Pour l'obtention du grade de
DOCTEUR DE L'UNIVERSITÉ DE POITIERS UFR des sciences fondamentales et appliquées
Institut de chimie des milieux et matériaux de Poitiers - IC2MP (Diplôme National - Arrêté du 7 août 2006)
École doctorale : Sciences pour l'environnement - Gay Lussac (La Rochelle) Secteur de recherche : Chimie organique, minérale et industrielle
Présentée par :
Amélie Martin
Utilisation de conditions superacides pour la mise en
évidence d'intermédiaires réactionnels glycosidiques inédits
Directeur(s) de Thèse : Yves Blériot, Sébastien Thibaudeau Soutenue le 10 novembre 2015 devant le jury
Jury :
Président Vincent Sol Professeur des Universités, Université de Limoges
Rapporteur Jean-Marie Beau Professeur des Universités, Université Paris Sud 11, Orsay
Rapporteur Matthieu Sollogoub Professeur des Universités, Université Pierre et Marie Curie, Paris 6 Membre Yves Blériot Professeur des Universités, Université de Poitiers
Membre Sébastien Thibaudeau Professeur des Universités, Université de Poitiers
Membre Thierry Brigaud Professeur des Universités, Université de Cergy-Pontoise
Pour citer cette thèse :
Amélie Martin. Utilisation de conditions superacides pour la mise en évidence d'intermédiaires réactionnels
glycosidiques inédits [En ligne]. Thèse Chimie organique, minérale et industrielle. Poitiers : Université de Poitiers,
THÈSE
Pou àl o te tio àduàg adeàde
DOCTEU‘àDEàL UNIVERSITÉ DE POITIERS
(Faculté des Sciences Fondamentales et Appliquées)
(Diplôme National – Arrêté du 7 août 2006)
Ecole doctorale S ie esàpou àl e i o e e tà– Gay-Lussac Secteur de recherche : Chimie Organique, Minérale et Industrielle
Présentée par
Amélie MARTIN
UTILISATION DE CONDITIONS SUPERACIDES POUR LA
MISE EN É
VIDENCE D’INTERMÉDIAIRES RÉACTIONNELS
GLYCOSIDIQUES INÉDITS
Soutenance le 10 Novembre 2015deva t la Co issio d’E a e
Pr Vincent SOL Professeur, Université de Limoges Président Pr. Jean-Marie BEAU Professeur, Université Paris-sud 11, Orsay Rapporteur Pr. Matthieu SOLLOGOUB Professeur, UPMC, Sorbonne Université Rapporteur Pr. Thierry BRIGAUD Professeur, Université de Cergy-Pontoise Examinateur Pr. Yves BLÉRIOT Professeur, Université de Poitiers Directeur de thèse Pr. Sébastien THIBAUDEAU Professeur, Université de Poitiers Directeur de thèse
‘EME‘CIEMENTS
Avant de présenter les travaux qui ont conduit à la réalisation de ce manuscrit je tiens à remercier toutesàlesàpe so esà uiào tà o t i u à àl a outisse e tàdeà eàt a ail.
J e p i eà aàp ofo deàg atitudeàauàP .àY esàBLE‘IOTàetàauàP .àS astie àTHIBáUDEáU,àtousàdeu à p ofesseu sà àl u i e sit àdeàPoitie sà uià o tàpe isàdeà éaliser et de mener à bien ce projet de th se.àJ aià eau oupàapp isà à osà ot sàetàjeàtie sà à ousà e e ie àpou àlaà o fia eà ueà ousà a ezà accordée durant ces trois dernières années.
J ad esseà gale e tà esàsi esà e e ie e tsàauàP .àJea -Marie BEAU,àp ofesseu à àl u i e sit à de Paris-Sud,à età auà P .à Matthieuà SOLLOGOUB,à p ofesseu à à l u i e sit à Pie eà età Ma ieà Cu ie-Sorbonne université,àpou àa oi àa ept àd t eà appo teu sàdeà esàt a au àdeàth se.
Je tiens également à remercier le Pr. Thierry BRIGAUD,à di e teu à deà l u it à deà ‘e he heà LCB à l u i e sit àdeàCe g -Pontoise et le Pr. Vincent SOL, directeur du LCSN àl u i e sit àdeàLi oges,àdeà
a oi àfaitàl ho eu àd a epte àdeàjuge à eàt a ail.
