Purification par affinité et marquage isotopique spécifique pour études d’ARN fonctionnels
148
0
0
Texte intégral
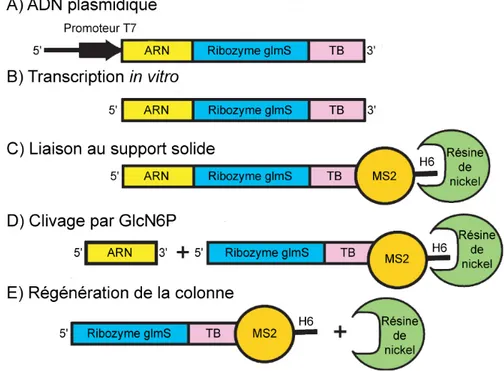
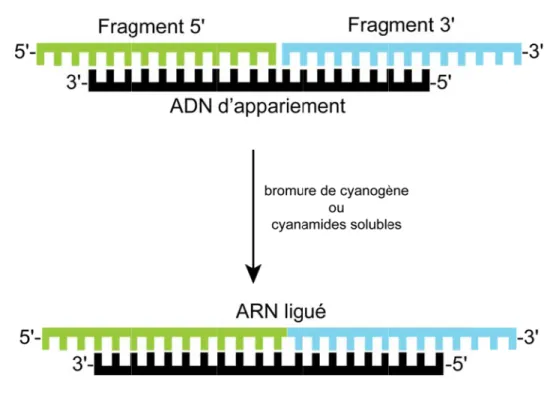
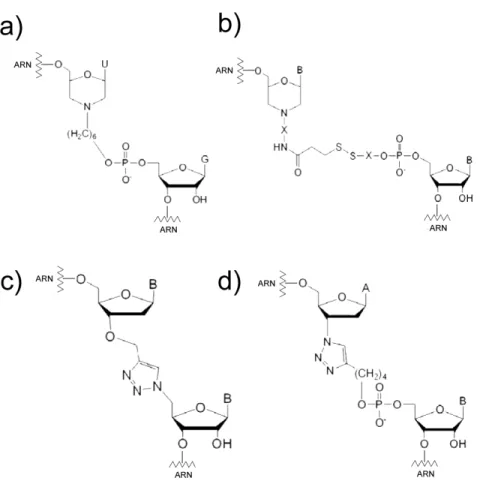
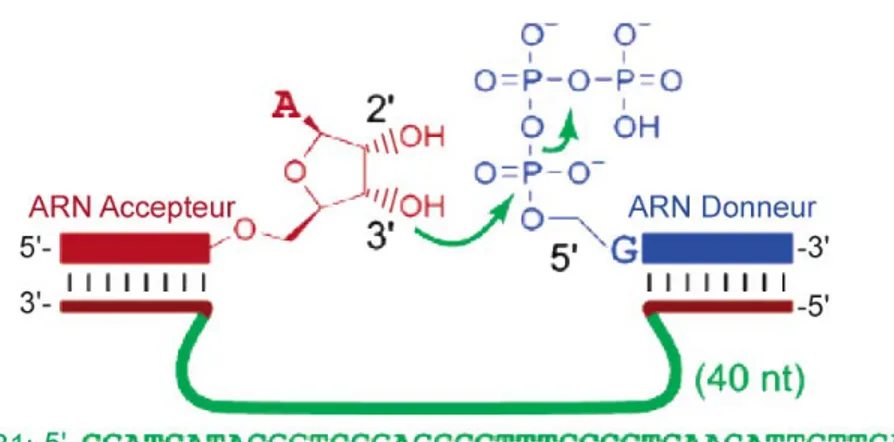
Figure
Documents relatifs