l’exon. La définition de l’exon est cependant rendue possible grâce à des séquences appelées amplificateurs d’épissage exoniques (ESE) qui recrutent des protéines riches en sérine et en arginine (protéines SR) permettant l’établissement d’un réseau d’interaction protéine-protéine dans l’exon, ce qui stabilise le complexe de définition décrit plus tôt (Hoffman and Grabowski 1992; Berget 1995; Reed 2000). Les protéines SR sont donc nécessaires à l’épissage constitutif, mais elles sont aussi obligatoires pour l’épissage alternatif.
Figure 2. Épissage de type U2 du pré-ARNm par le spliceosome
Étapes s’assemblage du spliceosome et des réactions de transestérification. Figure adaptée de Warzecha et Carstens 2012.
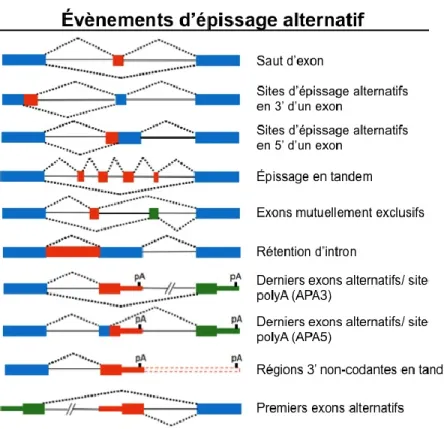
Le processus d’épissage alternatif se résume par une utilisation différentielle de sites d’épissage permettant l’utilisation d’une ou plusieurs réactions d’épissage. Ces réactions permettent dix évènements d’épissage différents (Figure 3) (Warzecha and Carstens 2012). Par exemple, deux types d’évènements d’épissage sont connus pour le gène UGT1A: premiers exons alternatifs et derniers exons alternatifs en 3’ du gène.
Figure 3. Évènements d'épissage alternatif Figure adaptée de Warzecha et al. 2012.
1.2. Régulation de l’épissage alternatif
Tout ce processus est contrôlé par des régulateurs en cis et en trans. Les plus connus en trans sont les protéines SR et les ribonucléoprotéines hnRNP (« heterogenous nuclear RNP »), et pour ce qui est des éléments en cis, on retrouve des amplificateurs d’épissage exoniques (ESE) et des amplificateurs d’épissage introniques (ISE) ou des régulateurs négatifs (ESS et ISS) (Smith and Valcarcel 2000; Wang and Burge 2008). Ces séquences sont, la plupart du temps, très courtes et sont liées par des protéines qui stimuleront ou inhiberont l’assemblage du spliceosome à différents sites d’épissage (Wang and Burge 2008). Les protéines SR sont des peptides qui s’accrochent à des séquences exoniques (ESE) activant par la suite le processus via le recrutement du spliceosome. Dans le cas des protéines hnRNP, celles-ci se lient aussi à l’ARNm, mais elles peuvent à la fois s’accrocher à des séquences introniques (ISS) ou exoniques (ESS) afin d’empêcher la liaison du spliceosome, ce qui inhibe l’épissage alternatif.
Étant donné que la transcription et l’épissage (constitutive et alternative) ne sont pas des évènements séquentiels mais, bien concomitants, de plus en plus d’études montrent que ces éléments peuvent s’influencer mutuellement (Corden and Patturajan 1997; Cramer et al. 1999; de la Mata et al. 2003; Ip et al. 2011). En effet, il a été démontré que l’ARN polymérase II peut contrôler la régulation de l’épissage alternatif selon sa vitesse d’élongation (de la Mata et al. 2003; Ip et al. 2011). Le recrutement de co-facteurs via des facteurs de transcription spécifiques au promoteur peut aussi influencer l’inclusion d’un exon : par exemple, le co-facteur PGC1, qui est connu à la fois pour co-activer des facteurs de transcription comme PPARα et NRF-1 ainsi que pour recruter des modificateurs de la chromatine comme CBP/300, a été identifié comme étant un modulateur de l’épissage alternatif (Wu et al. 1999; Monsalve et al. 2000; Vega et al. 2000). Cette protéine contient deux régions riches en sérine/arginine, ce qui est aussi une caractéristique des protéines SR (Shamoo et al. 1995). Le modèle proposé stipule que PGC-1 agirait en compétition avec les protéines SR, c’est-à-dire que lorsque PGC-1 est présent, les protéines SR ne peuvent plus s’attacher à un exon pour permettre son inclusion (Monsalve et al. 2000). D’autres hypothèses ont aussi été proposées quant à la régulation de l’épissage alternatif, par exemple, l’implication de l’épigénétique et du protéasome (Suh et al. 2010; Shukla et al. 2011). Des résultats récents laissent donc entrevoir l’importance du couplage transcription/épissage dans le contrôle de l’expression, mais cette régulation n’est pas complètement connue et il reste encore beaucoup de questions auxquelles répondre.
2. La glucuronidation
Pour se protéger, l’organisme possède une défense de première ligne contre les molécules hydrophiles : la membrane lipidique (van Meer et al. 2008). En effet, cette membrane agit comme une barrière contre les composés solubles. Grâce à elle, ces molécules ne peuvent pas pénétrer dans la cellule. La seule voie d’entrée pour celles-ci est d’emprunter des canaux protéiques ou des transporteurs. Toutefois, ces protéines possèdent une grande spécificité pour leurs substrats afin d’éviter une telle situation. Malheureusement, il n’en est pas de même pour les molécules hydrophobes. Grâce à leur hydrophobicité, il leur est plus facile de traverser la membrane lipidique et de s’accumuler
dans la cellule, entrainant un effet toxique. C’est donc pourquoi l’organisme s’est pourvu d’un mécanisme permettant l’élimination rapide de ces molécules : le métabolisme (Nebert 2006).