Je remercie tous ceux qui ont collaboré à la réalisation de ce projet de thèse : pour toute la partie modélisation moléculaire, le Pr. Jésús JIMENEZ-BARBERO, di e teu à s ie tifi ueà deà l i stitutà deà biologie CIC-BioGUNE à Bilbao (Espagne) et le Dr. Ana ARDA, chargée de recherche. De plus, je les remercie chaleureuse e tàpou àl a ueilà u ils o tà se àlo sàd u àstageàdeà uel uesàse ai esà au sein de leur institut et des bons moments partagés autour de délicieuses tapas. Je remercie également le Dr. Agnès MINGOT pour avoir été à mes côtés tout au long de ma thèse et tous nos échanges, notamment lo sàdesàa al sesà‘MN.àBie àplusà u u merci au Dr. Jérôme DESIRE pour son implication dans ce projet, pour son aide précieuse pour les synthèses et surtout pour toutes nos conversations quotidiennes en fin de journée et autres.
U à g a dà e ià à tousà esà oll guesà età a isà pou à laà o i ialit à età laà o eà hu eu à u ilsà o tà apportées à mon quotidien ; à mes copines Vivi, Fati, Toto etàCo aàpou àtousàlesà o sà o e tsà u o à a passés ensemble (sans oublier nos séances sportives) ; à mes camarades : NoNo, Anne-Ju, Alex, et Ugoà pou à laà o eà a ia eà u ilsà o tà a e e au labo ; à Jaufret pour sa gentilesse et ses imitations.
U à e iàpa ti ulie à àJoëlleàpou àl aideàp ieuseà u elleàaàpuà appo te àdu a tà esàt oisàa esà;à sans oublier Brigitte pour notre complicité mère-fille, à Isa pour son aide et sa bonne humeur surtout le vendredi matin, et à Patrick pour ses talents de mécanicien ; Jérôme G., Séb P., et Hélène pour la s pathieà u ilsào tàpuà eàt oig e .
Un merci à mes amis Claire et Sé ,àlesà ou eau àpa e tsàd u àpetitàJules, Camille, Ju et Alexis pour osàsou e i sàe eig sàetà ie àd aut es… ; à esàa isàd e fa e,àMa ieàetàFlo.
U àg a dà e ià à esàa isà eto sàetàENSCMulie sà esà o pag o sàd a e tu eàdans mes études et plus particulièrement à Jean-Gui, Titi, Cok, Anne et Morguy.
Un merci aussi à ma belle et grande famille sur qui je peux compter. Merci pour tous ces bons et merveilleux moments passés lors des réunions de famille. Je tiens à remercier Fanfan et Domi pour leu à te elleàjoieàdeà i eàetàgo tàpou àl a e tu eà u ils partagent par des anecdotes et présents.
Bie àplusà u u àmerci à mes grand-parents de Bazouges, à Mamie et bien sur Jean-Paul,àluià uià aà toujours soutenue dans mes choix.
Je ne saurais trop remercier mes pa e ts,à esàh os,à uià o t cessé de croire en moi. Merci pour votre amour, votre soutien et votre présence dans les moments de joie et très récemment de peine. G eà à ousà j aià puà f a hi à lesà o sta lesà età de e i à laà pe so eà ueà jeà suisàaujou d hui.à Jeà ousà
e e ieà gale e tàpou à a oi àapp isà àesti e àlaà ha eà ueàj ai.
U à o eà e ià à aàsœu ,àC li e,àpou àto àa ou àetàtaàp se e.àJeàsuisàfi eàdeà ot eà o pli it à et de pouvoir être là pour toi, eàsiàsou e tà est l i e se. A Lolo et Aline pour tous nos moments ensemble et surtout autour « des aventuriers du rail ». A mon frère, Nico, pour ton soutien et pour l ho eu à ue tu me fais de devenir Tata d u eàpetiteàád le.
