The role of SHIP-1 in T lymphocytes life and death
Geoffrey Gloire
1*, Christophe Erneux
2and Jacques Piette
11Center for Biomedical Integrated Genoproteomics (CBIG), Virology and Immunology Unit,
University of Liège, 4000 Liège, Belgium
2Interdisciplinary Research Institute (IRIBHM), University of Brussels, 1070 Brussels,
Belgium.
*Address for correspondence: Geoffrey Gloire, CBIG/GIGA, Virology and Immunology Unit Institute of Pathology B23, B-4000 Liège Belgium. Email: geoffrey.gloire@ulg.ac.be.
Tel: + 32 4 366 24 26, Fax: + 32 4 366 99 33
Key Words: SHIP-1, T lymphocyte, PI3K, NF-κB, apoptosis, proliferation.
ABBREVIATIONS
PI3K: phosphoinositide 3-kinase; ITIM: immuno-receptor tyrosine inhibition motif; ITAM: immuno-receptor tyrosine activation motif; Dok: downstream of tyrosine kinase; FcR: IgG Fc receptor; PLCγ: phospholipase C gamma; BTK: Burton’s tyrosine kinase; GSK-3β: glycogen synthase kinase 3 beta; KLF2: Krüppel-like factor 2; NF-κB: nuclear factor κB; IκBα : Inhibitor of κB alpha; TGFβ: transforming growth factor beta.
ABSTRACT
SHIP-1, an inositol 5-phosphatase expressed in hemopoietic cells, acts by hydrolysing the 5-phosphates from phosphatidylinositol-3,4,5-triphosphate
(PtdIns(3,4,5)P3) and
inositol-1,3,4,5-tetrakisphosphate (InsP4), thereby
negatively regulating the PI3K pathway. SHIP-1 plays a major role in inhibiting proliferation of myeloid cells. As a result,
SHIP-1-/- mice have an increased number
of neutrophils and monocytes/macrophages due to enhanced survival and proliferation of their progenitors. Although SHIP-1 contributes to PtdIns(3,4,5)P3 metabolism
in T lymphocytes, its exact role in this cell type is much less explored. Jurkat cells have recently emerged as an interesting tool to study SHIP-1 function in T cells because they do not express SHIP-1 at the protein level, thereby allowing reintroduction experiments in a relatively easy to use system. Data obtained from SHIP-1 reintroduction have revealed that
SHIP-1 not only acts as a negative player in T cell lines proliferation, but also regulate critical pathways, such as NF-κB activation, and also appear to remarkably inhibit T cell apoptosis. On the other hand, experiments using primary T cells from
SHIP-1-/- mice have highlighted a new role
for SHIP-1 in regulatory T cell development, but also emphasize that this protein is not required for T cell proliferation. In support of these results,
SHIP-1-/- mice are lymphopenic,
suggesting that SHIP-1 function in T cells differs from its role in the myeloid lineage.
INTRODUCTION
The PI3K pathway regulates many important cellular functions, such as survival, proliferation, cell activation and migration [1-3]. Upon activation, PI3K
generates
phosphatidylinositol-3,4,5-triphosphate (PtdIns(3,4,5)P3) from
PtdIns(4,5)P2. PtdIns(3,4,5)P3 attracts
pleckstrin homology (PH) domain-containing proteins to the plasma membrane, which allows their activation and subsequent signalling. PH domain-containing proteins include notably Akt, a key messenger in cellular proliferation and protection from apoptosis [4]. The negative regulation of the PI3K pathway is achieved notably by PTEN (phosphatase and tensin homologue deleted on chromosome 10), which hydrolyses
PtdIns(3,4,5)P3 back to PtdIns(4,5)P2 [5],
and by SH2-containing inositol-5-phosphatase-1 (SHIP-1), which hydrolyses
the 5-phosphates from
phosphatidylinositol-3,4,5-triphosphate
(PtdIns(3,4,5)P3) and
inositol-1,3,4,5-tetrakisphosphate (InsP4) [6]. SHIP-1, a
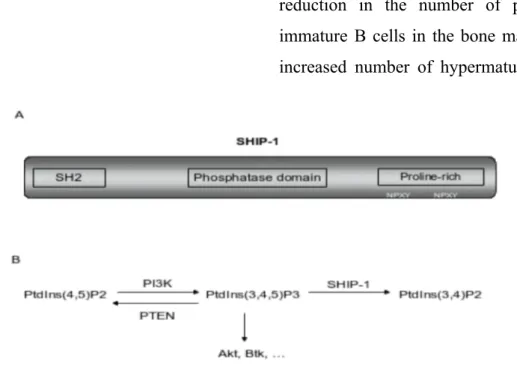
145 kDa protein expressed in hemopoietic cells, contains a central phosphatase domain, an N-terminal SH2 domain and a
C-terminal domain containing