Lors du métabolisme, on retrouve trois phases de bio-transformation : l’oxydation (phase I), la conjugaison (phase II) et l’élimination (phase III) (Croom 2012). Les enzymes de phase I ont une activité de fonctionnalisation, c’est-à-dire qu’elles catalysent des réactions d’hydrolyse, de réduction et d’oxydation. Le but principal de cette phase est d’introduire un groupement polaire pour rendre les molécules plus solubles et/ou pour faciliter leur conjugaison (Nebert 2006; Chen et al. 2012). Les enzymes les plus connues pour ce groupe sont les cytochromes P450 (Nebert 2006; Zhou et al. 2009; Lee and Kim 2011). Chez l’humain, on retrouve plus de 57 gènes permettant le codage de ces enzymes qui peuvent à la fois métaboliser des molécules endogènes et exogènes comme les stéroïdes, la clozapine et le bortezomibe (Zhou et al. 2009; Lee and Kim 2011). La seconde phase est l’étape de conjugaison d’un xénobiotique ou d’un métabolite provenant de la phase I. Cette étape est catalysée par des enzymes qui permettent des réactions de transfert (Guillemette 2003; Chen et al. 2012; Naik et al. 2013). On retrouve plusieurs enzymes pouvant catalyser cette phase, notamment les uridines 5’-diphospho-glucuronosyltransférases (UGT), les glutathiones S-transférases (GST), les N-acétyltransférases (NAT) et les sulfotransférases (SULT) (Evans and Relling 1999; Guillemette 2003). La réaction qui découle de ces enzymes permet d’augmenter la polarité des molécules autant exogènes qu’endogènes afin de permettre leur élimination via la bile ou l’urine (Dutton 1980). Pour ce faire, lorsque les molécules sont conjuguées, elles doivent être transportées hors de la cellule à l’aide de transporteurs (phase III) (Chen et al. 2012; Naik et al. 2013). L’une des familles de transporteurs les plus connues est celle des « ATP-binding cassettes » (ABC). Ces protéines sont connues pour exporter contre le gradient un grand nombre de molécules hors de la cellule comme le cholestérol, les acides gras et les stéroïdes (Falasca and Linton 2012). Le « organic anion transporting polypeptide » (OATP) est aussi un transporteur bien connu. Il permet de transporter des molécules telles que des antibiotiques, des stéroïdes conjugués, des hormones et de la bilirubine (Konig et al. 2006).
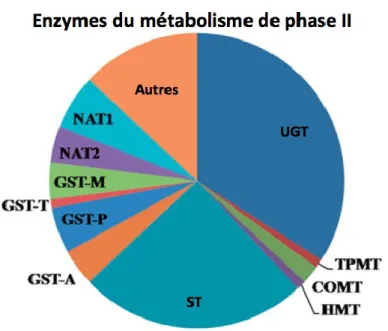
Figure 4. Principales enzymes responsables du métabolisme de phase II
Énumération des enzymes impliquées dans le métabolisme de phase II où chaque segment représente leur fréquence d’utilisation. Les UGT sont celles les plus impliquées suivies par les ST (SULT). Figure adaptée de Tripathi et al. 2013.
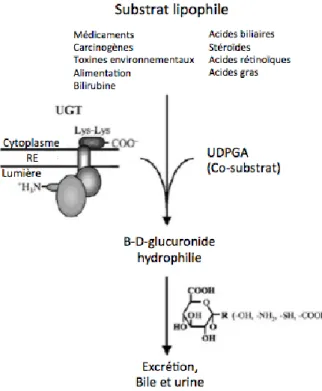
La glucuronidation est l’une des réactions les plus fréquentes dans le métabolisme de phase II (Figure 4) (Tripathi et al. 2013). Elle est impliquée dans environ 35 % du métabolisme des médicaments (Evans and Relling 1999; Kaivosaari et al. 2011). Elle consiste à ajouter un groupement acide glucuronique provenant d’un co-substrat, l’acide uridine diphosphate glucuronique (UDPGA), à un groupement fonctionnel de la molécule comme un hydroxyle (-OH), un carboxyle (-COOH), un sulfure (-SH) ou un amine (-NH2),
ce qui entraine une augmentation de la polarité de cette dernière l’amenant ainsi à son élimination (Figure 5) (Tukey and Strassburg 2000; Guillemette 2003). Il est à noter que la glucuronidation peut aussi amener à augmenter l’effet pharmacologique ou rendre une molécule active pharmacologiquement. Par exemple, la morphine-6-glucuronide est la forme active issue de la glucuronidation de la morphine par l’UGT2B7 qui permet d’augmenter le pouvoir actif de la molécule de plus de 20 fois comparativement à la forme non-conjuguée (Coffman et al. 1997). Cette réaction métabolique de phase II est rendue possible grâce aux enzymes UDP-glucuronosyltransférases, qui sont très conservées dans la nature. En effet, elles sont présentes chez les plantes, les bactéries et les mammifères (Guillemette 2003; Bolam et al. 2007; Brazier-Hicks et al. 2007). Les UGT possèdent trois
grands rôles (Guillemette 2003). Tout d’abord, elles permettent le métabolisme de toxines (environnementales et alimentaires) et de carcinogènes (Bartsch et al. 1992; Orzechowski et al. 1994; Nowell et al. 1999). Les UGT permettent aussi l’homéostasie de nombreuses molécules endogènes comme la bilirubine, les acides gras, les hormones stéroïdiennes et thyroïdiennes, et les acides biliaires (Pillot et al. 1993; Bosma et al. 1994; Liu et al. 1995; Belanger et al. 1998). Finalement, les UGT sont impliquées dans le métabolisme ou l’activation des médicaments (Herman et al. 1994; Iyer et al. 1998; Evans and Relling 1999).