A toi, Guigui, ma moitié, pour ton amour, ton soutien de tous lesàjou sàdepuisàta tàd a es,àto àaideà da sàlesà o e tsàdiffi iles,àto àg a dà œu ,àtaàpatie e,àto à ou age…et tellement plus…
A
iatio s
Dans ce manuscrit, plusieurs abréviations seront utilisées dont les significations sont présentées ci-dessous : Pou des aiso s p ati ues e tai s te es a glais ’o t pas t t aduits e f a çaisA
Ac : Acétyle ACN : Acétonitrile
Aq. : aqueous Ar : Aromatique
B
B : Conformation Bateau Bn : Benzyle
Bu : Butyle
C
C : Conformation Chaise CAN : Nitrate de cérium et d'ammonium
CCM : Chromatographie sur Couche Mince CID : Collision-Induced dissociation
CIP : Contact Ion Pair COSY : Correlation Spectroscopy
D
d : Doublet δ : Déplacement chimique
DAST : Trifluorure de diéthylaminosulfure DBU : 1,8-diaza-7-bicyclo-undécène
DCM : Dichlorométhane dd : Doublet de doublet
ddd : Doublet de doublet dédoublé dddd : Doublet de doublet de doublet dédoublé
ddt : Doublet de doublet de triplet DEPT : Distortionless Enhancement by Polarisation Transfert
DFT : Density Functional Theory DMF : N,N-Diméthylformamide
DMP : Périodinane de Dess-Martin dq : Doublet de quadruplet
dt : Doublet de triplet DTBMP : 2,6-di-tert-butyl-4-méthylpyridine
E
E : Conformation Enveloppe EI : Electronic Impact
EIC : Effet Isotopique Cinétique ELSD: Evaporative Light Scattering Detector
Eq. : Equivalent Et : Ethyle
EtOAc :àá tateàd Eth le
G
Glc : Glucose Gal : Galactose
H
H : Conformation demi-chaise H0 :àFo tio àd a idit àdeàHa et
HRMS : High Resolution Mass Spectrometry HSQC : Heteronuclear Single Quantum Coherence
M
m : Multiplet Man : Mannose
Me : Méthyle MS : Mass Spectrometry
N
NBO : Natural Bond Orbital NBS : N-bromosuccinimide
NCS : N-chlorosuccinimide NIS : N-iodosuccinimide
NMR : Nuclear Magnetic Resonance
P
Ph : Phényl Phth : Phtalimide
Pr : Propyle
Q
Quant. : quantitatif
R
Rf : Retention Factor RMN : Résonance Magnétique Nucléaire
Rt : Room temperature
S
s : Singulet S : Conformation bateau-croisé
Sat. : Saturated SN : Substitution Nucléophile
SSIP : Solvent Separated Ion Pair
T
t : Triplet T : Conformation Twist
T.A. : Température Ambiante TBDMS : tert-Butyldiméthylsilyle
TBAF : Fluorure de tétra-N-butylammonium Tf : Triflate
TFA : Trifluoroacétamide THF : Tétrahydrofurane
TMS : Triméthylsilyle TLC : Thin Layer Chromatography
TTBP : 2,4,6-tri-tert-butylpyrimidine
U
UV : Ultraviolet
X
Ta le des ati es
Introduction générale
1
Les glycosciences : de la chimie à la biologie __________________________________ 3
2
La glycosylation _________________________________________________________ 4
2.1 Généralités ______________________________________________________________ 4 2.2 Les différentes méthodes de O-glycosylation ___________________________________ 5
2.2.1 Les pionniers ____________________________________________________________________ 5
2.2.2 Nouvelles méthodes de O-glycosylation ______________________________________________ 6
2.3 Le mécanisme de la glycosylation ____________________________________________ 7
2.3.1 Différents types de mécanismes _____________________________________________________ 7 2.3.2 Deux voies réactionnelles __________________________________________________________ 8
2.4 Fa teu s i flue ça t l’effi a it et la st os le tivit de la gl os latio ____________ 9
2.4.1 L effetàa o e __________________________________________________________________ 9
2.4.2 La structure du donneur de glycosyle _______________________________________________ 10 2.4.3 L effetàdeàsol a t ________________________________________________________________ 11
3
Etat de l a t su l e iste e d u atio gl os le _____________________________ 12
3.1 Etude des intermédiaires cationiques ________________________________________ 12
3.1.1 Historique _____________________________________________________________________ 12
3.1.2 Les ions alkoxycarbénium en milieu superacide _______________________________________ 14
3.1.3 G atio àd io sàalko a iu àpa ào datio à le t o hi i ue _______________________ 14
3.1.4 Laàdu eàdeà ieàdeàl io ào o a iu ______________________________________________ 17