phosphorylable NPXY sites and proline-rich sequences (Figure 1) [7]. SH2 domain and NPXY sites mediate the binding of SHIP-1 with a series of consensus motifs or adapter proteins, like immunoreceptor signalling motifs (ITIM and ITAM) and various Doks [8, 9]. Through its inositol phosphatase activity, SHIP-1 influences
PtdIns(3,4,5)P3 levels in many cell types,
which has a crucial impact in the negative regulation of cellular proliferation and apoptosis. In support of this, SHIP-1-/- mice
have an increased number of myeloid cells due to enhanced survival and proliferation of their progenitors, thereby resulting in splenomegaly and myeloid infiltration in various organs, including lungs [10, 11].
SHIP-1-/- myeloid cells exhibit furthermore
decreased susceptibility to apoptotic cell death, an observation that has to be linked to the constitutive Akt activation in these cells, leading to enhanced survival and proliferation [11]. This observation is
Figure 1. A. Structure of the SHIP-1 protein. B. Through its lipid phosphatase activity, SHIP-1
dephosphorylates PtdIns(3,4,5)P3 to PtdIns(3,4,)P2, thereby negatively regulating the PI3K pathway.
PTEN exerts the same function, but catalyzes PtdIns(3,4,5)P3 back into PtdIns(4,5)P2. See text for
details.
reinforced by the demonstration that a dominant-negative mutation reducing catalytic activity of SHIP-1 was found to be associated with acute myeloid leukaemia [12].
SHIP-1 also acts at different levels
in B cell biology. SHIP-1-/- mice exhibit a
reduction in the number of pre-B and immature B cells in the bone marrow, but increased number of hypermature B cells
in the spleen. B cells from SHIP-1-/- mice
are also hypersensitive to BCR stimulation, but less sensitive to apoptosis [10, 13-16]. These data reflect that the inhibitory signals mediated by SHIP-1 have positive or negative impacts depending on the stage of B cell development. The molecular mechanisms underlying SHIP-1-mediated inhibition of B cell proliferation are now well understood. Signalling pathways induced by BCR activation are inhibited by co-aggregation between BCR and FcγRIIB, a low affinity Fc receptor. This co-aggregation occurs when
IgG-containing immune complexes bind simultaneously the BCR and FcγRIIB and trigger tyrosine phosphorylation within immunoreceptor tyrosine-based inhibitory motifs (ITIM) of the cytoplasmic tail of FcγRIIB [17]. This phosphorylation induces the recruitment of SHIP-1 to the tyrosine phosphorylated ITIM of FcγRIIB via its SH2 domain. SHIP-1 then dephosphorylates PtdIns(3,4,5)P3, leading
to inhibition of PLCγ, Akt and Btk, thereby inhibiting proliferation and calcium mobilisation ([18-20] and [13] for review). SHIP-1 also functions in FcγRIIB-mediated inhibitory signals upon FcεR1-induced degranulation in mast cells ([21-23] and [9] for review).
ROLE OF SHIP-1 IN T LYMPHOCYTES
Data obtained from T cell lines
Although the role of SHIP-1 has been intensely studied in myeloid and B cells, its exact biological function in T cells remains poorly understood. Pioneer works have demonstrated a tyrosine phosphorylation of SHIP-1 upon CD3 and CD28 stimulation, and redistribution of its catalytic activity at the plasma membrane [24, 25], without highlighting a clear biological role of SHIP-1 in the regulation of T cell activation. SHIP-1 was also reported to interact with the ξ chain of the
TCR, at least in vitro [26]. An embryonic work has also demonstrated that FcγRIIB co-aggregation with the TCR results in tyrosine phosphorylation of SHIP-1, indicating that, like in B cells, SHIP-1 would be responsible for FcγRIIB-elicited inhibitory signalling in T cells [27]. Finally, SHIP-1 was demonstrated to play a role in LFA-1-dependent adhesion [28].