Figure 5. Schématisation de la réaction de glucuronidation
Afin qu’un substrat hydrophobe soit éliminé par l’urine ou la bile, celui-ci doit être conjugué via le transfert d’un acide glucuronique d’un co-substrat UDPGA à l’aide d’une enzyme UDP-glucuronosyltransférase. Figure adaptée de Guillemette 2003.
Pour que la réaction de glucuronidation soit possible, le co-substrat UDPGA et le substrat doivent se localiser dans le site actif de l’UGT (Yin et al. 1994). Généralement, deux réactions de glucuronidation sont possibles : N-glucuronidation et O-glucuronidation (Chiu and Huskey 1998; Tukey and Strassburg 2000). Les alcools, les phénols et les acides carboxyliques permettent une réaction de O-glucuronidation tandis que les amines
primaires, secondaires, tertiaires, hétérocycliques et les amides permettent la réaction de N-glucuronidation (Tukey and Strassburg 2000; Kaivosaari et al. 2011). Il est à noter qu’il existe d’autres réactions plus rares comme la S-glucuronidation et la C-glucuronidation qui nécessitent un thiol et un carbonyle, respectivement (Tukey and Strassburg 2000). Une fois conjuguée, la molécule devient plus polaire et ionisée car, l’acide carboxylique (pKa ~4) s’ionise au pH physiologique (pH ~7,4), ce qui procure une charge négative à la molécule, augmentant ainsi sa solubilisation et son élimination via l’urine ou la bile (Remmel RP 2008).
2.1. Les UDP-glucuronosyltransférases
2.1.1. Nomenclature et classification des différentes familles UGT
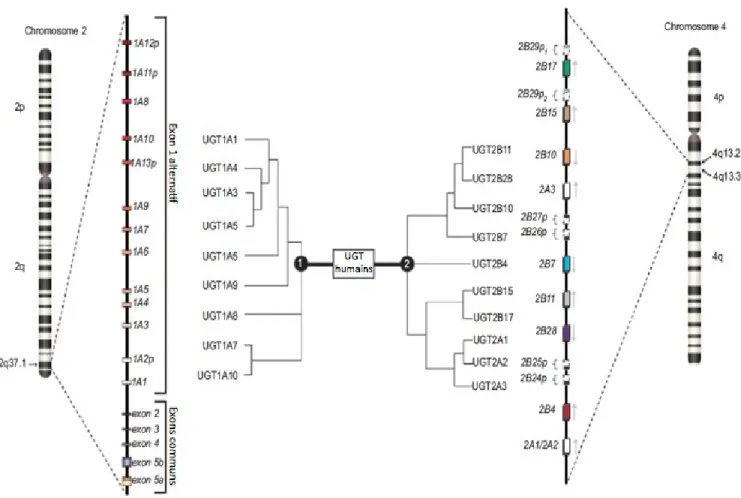
Les enzymes UGT se divisent en quatre grandes familles selon leur homologie de séquence (UGT1, UGT2, UGT3 et UGT8) où les UGT1 et UGT2 sont les plus connues grâce à leurs activités métaboliques via la conjugaison d’un acide glucuronique sur leurs substrats (Mackenzie et al. 2005; Guillemette et al. 2010) (Figure 6). Il est à noter qu’il est aussi possible d’observer une utilisation de l’UDP-xylose et de l’UDP-glucose comme co-substrat même si la majorité des catalyses se fait à l’aide de l’acide glucuronique (Senafi et al. 1994; Mackenzie et al. 2003). La famille UGT1 comporte une seule sous-famille, les UGT1A. Elles sont issues d’un seul gène se trouvant sur le chromosome deux en position q37.1 permettant la synthèse de 9 enzymes actives (Gong et al. 2001). L’évolution de ce gène est issue de la duplication des exons, des délétions et des conversions (Mackenzie et al. 2005). Quant à la famille UGT2, elle est divisée en deux sous-familles, 2A et 2B, où l’on retrouve trois et douze membres, respectivement (Mackenzie et al. 2005; Guillemette et al. 2010). Les UGT2B sont issues de duplications de gènes, ce qui implique que chaque membre possède son propre gène se situant sur le chromosome 4q13.2 (Meech et al. 2012). Pour ce qui est des UGT2A, le gène unique de 2A3 se situe au même locus que ceux des 2B (Court et al. 2008). Toutefois, les 2A1 et 2A2 sont issues du même gène (4q13.3) via des exons 1 alternatifs (Mackenzie et al. 2005).
Figure 6. Arbre phylogénétique des principaux membres de la superfamille UGT Les UGT se divisent principalement en deux grandes familles : UGT1 et UGT2, où une grande homologie de séquence est observable. La famille UGT1 se divise en une sous-famille appelée UGT1A. Elles sont transcrites à partir d’un seul gène situé sur le chromosome 2q37.1. Pour ce qui est de la famille UGT2, elle se divise en deux sous-familles, les UGT2A et les UGT2B (4q13.2 et 4q13.3). Chacun des différents membres est transcrit à partir d’un gène unique contrairement aux UGT2A1 et 2A2 qui sont issues du même gène. Figure adaptée de Guillemette et al. 2010.
Contrairement aux deux familles précédentes, les UGT3 et UGT8 sont moins connues. La famille UGT3 est constituée de deux membres, l’UGT3A1 et l’UGT3A2, exprimés à l’aide de gènes se situant au chromosome 5.p13.2 (Meech and Mackenzie 2010). À l’inverse des UGT1 et des UGT2, les UGT3 n’utilisent pas l’acide glucuronique pour conjuguer leurs substrats. En effet, l’UGT3A1 utilise l’UDP-N-acétylglucosamine et l’UGT3A2 utilise l’UDP-xylose et l’UDP-glucose (Mackenzie et al. 2008; MacKenzie et al. 2011). Pour ce qui est de l’UGT8, elle est composée d’un seul membre, l’UGT8A1
(Mackenzie et al. 2005). Cette enzyme possède toutefois un rôle qui est différent de celui des autres UGT, car l’UGT8A1 joue un rôle dans la biosynthèse pour le système nerveux (Bosio et al. 1996). Son locus se situe au chromosome 4q26 et elle catalyse le transfert d’un galactose via un UDP-galactose afin de conjuguer un céramide, ce qui est une étape importante dans la synthèse des lipides pour la gaine de myéline du système nerveux (Bosio et al. 1996; Marcus and Popko 2002).