3.2 Etude du mécanisme de la glycosylation par mesure des effets isotopiques cinétiques 17
3.2.1 Laà otio àd effetàisotopi ueà i ti ue _______________________________________________ 17 3.2.2 Les effets isotopiques cinétiques et la réaction de glycosylation __________________________ 19
3.3 Etude o fo atio elle du atio gl os le à l’aide de al uls th o i ues __________ 23
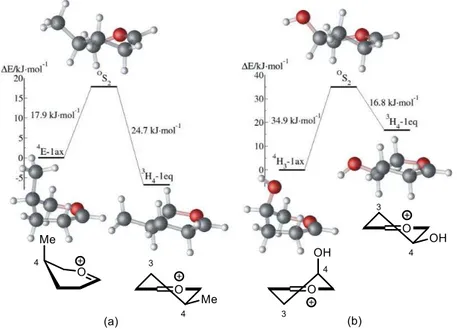
3.3.1 Etudeà o fo atio elleàd io sàgl os lesào o a iu à o àsu stitu s __________________ 23 3.3.2 Etudeà o fo atio elleàd io sào o a iu à o o-, di- ou tri-substitués _________________ 24
3.4 Mise en évidence du cation glycosyle par spectrométrie de masse _________________ 30 3.5 Etudes des intermédiaires réactionnels de la glycosylation par RMN à basse température
31
3.5.1 L i te diai eà atio à a op a os leàsta ilis àpa àl io àt iflate ________________________ 32
4
Les milieux superacides __________________________________________________ 35
4.1 Les origines _____________________________________________________________ 35 4.2 Acidité des milieux superacides _____________________________________________ 36 4.3 Structure et propriétés de HF anhydre et du mélange HF/SbF5 ____________________ 37
4.3.1 HF anhydre ____________________________________________________________________ 37
4.3.2 Mélange HF/SbF5 ________________________________________________________________ 37
4.4.1 Les espèces transitoires en milieu superacide _________________________________________ 38 4.4.2 Les réactions en milieu superacide __________________________________________________ 40
4.5 Les superacides et les sucres _______________________________________________ 43
4.5.1 Les superacides usuels ___________________________________________________________ 43 4.5.2 Cyclisation intramoléculaire en milieu superacide _____________________________________ 43 4.5.3 Dépolymérisation de la cellulose en milieu superacide __________________________________ 45
5
Deux chimies
– Deu te h i ues d a al se __________________________________ 46
Résultats et discussion
Chapitre 1
1
Mise au point de la méthode _____________________________________________ 51
1.1 Mat iels et i st u e ts d’a alyse __________________________________________ 51
1.1.1 Réaction en milieu superacide _____________________________________________________ 51
1.1.2 Analyse par RMN ________________________________________________________________ 51
1.2 Mise en place du protocole ________________________________________________ 52
1.2.1 Conditions opératoires ___________________________________________________________ 52 1.2.2 Analyse par RMN in situ à basse température _________________________________________ 52 1.2.3 Calculs théoriques : analyse conformationnelle _______________________________________ 53
2
Preuve du concept ______________________________________________________ 54
2.1 Dénomination des ions carboxonium/oxocarbénium____________________________ 54 2.2 E e ples d’io s alko a iu li ues a a t is s ________________________ 54
2.2.1 Fo atio àd u àio ào o a iu àe à ilieuàsupe a ide ________________________________ 54
2.2.2 Fo atio àd u àio ào o a iu àpa ào datio à le t o hi i ue ________________________ 55
2.3 G atio d’u io o o a iu e ilieu supe a ide ________________________ 56
2.3.1 Synthèse et étude de la réactivité en milieu superacide _________________________________ 56
Chapitre 2
1
Fo atio d esp es pol p oto es sta les e ilieu supe a ide ________________ 61
1.1 Evaluation de la réactivité du 1,2,3,4,6-penta-O-acétyl-α-D-glucopyranose __________ 61
1.1.1 Etude RMN en milieu superacide ___________________________________________________ 61 1.1.2 Analyse conformationnelle ________________________________________________________ 63
1.2 Evaluation de la réactivité du 2-acétamido-1,3,4,6-tétra-O-acétyl-2-déoxy-
α-D-glucopyranose _________________________________________________________________ 64
1.2.1 Synthèse et étude en milieu superacide _____________________________________________ 64 1.2.2 Analyse conformationnelle ________________________________________________________ 66