The finding that the Jurkat T cell line was defective in SHIP-1 (and PTEN) expression [29-31] has opened up new possibilities for studying SHIP-1 biological role in T cells by complementation experiments. Because of their deficiency in SHIP-1 expression, Jurkat cells express
a very high level of PtdIns(3,4,5)P3 and a
subsequent constitutive Akt activation compared to other leukemic cell lines (such as CEM, MOLT-4 or HUT78) or human PBLs [32]. Restoration of SHIP-1 activity in Jurkat cells was first carried out using a Tet-Off regulated system encoding the lipid phosphatase core of SHIP-1 fused to the transmembrane region of rat CD2. Expression of this membrane-localized SHIP-1 in Jurkat was sufficient to reduce Akt constitutive activation and the activity of downstream PI3K effector, constituting the first evidence that SHIP-1 is capable of modulating important signalling pathways in T cells [32]. Ectopic expression of the complete SHIP-1 protein was also achieved in Jurkat cells by Horn et al.
SHIP-1 expressing Jurkat cells have
reduced PtdIns(3,4,5)P3 level compared to
the parental clone, and subsequent diminution of Akt phosphorylation on S473 and T308. Phosphorylation of GSK-3β, an Akt substrate, is also down regulated upon SHIP-1 reexpression [33]. Interestingly, expression of SHIP-1 in Jurkat also reduces cellular proliferation, notably by affecting the G1 cell cycle
regulators Rb and p27Kip1 [33] and KLF2, a
factor implicated in T cell quiescence [34].
Altogether, these reintroduction
experiments have highlighted a role for SHIP-1 in negatively regulating T cell lines proliferation. In support of this, down-regulation of SHIP-1 expression has been described in adult T cell leukaemia (ATLL) [35].
Using a lentiviral system, we have also express SHIP-1 in Jurkat cells and have furthermore positioned this protein as a key adaptor in the activation of NF-κB upon oxidative stress. After exposure to
H2O2, parental Jurkat cells activate NF-κB
through phosphorylation of the inhibitor IκBα on Y42, which triggers its calpain-mediated degradation and allows the freed NF-κB to migrate into the nucleus [36, 37]. On the contrary, Jurkat expressing SHIP-1 activate NF-κB through IκB Kinase (IKK)
activation and phosphorylation of the
inhibitor IκBα on S32 and 36 upon H2O2
stimulation. Cell lines that express SHIP-1 naturally (i.e. CEM or the subclone Jurkat JR) also follow the same signalling pathway [38]. Importantly, we have also shown that Jurkat expressing SHIP-1 are much more resistant to H2O2 and
FasL-induced apoptosis than the parental cells ([38] and manuscript under preparation). This result may appear discrepant with previous data obtained with other cell types proposing that SHIP-1 increases apoptosis by antagonising the anti-apoptotic function of PI3K. Indeed, a proapoptotic role of SHIP-1 was described in several cell types, including B, myeloid and erythroid cells [11, 14, 39-41]. On the contrary, some authors have reported that SHIP-1 recruitment attenuates B cell apoptosis induced by FcγRIIB independently of BCR coligation, and that failure to recruit SHIP-1 increases the
apoptotic phenotype [42, 43].
Furthermore, it is worth noting that SHIP-1 deficient mice are lymphopenic [10, 11, 44], suggesting that, depending on the cell-type and the biological context, SHIP-1 can exert either pro- or anti-apoptotic functions (Table I).