2.2.1. La famille UGT1A
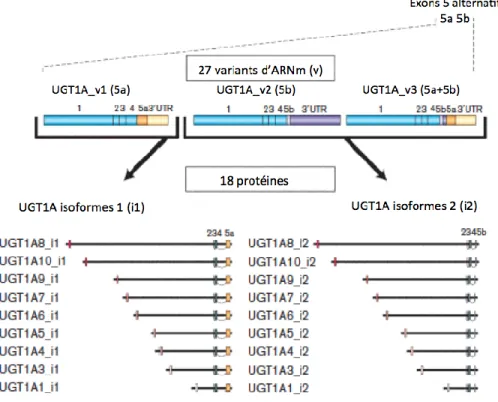
Le locus du gène UGT1A contient un total de 18 exons. Treize de ceux-ci sont des exons 1 alternatifs dont quatre d’entre eux sont des pseudogènes (1A2p, 1A11p, 1A12p et 1A13p). Donc, neuf exons 1 alternatifs sont disponibles pour la synthèse des enzymes : 1A1, 1A3, 1A4, 1A5, 1A6, 1A7, 1A8, 1A9 et 1A10 (Ritter et al. 1992; Gong et al. 2001). Il est à noter que chacun des exons 1 possèdent son propre promoteur, ce qui garantit une diversité chez les UGT1A. Le reste du gène est quant à lui composé de quatre exons communs, exon 2-5 (Ritter et al. 1992; Gong et al. 2001). Il a aussi été démontré qu’il y a un évènement d’épissage en 3’ du gène. En effet, un exon alternatif appelé 5b a été découvert dans l’intron 4, soit entre l’exon 4 et l’exon 5 classique maintenant nommé 5a (Levesque et al. 2007). L’exon 5a est connu pour être responsable de la partie transmembranaire de l’enzyme tandis que l’exon 5b est dépourvu de ce domaine (Levesque et al. 2007). Afin de différencier chacun de ces évènements d’épissage en 3’, une nouvelle nomenclature a été suggérée. Les enzymes issues de l’exon 5 classique, c’est-à-dire 5a, se nomment isoformes 1 (i1) et ceux de l’exon 5b se nomment isoformes 2 (i2). Il est donc possible d’obtenir 27 transcrits différents qui permettent la synthèse de 18 enzymes, soit neuf enzymes classiques (i1) et neuf enzymes i2 (Girard et al. 2007; Levesque et al. 2007) (Figure 7). Par exemple, il est possible d’obtenir trois variants pour l’UGT1A1 (Girard et al. 2007). Premièrement, il y a le variant 1 (v1) qui contient l’exon 5a et qui permet la synthèse de l’isoforme 1. Il y a ensuite le variant 2 (v2), c’est-à-dire celui qui contient l’exon 5b. Puis, le variant 3 (v3) qui contient à la fois l’exon 5a et 5b. Comme l’exon 5b contient un codon stop, la protéine issue de ce dernier variant est la même que celle du v2, c’est-à-dire l’isoforme 2. Les premières études fonctionnelles des i2 ont permis de démontrer qu’elles ne possèdent pas d’activité enzymatique avec l’UDPGA même si elles
conservent la capacité de se lier aux substrats et aux co-substrats. De plus, les i2 possèdent un rôle de modulateur négatif sur l’activité de glucuronidation de la forme active (Girard et al. 2007; Levesque et al. 2007; Bellemare et al. 2010c). Des études subséquentes ont aussi démontré que l’activité inhibitrice de i2 n’est pas le fruit d’une compétition entre la forme active et inactive pour le co-substrat et le substrat, mais plutôt le résultat d’une interaction protéine-protéine et que chaque isoenzyme des UGT1A peut former un oligomère avec toutes les isoformes i1 et i2 des différents membres UGT1A (Bellemare et al. 2010a; Bellemare et al. 2010b). Toutefois, le(s) domaine(s) impliqué(s) dans cette oligomérisation n’est(ne sont) pas encore connu(s).
Figure 7. Évènements d'épissage en 3’ au locus UGT1A
L’évènement d’épissage en 3’ mène à trois transcrits différents : v1 (exon 5a), v2 (exon 5b) et v3 (exon 5a+5b). Ces différents transcrits permettent la synthèse de deux isoformes, c’est-à-dire la protéine classique ou isoforme 1 (i1) qui est issue de l’exon 5a et l’isoforme 2 (i2) qui correspond à la forme inactive provenant de l’exon 5b. Il est à noter que l’exon 5b possède un codon stop à son extrémité 3’, ce qui explique que le v2 et v3 permettent la synthèse de l’isoforme i2. Au total, 27 transcrits d’épissage sont possibles permettant la synthèse de 18 protéines. Figure adaptée de Girard et al. 2007.