1.3 Evaluation de la réactivité du 1,3,4,6-tétra-O-acétyl-2-déoxy-2-trifluoroacétamido- α-D-glucopyranose _________________________________________________________________ 67
1.3.1 Synthèse et étude en milieu superacide _____________________________________________ 67 1.3.2 Analyse conformationnelle ________________________________________________________ 69
2
G
atio et a al se d io s gl os les sta ilis s pa pa ti ipatio d u g oupe e t
voisin ____________________________________________________________________ 70
2.1 Etude de l’io dio al iu _________________________________________________ 70
2.1.1 Découverte et première caractérisation _____________________________________________ 70 2.1.2 Caractérisation des ions dioxalénium générés en milieu superacide HF/SbF5 ________________ 72
2.2 Les ions oxazolinium ______________________________________________________ 80
2.2.1 Découverte et première caractérisation _____________________________________________ 80 2.2.2 Evaluation de la réactivité du 2-acétamido-3,4,6-tri-O-acétyl-2-déoxy-1-fluoro-α-D-glucopyranose
81
2.2.3 Evaluation de la réactivité du 1,3,4,6-tétra-O-acétyl-2-trifluoroacétamido-2-déoxy-
β-D-glucopyranose _________________________________________________________________________ 84
Chapitre 3
1
Evaluation de la réactivité de la 1,3,4,6-tétra-O-acétyl-
β-
D-glucosamine en milieu
superacide ________________________________________________________________ 91
1.1 Synthèse et étude en milieu superacide ______________________________________ 91 1.2 Analyse conformationnelle ________________________________________________ 92
2
Evaluation de la réactivité du 3,4,6-tétra-O-acétyl-2-azido-2-déoxy-1-fluoro-
D-glucopyranose _____________________________________________________________ 92
2.1 Synthèse et étude en milieu superacide ______________________________________ 93 2.2 Analyse conformationnelle ________________________________________________ 96
3
Evaluation de la réactivité du 3,4,6-tri-O-acétyl-1-fluoro-
D-glucopyranose _________ 97
3.1 Synthèse _______________________________________________________________ 97 3.2 Etude en milieu superacide ________________________________________________ 98
4
Evaluation de la réactivité du 3,4,6-tri-O-acétyl-2-O-méthyl-1-fluoro-
D-glucopyranose
100
4.1 Synthèse ______________________________________________________________ 100 4.2 Réactivité en milieu superacide ____________________________________________ 100
5
Etude de la réactivité du 3,4,6-tri-O-acétyl-2-O-benzyl-1-fluoro-
D-glucopyranose __ 102
5.1 Synthèse ______________________________________________________________ 102 5.2 Etude en milieu superacide _______________________________________________ 102
Chapitre 4
1
Etude des dérivés 2-déoxy-
D-glycopyranose en milieu superacide _______________ 105
1.1 Introduction ___________________________________________________________ 105 1.2 Evaluation de la réactivité du 1,3,4,6-tétra-O-acétyl-2-déoxy-β-D-glucopyranose ____ 105
1.2.1 Synthèse et étude en milieu superacide ____________________________________________ 105 1.2.2 Analyse conformationnelle _______________________________________________________ 107
1.3 Evaluation de la réactivité du 1,3,4,6-tétra-O-acétyl-2-déoxy-D-galactopyranose ____ 108
1.3.1 Synthèse et étude en milieu superacide ____________________________________________ 108 1.3.2 Analyse conformationnelle _______________________________________________________ 109
2
Etude des dérivés 2-déoxy-2-halogéno-D-gluco-pyranoses en milieu superacide ___ 110
2.1 Introduction ___________________________________________________________ 110 2.2 Evaluation de la réactivité du 3,4,6-tri-O-acétyl-2-bromo-2-déoxy-1-fluoro-β-D
-glucopyranose ________________________________________________________________ 111
2.2.1 Synthèse et étude en milieu superacide ____________________________________________ 111 2.2.2 Analyse conformationnelle _______________________________________________________ 112
2.3 Evaluation de la réactivité du 3,4,6-tri-O-acétyl-2-chloro-2-déoxy-1-fluoro-β-D
-glucopyranose ________________________________________________________________ 114