Cell type Apoptosis inducer Role of SHIP-1 References
cycloheximide, anisomycin A, anti-Fas antibody, TNF-α
Erythroid cells EPO deprivation Pro-apoptotic [39]
B cells Aggregation of the
BCR Pro-apoptotic [14] Activin/TGF-β Pro-apoptotic [41] Clustering of FcγRIIB independently of BCR coligation Anti-apoptotic [42] T cells H2O2 Anti-apoptotic [38]
Table I: Pro- or anti-apoptotic role of SHIP-1 in different hemopoietic cells and biological conditions.
Data obtained from primary T cells
As described above, generation of SHIP-1 deficient mice have considerably increased our understanding of its biological role. These mice have notably revealed that SHIP-1 does not function similarly in the lymphoid than in the myeloid lineage. Indeed, whereas SHIP-1 KO mice have more myeloid cells than wt mice, they exhibit a reduction in the T cell population [10, 11]. An elegant study carried out by the Rothman’s group recently addressed SHIP-1’s role in primary mouse T cells [44]. These authors have demonstrated that, like in B cells, SHIP-1 appears to play a biphasic role in T
cell physiology. First, SHIP-1, together with the adapter protein Dok-1, plays a key role in T cell development, since mice
deficient for both SHIP-1 and Dok-1 exhibit an important reduction in the
percentage of CD4+ and CD8+ double
positive, together with an increased
number of CD4- and CD8- double
negative cells [44]. On the other hand, there is a reduced number of T cells in the spleen from SHIP-1 KO mice, due to a
severe reduction of CD8+ T cells,
suggesting that SHIP-1 is important in
maintaining CD4+/CD8+ ratio in the
periphery. But the most intriguing feature
of CD4+ T cells from SHIP-1 KO mice is
that they are phenotypically and functionally regulatory T cells (Tregs) [44]. Indeed, splenic T cells from SHIP-1 KO mice are activated, but do not respond to TCR stimulation or produce cytokines in response to PMA stimulation. They exhibit furthermore Tregs markers (such as high TGFβ production and expression of the transcription factor
Foxp3) and are able to suppress IL-2 production by CD4+ CD25- T cells upon
CD3 stimulation. T cells from mice deficient for both SHIP-1 and Dok-1 share the same phenotype, highlighting the crucial role of the adaptor Dok-1 for SHIP-1 activity [44]. An analogous cooperation of Dok-1 with SHIP-1 has already been demonstrated in FcγRIIB signalling in B cells, where SHIP-1 serves as an adaptor protein to recruit Dok-1 to the receptor, which in turn, inhibits the Ras/MAPK pathway [45].
Altogether, these data prompt us to conclude that SHIP-1 functions differently in T cells than in myeloid or B cells for at least three reasons: i) SHIP-1 does not seem to inhibit T cells proliferation (at
least primary T cells), but instead its absence creates an anergy to TCR stimulation. ii) Whereas SHIP-1 is clearly a pro-apoptotic protein in myeloid and, under certain conditions, in B cells, it appears to play an anti-apoptotic role in T cell lines. iii) Moreover, the fact that T cell specifc loss of PTEN produces some opposite phenotypes to SHIP-1 KO (i.e. T cells hyperproliferation) suggests that the signalling role of SHIP-1 in T cells is not restricted to its lipid phosphatase activity with respect to PtdIns(3,4,5)P3, the
common substrate of the two enzymes [46]. The mechanisms underlying these opposite roles are currently under investigation in our laboratory.
AKNOWLEDGEMENTS
This work was supported by grants from the Belgian National Fund for Scientific Research (FNRS, Brussels, Belgium), the Interuniversity Attraction Pole (IAP5/12, Brussels, Belgium), and the ARC program (ULG 04/09-323, Belgium). GG is a PhD student from the Télévie program (FNRS, Belgium), EC is supported by the FRIA (FNRS, Brussels) and JP is Research Director from the FNRS.