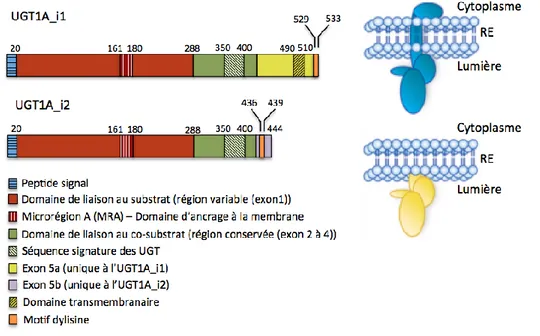
2.1.2.1. Structure et localisation
Les UDP-glucuronosyltransférases sont des enzymes transmembranaires de type 1, c’est-à-dire qu’elles sont ancrées dans la membrane via une hélice α et que leur domaine N-terminal est situé dans la lumière de l’organelle contrairement au domaine C-N-terminal qui se situe dans le cytoplasme. Elles se localisent principalement dans la lumière du réticulum endoplasmique, mais elles sont aussi connues pour se localiser à la membrane nucléaire (Tephly and Burchell 1990; Ouzzine et al. 1999). Leur structure primaire se divise en deux parties, une partie variable en N-terminale qui est responsable de la liaison du substrat et une partie commune en C-terminale qui permet la liaison du co-substrat (Figure 8). Lors de leur synthèse, les UGT1A sont composées de 530 acides aminés. Toutefois, dès leur entrée au réticulum endoplasmique, une courte séquence est coupée, le peptide signal (PS), permettant d’avoir une séquence finale d’environ 505 acides aminés pour une masse moléculaire de 50-60 kDa (Mackenzie and Owens 1984; Finel and Kurkela 2008). Le PS est la séquence se trouvant à l’extrémité N-terminale. Cette courte séquence d’environ 20 acides aminés est connue pour permettre la localisation d’une protéine à son compartiment. Cependant, il a été démontré que le peptide signal peut aussi être impliqué dans le repliement des UGT. En effet, Seppen et collaborateurs ont démontré qu’un polymorphisme d’un seul nucléotide (SNP) localisé dans la région responsable de la traduction du PS affecte l’activité enzymatique de l’UGT1A1 (Seppen et al. 1996). Quelques années plus tard, des chercheurs ont démontré que cette UGT1A1 mutante est bel et bien localisée au réticulum endoplasmique mais, via la face externe, ce qui amène la protéine à être dégradée (Ohnishi and Emi 2003). Un domaine de liaison au substrat issu de l’exon 1 est aussi localisé en N-terminal. De plus, un motif hydrophobe d’environ 20 acides aminés a été découvert dans cette région. Ce microdomaine appelé microrégion A (MRA) a été découvert à l’aide d’analyses in silico qui ont permis de situer une hélice alpha entre les résidus 159 et 177 de l’UGT1A1 (Ciotti et al. 1998). Des études subséquentes ont permis de démontrer que ce motif est essentiel à la glucuronidation de la bilirubine par l’UGT1A1 (Ritter et al. 1993; Ciotti et al. 1998; Ghosh et al. 2005). Il a aussi été démontré que le MRA est conservé chez d’autres UGT comme l’UGT1A3, 1A4, 1A5 et 1A6 (Ciotti et al. 1998).
On retrouve dans la région C-terminale une séquence peptidique qui est conservée chez les UGT1A, c’est-à-dire un domaine de liaison au co-substrat (Meech and Mackenzie 1997a). Ce domaine possède aussi une séquence de 50 acides aminés représentant la séquence signature. Finalement, les exons 5a ou 5b terminent la partie C-terminale de la structure des UGT1A (Girard et al. 2007; Levesque et al. 2007). L’usage de l’exon 5a ou de 5b permet l’obtention d’une séquence différente en C-terminal. L’exon 5b engendre une séquence de 10 acides aminés (RKKQQSGRQM) contrairement à l’exon 5a qui est de 99 acides aminés (Levesque et al. 2007). Cette forme tronquée de i1 ne possède pas de domaine transmembranaire. Toutefois, les UGT1A_i2 possèdent un motif dilysine (KKXX) tout comme les UGT1A_i1 (KXKXX), ce qui permet une rétention au réticulum endoplasmique (Kinosaki et al. 1993; Meech and Mackenzie 1998; Girard et al. 2007; Magdalou et al. 2010).
Figure 8. Structures primaires et tertiaires des isoformes i1 et i2
La séquence primaire des UGT1A est divisée en deux parties, une partie N-terminale qui correspond à la région variable, plus précisément le domaine de liaison aux substrats et une partie C-terminale correspondant à la région conservée, c’est-à-dire le domaine de liaison aux co-substrats. À l’extrémité de cette dernière, une séquence variable est observable selon l’isoforme i1 ou i2. Dans le cas de l’isoforme i1, une séquence de 99 acides aminés est présente contenant un domaine transmembranaire, contrairement à l’isoforme i2 qui est
seulement composé de 10 acides aminés, et ce, sans domaine transmembranaire. Figure adaptée de Girard et al. 2007.
2.1.2.2. Expression tissulaire
Les UGT se localisent dans de nombreux tissus mais, principalement dans le foie, un centre de détoxification majeur, mise à part l’UGT1A8, 1A7 et 1A10 que l’on retrouve seulement dans les tissus extrahépatiques comme le système gastro-intestinal (Strassburg et al. 1997; Strassburg et al. 1998). Les différents résultats obtenus au courant des dernières années ont démontré que les différents UGT1A possèdent un patron d’expression différentiel selon le tissu, et ce, grâce au promoteur unique de chaque exon 1 alternatif (Beaulieu et al. 1998; Strassburg et al. 1998; Strassburg et al. 1999; King et al. 2000; Nakamura et al. 2008; Ohno and Nakajin 2009; Court et al. 2012; Harbourt et al. 2012) (Figure 11). Par exemple, l’UGT1A9 est celle qui est la plus exprimée dans le rein contrairement à l’UGT1A1 qui est absente des reins, mais fortement exprimée dans le foie. Il est donc possible d’observer une expression différentielle de chaque UGT1A selon le tissu. Il en est de même pour chaque isoforme i1 et i2 des UGT1A. En effet, Girard et collaborateurs ont démontré que le patron d’expression des i1 est différent de celui de i2 (Girard et al. 2007). Par exemple, l’UGT1A6_i2 est retrouvée dans le rein tout comme l’isoforme i1. Toutefois, dans le tissu colorectal et le tissu hépatique, la forme i2 n’est pas présente. Il en est aussi de même pour l’UGT1A3 où l’isoforme i2 est absente du tissu intestinal et de l’œsophage tandis qu’elle est présente dans le foie et les reins. En somme, chaque UGT semble jouer un rôle qui lui est spécifique comme le montrent les différents patrons d’expression. Toutefois, contrairement à chaque isoenzyme qui possède une expression tissulaire différentielle grâce à son promoteur, rien n’est encore connu en ce qui a trait à l’expression ou à l’induction différentielle des formes d’épissage des UGT1A.
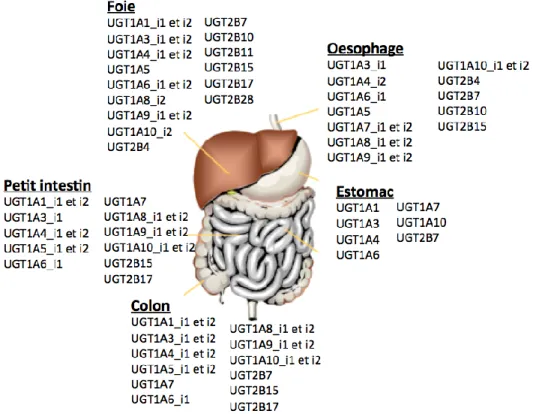
Figure 9. Distribution tissulaire des différents membres UGT1A et UGT2B
Les UGT1A et les UGT2B sont fortement exprimées dans le système gastro-intestinal et dans le foie, le principal centre de détoxification de l’organisme.
3. Oligomérisation des UGT
Avec les récentes découvertes dans le domaine de la protéomique, il est de plus en plus démontré que les protéines fonctionnent par interaction protéine-protéine, et les UGT n’y échappent pas. Les premiers travaux portant sur ce sujet ont constaté un complexe d’UGT dans des microsomes de foie de rat ayant une masse de 316 kDa tout en ayant une activité de glucuronidation (Matern et al. 1982). D’autres études ont suivi mais, avec la difficulté de purification des UGT, les chercheurs ont plutôt opté pour une méthode de radiation qui permet d’inactiver les protéines (Peters et al. 1984; Gschaidmeier and Bock 1994). La radiation permet d’informer sur la masse moléculaire et la structure quaternaire d’une enzyme. Par exemple, de gros complexes nécessiteront moins d’énergie pour être inactivés qu’un simple monomère. Ces différents travaux ont démontré que les UGT peuvent former des monomères (environ 55 kDa) et des dimères (environ 90 kDa). De plus, ils ont établi l’hypothèse que l’obtention d’une di-glucuronidation nécessiterait un
complexe d’enzyme contrairement à une mono-glucuronidation. Par la suite, en 1997, Meech et Mackenzie ont fait une découverte importante (Meech and Mackenzie 1997b). À l’aide de différents mutants de l’UGT2B1, ils ont analysé les différentes possibilités d’oligomérisation ainsi que leurs activités. Ils ont découverts que les deux mutants inactifs peuvent former un complexe et que la co-expression d’un mutant inactif avec le sauvage peut aussi former un oligomère. La découverte la plus importante est toutefois lorsqu’ils ont démontré que l’interaction du mutant avec le sauvage rend le complexe inactif. Ils ont aussi créé des protéines tronquées afin de déterminer si les régions N-terminales et C-terminales sont impliquées dans l’hétéro-oligomérisation. Ces expériences ont permis de démontrer que la région N-terminale est essentielle à l’oligomérisation contrairement au C-terminale. Toutefois, ces deux domaines sont nécessaires à l’activité enzymatique. Quelques années plus tard, Ghosh et son équipe ont démontré la présence d’homo-oligomères et l’implication de la région N-terminale dans les interactions protéine-protéine entre les UGT. En effet, à l’aide d’un système double hybride, ils ont démontré que les UGT1A1 peuvent former un homo-oligomère et que la microrégion MRA est importante pour l’interaction protéique. Toutefois, une délétion dans la région C-terminale n’a eu aucune influence sur la formation du complexe. Ces résultats appuient donc le rôle essentiel de la région N-terminale dans les interactions d’homo-oligomérisation (Ghosh et al. 2001). Ils ont aussi démontré qu’il existe des ponts disulfures entre les UGT en effectuant des électrophorèses sur gel de polyacrylamide en présence de dodécylsulfate de sodium (SDS-PAGE) en conditions non-réductrices, suggérant une implication de ces liaisons covalentes dans les interactions protéiques des UGT. Les UGT possèdent plusieurs résidus cystéines conservés, où l’un d’eux se situant dans le PS est éliminé lors de la maturation de la protéine (Ghosh et al. 2005). Ces acides aminés sont importants pour l’activité, mais certains d’entre eux semblent aussi être nécessaires à l’oligomérisation, soit la cystéine 186 pour l’UGT1A9 et la cystéine 223 pour l’UGT1A1 (Ghosh et al. 2001; Olson et al. 2009).
Quelques années plus tard, l’homo-oligomérisation de l’UGT1A9 a été démontrée (Kurkela et al. 2003). Un an plus tard, la même équipe a constaté que l’UGT1A9 et l’UGT1A4 peuvent former un complexe (Kurkela et al. 2004a). Afin de confirmer ce résultat, ils ont créé un UGT1A9 mutant, c’est-à-dire une enzyme dépourvue de son
domaine transmembranaire en C-terminal et ils ont ensuite transfecté ce vecteur d’expression avec celui d’un 1A4 sauvage (Kurkela et al. 2004b). Ils ont ainsi observé que l’activité de 1A4 diminue, ce qui confirme l’interaction entre 1A4 et 1A9, car il était déjà connu qu’une forme inactive d’une UGT peut moduler négativement l’activité d’une autre UGT lorsqu’elles forment un complexe (Meech and Mackenzie 1997a). Une autre information importante a aussi été révélée à l’aide de ce résultat : la structure C-terminale ne semble pas être nécessaire à l’interaction des UGT, ce qui suggère l’implication de la région N-terminale.