REFERENCES
1 Krystal, G. (2000) Semin Immunol
12, 397-403
2 Ward, S. G. (2006) Trends
Immunol 27, 80-7
3 Ward, S. G. and Cantrell, D. A.
(2001) Curr Opin Immunol 13, 332-8
4 Abraham, E. (2005) Crit Care Med
33, S420-2
5 Stambolic, V., Suzuki, A., de la
Pompa, J. L., Brothers, G. M., Mirtsos, C., Sasaki, T., Ruland, J., Penninger, J. M., Siderovski, D. P. and Mak, T. W. (1998) Cell 95, 29-39
6 Damen, J. E., Liu, L., Rosten, P.,
Humphries, R. K., Jefferson, A. B., Majerus, P. W. and Krystal, G. (1996) Proc Natl Acad Sci U S A
93, 1689-93
7 Backers, K., Blero, D., Paternotte,
N., Zhang, J. and Erneux, C. (2003) Adv Enzyme Regul 43, 15-28
8 Pesesse, X., Backers, K., Moreau, C., Zhang, J., Blero, D., Paternotte, N. and Erneux, C. (2006) Adv Enzyme Regul
9 Rauh, M. J., Kalesnikoff, J.,
Hughes, M., Sly, L., Lam, V. and Krystal, G. (2003) Biochem Soc Trans 31, 286-91
10 Helgason, C. D., Damen, J. E.,
Rosten, P., Grewal, R., Sorensen, P., Chappel, S. M., Borowski, A., Jirik, F., Krystal, G. and
Humphries, R. K. (1998) Genes Dev 12, 1610-20
11 Liu, Q., Sasaki, T., Kozieradzki, I.,
Wakeham, A., Itie, A., Dumont, D. J. and Penninger, J. M. (1999) Genes Dev 13, 786-91
12 Luo, J. M., Yoshida, H., Komura,
S., Ohishi, N., Pan, L., Shigeno, K., Hanamura, I., Miura, K., Iida, S., Ueda, R., Naoe, T., Akao, Y., Ohno, R. and Ohnishi, K. (2003) Leukemia 17, 1-8
13 Brauweiler, A. M., Tamir, I. and
Cambier, J. C. (2000) Immunol Rev 176, 69-74
14 Brauweiler, A., Tamir, I., Dal
Porto, J., Benschop, R. J.,
Helgason, C. D., Humphries, R. K., Freed, J. H. and Cambier, J. C. (2000) J Exp Med 191, 1545-54
15 Helgason, C. D., Kalberer, C. P.,
Damen, J. E., Chappel, S. M., Pineault, N., Krystal, G. and Humphries, R. K. (2000) J Exp Med 191, 781-94
16 Liu, Q., Oliveira-Dos-Santos, A. J.,
Mariathasan, S., Bouchard, D., Jones, J., Sarao, R., Kozieradzki, I., Ohashi, P. S., Penninger, J. M. and Dumont, D. J. (1998) J Exp Med