Tableau 1. Les domaines protéiques des UGT et leurs implications dans l'oligomérisation, la localisation et l’activité
Domaine de la protéine UGT
Oligomérisation Localisation Activité Références
Protéine sauvage + Réticulum
endoplasmique + Ishii et al., 2010; Ishii et al., 2005; Finel et al., 2008 ΔPS (1-26) N.D. - - Seppen et al., 1996; Ohnishi et al., 2003; Ouzzine et al., 1999
ΔMRA (152-180) - N.D. - Ciotti et al., 1998;
Ouziine et al., 1999; Ghosh et al., 2001 ΔTMD (490-510) + + - Meech et al., 1997; Ouzzine et al., 1999; Meech et al., 1996; Kurkela et al., 2004; Kurkela et al., 2004
ΔN-ter (24-276) - + - Meech et al., 1997
ΔC-ter (291-534) + + - Meech et al., 1997;
Ouziine et al., 1999; Ghosh et al., 2001; Koiwai et al., 1996 N.D. : Non déterminé
Jusqu’à maintenant, il n’y avait aucune étude portant sur l’oligomérisation de toutes les UGT1A. Il a fallu attendre jusqu’en 2007 où Operana et Tukey ont été capable de démontrer l’homo-oligomérisation de tous les UGT1A ainsi que l’interaction entre
l’UGT1A1 et tous les autres UGT1A à l’aide de transferts d’énergie par résonance en fluorescence (FRET) et de co-immunoprécipitations (Operana and Tukey 2007). Ils avaient même émis l’hypothèse que l’interaction entre les UGT se ferait plutôt via le C-terminal et non par le N-terminal comme le proposait déjà la littérature. Fujiwara et al. ont, par la suite, montré à l’aide de gel natif que l’UGT2B7 peut d’abord former un homo-oligomère, mais qu’elle peut aussi former un complexe avec les UGT1A1, UGT1A4, UGT1A6 et UGT1A9, les principales enzymes UGT1A se localisant au foie (Fujiwara et al. 2010).
De plus, des interactions entre différentes enzymes du métabolisme et les UGT ont aussi été rapportées. En effet, une première étude en 2000 a démontré une interaction entre les CYP1A1 et les UGT, sans toutefois être en mesure d’identifier quelles UGT étaient impliquées (Taura et al. 2000). Par la suite, il a été établi à l’aide d’immunoprécipitation que l’UGT1A1, 1A6 et l’UGT2B7 peuvent former un complexe avec CYP3A4, où l’activité de 2B7 diminue lorsqu’il est en interaction avec le CYP3A4 (Fremont et al. 2005; Takeda et al. 2005; Takeda et al. 2009). Mise à part les cytochromes P450, les UGT peuvent aussi avoir des interactions avec d’autres enzymes impliquées dans le métabolisme de phase II. En effet, une interaction entre GSTA1 et l’UGT2A1 est connue, sans toutefois avoir d’interaction avec GSTT1 (Akizawa et al. 2008). Les GST, plus particulièrement GSTP1, peuvent aussi avoir une interaction avec JNK-1, une protéine de signalisation, ce qui impliquerait une fonction biologique autre que pour le métabolisme (Adler et al. 1999; Romero et al. 2006). Il serait donc intéressant de déterminer si les UGT possèdent elles aussi un tel rôle.
Finalement, avec la mise à jour en 2007 des nouvelles isoformes i2 des UGT1A, la même question a été abordée pour les isoformes i1 et i2. Est-ce que ces isoformes oligomérisent? Notre laboratoire a tenté de répondre à cette question et la réponse s’est avérée positive. En effet, à l’aide de modèles de surexpression et de co-IP, il a été démontré que les isoformes i1 et i2 d’un même UGT1A peuvent former un homo-oligomère, mais qu’il est aussi possible pour toutes les i1 et i2 de chaque isoenzyme de former un hétéro-oligomère (Bellemare et al. 2010a). Ces résultats ont permis de proposer que la capacité de modulateur négatif de i2 est rendue possible grâce à des interactions protéine-protéine entre
les isoformes d’UGT1A. Toutefois, rien n’est connu quant aux domaines d’interaction impliqués dans les formes i1 et i2, ce qui pourrait être une étape importante dans la compréhension du rôle de i2 comme régulateur de l’activité de glucuronidation et dans le développement potentiel de nouveaux outils pharmacologiques ciblant ces interactions.
3.1. Interaction protéine-protéine
Les interactions protéine-protéine sont essentielles pour le fonctionnement de la cellule, car elles sont impliquées dans de nombreux processus cellulaires comme la régulation génique, la réponse immunitaire et les voies de signalisation (Veselovsky et al. 2002; Cho et al. 2005; Tuncbag et al. 2008). Elles peuvent avoir lieu entre des protéines identiques ou non-identiques (homo-oligomères ou hétéro-oligomères). Les oligomères impliquant des protéines identiques ou homologues peuvent être divisés selon deux types de liaison, c’est-à-dire une association qui implique la même surface pour les deux protéines ou qui implique des surfaces différentes (Goodsell and Olson 2000). Ensuite, ces oligomères peuvent être divisés selon le type de complexe qu’ils forment. Deux possibilités sont possibles: il peut s’agir d’un complexe obligatoire ou non obligatoire (Amoutzias 2010). Pour qu’un complexe soit obligatoire, il ne faut pas que les protéines impliquées soient retrouvées sous une forme stable lorsqu’elles sont sous une forme monomérique. Plusieurs complexes protéiques se catégorisent dans cette classe comme les complexes de signalisation intracellulaire et les complexes antigènes-anticorps (Nooren and Thornton 2003). Pour ce qui est des complexes non-obligatoires, c’est l’inverse : les protéines sont stables lorsqu’elles sont sous une forme monomérique (Amoutzias 2010). Dans le cas des UGT, tout porte à croire qu’elles forment des complexes non-obligatoires (Fujiwara et al. 2007; Operana and Tukey 2007).
Les complexes peuvent ensuite être distingués selon leur durée de vie. Pour ce faire, on retrouve des interactions permanentes qui sont très stables et qui impliquent des protéines existant seulement sous la forme de complexe, et des interactions transitoires qui se résument par une association ou une dissociation des protéines impliquées (Nooren and Thornton 2003; Brown and Jurisica 2007). Le complexe hétéromère de la protéine G est un
excellent exemple d’interaction transitoire. En effet, ce complexe comporte trois unités qui peuvent être associées lors d’une activation par un récepteur pour finalement être dissociées : le récepteur couplé aux protéines G, la sous-unité Gα et le complexe Gβγ
(Winzell and Ahren 2007). Il est à noter que les complexes sont dynamiques, donc il est possible de retrouver un équilibre entre des liaisons obligatoires ou non, car la stabilité des complexes est sous l’influence d’un grand nombre de facteurs physicochimiques comme la concentration d’ions, le pH et la température (Archakov et al. 2003; Amoutzias 2010). De plus, les interactions permanentes sont souvent obligatoires et conservées tandis que les interactions transitoires peuvent être obligatoires ou non et sont généralement peu conservées, mais dominantes chez l’homme (Mintseris and Weng 2005; Brown and Jurisica 2007).
La structure primaire est aussi importante pour la formation d’interaction protéine-protéine. En effet, des auteurs ont démontré que certains résidus étaient plus abondants dans les surfaces d’interaction que d’autres. On retrouve des résidus tels que l’arginine, l’histidine, l’asparagine, le tryptophane, la tyrosine et la sérine ainsi que des acides aminés aromatiques et hydrophobes (Davies and Cohen 1996; Jones and Thornton 1996; Stites 1997). Ces molécules impliquent plusieurs facteurs biophysiques qui permettent des liaisons non-covalentes. Les plus importants sont les forces hydrophobes, les forces électrostatiques et les liaisons hydrogènes (Veselovsky et al. 2002; Pechmann et al. 2009).
Pour commencer, l’interaction hydrophobe est la force qui contribue le plus aux interactions protéine-protéine (Eisenhaber and Argos 1996; Wells 1996). Cette force non polaire d’environ 200-400 Å est très importante chez les complexes permanents contrairement aux complexes non obligatoires, car ces derniers sont assemblés dans un environnement riche en eau, ce qui est défavorable énergétiquement pour les surfaces hydrophobes (Jones and Thornton 1996; Lijnzaad and Argos 1997). Les forces électrostatiques sont les forces les plus importantes après les forces hydrophobes (Gong et al. 2000). Pour qu’il y ait des interactions entre deux protéines via ces forces, il faut que les charges électriques à leurs surfaces soient complémentaires (Veselovsky et al. 2002). D’ailleurs, plus cette force est présente, plus la formation du complexe sera rapide
(Gabdoulline and Wade 1999). Un autre facteur important impliqué est la liaison hydrogène. Cette liaison de type dipôle-dipôle d’environ 110-200 pm est de faible énergie et elle implique un hydrogène et un élément polaire qui est majoritairement l’oxygène (Jones and Thornton 1996; Xu et al. 1997). Certes, cette liaison implique peu d’énergie, mais leur grand nombre résulte à une force globale très importante. En somme, les interactions protéine-protéine sont le résultat de plusieurs liaisons non covalentes via des résidus spécifiques présents à la surface de chacune des protéines.
Malgré le fait que ces forces permettent la création des complexes, elles peuvent aussi être responsables de la formation d’agrégats indésirables qui peuvent causer des maladies importantes, par exemple la maladie d’Alzheimer et la maladie des prions (Chiti and Dobson 2006). Afin d’éviter de telles situations, la cellule a développé des mécanismes de contrôle de qualité comme les protéines « heat shock » afin d’empêcher les protéines avec une mauvaise conformation de former, entre autres, des agrégats avec d’autres protéines qui normalement ne sont pas partenaires (Pechmann et al. 2009). Il existe toutefois des résidus spécifiques qui agissent comme des résidus protecteurs contre des repliements indésirables (Fass 2012). En effet, Pechman et son équipe ont démontré l’existence de résidus permettant des interactions intramoléculaires près de l’interface de la protéine afin d’empêcher cette dernière d’adopter une mauvaise conformation après une perturbation et ainsi former un agrégat (Pechmann et al. 2009). Donc, ces résidus empêchent la formation de certaines zones hydrophobes à la surface de la protéine qui n’existe pas normalement. Dans 92 % des complexes qu’ils ont étudiés, ils ont découvert la présence de ponts disulfures et de ponts ioniques en proximité des interfaces d’interaction. Ces liaisons permettent la stabilité des protéines et permettent de prévenir des changements de conformation lors d’un changement dans la physicochimie de la cellule.
L’étude de ces interactions est essentielle dans l’élaboration de nouvelles drogues synthétiques. Plusieurs interactions non spécifiques sont connues pour causer des maladies, ce qui représente des cibles thérapeutiques intéressantes (Chiti and Dobson 2006). Dans le cas présent, l’identification des domaines d’interaction entre les isoformes i1 et i2 permettrait le développement éventuel de molécules synthétiques permettant de moduler la
voie de glucuronidation. Pour ce faire, il s’agit de créer une molécule capable de mimer cette interaction, mais pour que cela soit possible, il faut d’abord connaitre les domaines impliqués dans l’interaction, et ce, à l’aide de différentes techniques comme la co-immunoprécipitation et l’élaboration de modèles comportant des mutations (Miernyk and Thelen 2008).