188, 1333-42
17 Muta, T., Kurosaki, T., Misulovin,
Z., Sanchez, M., Nussenzweig, M. C. and Ravetch, J. V. (1994) Nature
369, 340
18 Scharenberg, A. M., El-Hillal, O.,
Fruman, D. A., Beitz, L. O., Li, Z., Lin, S., Gout, I., Cantley, L. C.,
Rawlings, D. J. and Kinet, J. P. (1998) Embo J 17, 1961-72
19 Aman, M. J., Lamkin, T. D.,
Okada, H., Kurosaki, T. and Ravichandran, K. S. (1998) J Biol Chem 273, 33922-8
20 Bolland, S., Pearse, R. N.,
Kurosaki, T. and Ravetch, J. V. (1998) Immunity 8, 509-16
21 Ono, M., Bolland, S., Tempst, P.
and Ravetch, J. V. (1996) Nature
383, 263-6
22 Huber, M., Helgason, C. D.,
Damen, J. E., Liu, L., Humphries, R. K. and Krystal, G. (1998) Proc Natl Acad Sci U S A 95, 11330-5
23 Gimborn, K., Lessmann, E.,
Kuppig, S., Krystal, G. and Huber, M. (2005) J Immunol 174, 507-16
24 Lamkin, T. D., Walk, S. F., Liu, L.,
Damen, J. E., Krystal, G. and Ravichandran, K. S. (1997) J Biol Chem 272, 10396-401
25 Edmunds, C., Parry, R. V.,
Burgess, S. J., Reaves, B. and Ward, S. G. (1999) Eur J Immunol
29, 3507-15
26 Osborne, M. A., Zenner, G.,
Lubinus, M., Zhang, X., Songyang, Z., Cantley, L. C., Majerus, P., Burn, P. and Kochan, J. P. (1996) J Biol Chem 271, 29271-8
27 Jensen, W. A., Marschner, S., Ott,
V. L. and Cambier, J. C. (2001) Biochem Soc Trans 29, 840-6
28 Mazerolles, F., Barbat, C., Trucy,
M., Kolanus, W. and Fischer, A. (2002) J Biol Chem 277, 1276-83
29 Bruyns, C., Pesesse, X., Moreau,
C., Blero, D. and Erneux, C. (1999) Biol Chem 380, 969-74
30 Sakai, A., Thieblemont, C.,
Wellmann, A., Jaffe, E. S. and Raffeld, M. (1998) Blood 92, 3410-5
31 Astoul, E., Edmunds, C., Cantrell,
D. A. and Ward, S. G. (2001) Trends Immunol 22, 490-6
32 Freeburn, R. W., Wright, K. L.,
D. A. and Ward, S. G. (2002) J Immunol 169, 5441-50
33 Horn, S., Endl, E., Fehse, B.,
Weck, M. M., Mayr, G. W. and Jucker, M. (2004) Leukemia 18, 1839-49
34 Garcia-Palma, L., Horn, S., Haag,
F., Diessenbacher, P., Streichert, T., Mayr, G. W. and Jucker, M. (2005) Br J Haematol 131, 628-31
35 Fukuda, R., Hayashi, A.,
Utsunomiya, A., Nukada, Y., Fukui, R., Itoh, K., Tezuka, K., Ohashi, K., Mizuno, K., Sakamoto, M., Hamanoue, M. and Tsuji, T. (2005) Proc Natl Acad Sci U S A
102, 15213-8
36 Takada, Y., Mukhopadhyay, A.,
Kundu, G. C., Mahabeleshwar, G. H., Singh, S. and Aggarwal, B. B. (2003) J. Biol. Chem. 278, 24233-24241
37 Schoonbroodt, S., Ferreira, V.,
Best-Belpomme, M., Boelaert, J. R., Legrand-Poels, S., Korner, M. and Piette, J. (2000) J Immunol
164, 4292-300
38 Gloire, G., Charlier, E., Rahmouni,
S., Volanti, C., Chariot, A., Erneux, C. and Piette, J. (2006) Oncogene
39 Boer, A. K., Drayer, A. L. and
Vellenga, E. (2001) Leukemia 15, 1750-7
40 Gardai, S., Whitlock, B. B.,
Helgason, C., Ambruso, D., Fadok,
V., Bratton, D. and Henson, P. M. (2002) J Biol Chem 277, 5236-46
41 Valderrama-Carvajal, H.,
Cocolakis, E., Lacerte, A., Lee, E. H., Krystal, G., Ali, S. and Lebrun, J. J. (2002) Nat Cell Biol 4, 963-9
42 Pearse, R. N., Kawabe, T., Bolland,
S., Guinamard, R., Kurosaki, T. and Ravetch, J. V. (1999) Immunity 10, 753-60
43 Ono, M., Okada, H., Bolland, S.,
Yanagi, S., Kurosaki, T. and Ravetch, J. V. (1997) Cell 90, 293-301
44 Kashiwada, M., Cattoretti, G.,
McKeag, L., Rouse, T., Showalter, B. M., Al-Alem, U., Niki, M., Pandolfi, P. P., Field, E. H. and Rothman, P. B. (2006) J Immunol
176, 3958-65
45 Tamir, I., Stolpa, J. C., Helgason,
C. D., Nakamura, K., Bruhns, P., Daeron, M. and Cambier, J. C. (2000) Immunity 12, 347-58
46 Suzuki, A., Yamaguchi, M. T.,
Ohteki, T., Sasaki, T., Kaisho, T., Kimura, Y., Yoshida, R.,
Wakeham, A., Higuchi, T.,
Fukumoto, M., Tsubata, T., Ohashi, P. S., Koyasu, S., Penninger, J. M., Nakano, T. and Mak, T. W. (2001) Immunity 14, 523-34