2.3.1 Synthèse et réactivité en milieu superacide _________________________________________ 114 2.3.2 Analyse conformationnelle _______________________________________________________ 115
2.4 Evaluation de la réactivité des dérivés 2-déoxy-2-fluoro-D-glucopyranoses _________ 116
2.4.1 Synthèse et réactivité en milieu supeacide __________________________________________ 116 2.4.2 Analyse conformationnelle _______________________________________________________ 117
2.5 Réactivité du dérivé 1,3,4,6-tétra-O-acétyl-2-déoxy-2-iodo-β-D-glucopyranose en milieu superacide ___________________________________________________________________ 121
2.5.1 Synthèse et étude en milieu superacide ____________________________________________ 121 2.5.2 Analyse conformationnelle _______________________________________________________ 122
Chapitre 5
1
Modification des conditions expérimentales ________________________________ 125
1.1 Influence de la température su la sta ilit de l’io o o a iu _______________ 125 1.2 Sta ilit de l’io o o a iu au ou s du te ps ____________________________ 125 1.3 Influence de la composition du milieu sur la stabilité cation glycosyle _____________ 126
2
Piégeage in situ des cations glycosyles ____________________________________ 127
2.1 Méthanolyse ___________________________________________________________ 127 2.2 Deutération ____________________________________________________________ 128
Partie expérimentale
1
Outils et méthodes ____________________________________________________ 137
1.1 Matériels ______________________________________________________________ 137 1.2 Suivi de réaction et purifications ___________________________________________ 137 1.3 Outils analytiques _______________________________________________________ 137 1.4 Manipulation de HF et SbF5 : préparation du mélange HF/SbF5 ___________________ 137 1.5 Procédure générale et analyses RMN in situ à basse température ________________ 138 1.6 La partie expérimentale __________________________________________________ 138
2
Réactifs de départ et intermédiaires réactionnels ____________________________ 139
2.1 Chapitre 1 _____________________________________________________________ 139 2.2 Chapitre 2 _____________________________________________________________ 141
2.3 Chapitre 3 _____________________________________________________________ 160 2.4 Chapitre 4 _____________________________________________________________ 180 2.5 Chapitre 5 _____________________________________________________________ 196
Annexes
1
Fiche Sécurité
: Pe taflu o u e d a ti oi e ________________________________ 205
2
Fi he s u it de l a ide fluo h d i ue _____________________________________ 206
3
Références des logiciels Maestro et Gaussian _______________________________ 208
1 Les glycosciences : de la chimie à la biologie
Les sucres ou hydrates de carbone sont omniprésents et essentiels pour toute vie sur Terre.àD u eà part, dans le règne végétal la réaction la plus importante est la photosynthèse. En effet, les plantes, les algues et tout autre organisme similaire utilisent l e gieàsolai eàpou àt a sfo e àleàdio deàdeà a o eàetàl eauàe à o pos sàgl osidi ues.àD aut eàpa t,àleà ta olis eà deàl ho e etàd aut esà organismes vivants utilisent cette source glycosidique comme énergie transformant ainsi le glucose en dioxyde de carbone et en eau permettant de réaliser à nouveau la photosynthèse.1
Les glycosciences, qui regroupent toutes les disciplines étudiant les glycanes, sont sans cesse en plein essor. Les champs d appli atio àdesàgl os ie ces vont de la conception de nouveaux matériaux, au développement de nouvelles énergies renouvelables jus u au domaine de la santé, notamment avec l tudeàduà ôle des sucres et de leurs conjugués en biologie et leur exploitation en médecine (Figure 1).
Figure 1 : Do ai es d appli atio des gl os ie es
Ceàdo ai eàdeà e he heà àl i te fa eàdeàlaà hi ieàet de la biologie aà ess àdeà oit eàauà ou sàdesà dernières décennies. C est ainsi que le terme glycobiologie est apparu à la fin des années 1980 eg oupa tàlaà hi ieàdesàsu es,àl e z ologie impliquée dans la formation et de la dégradation des glycanes, la reconnaissance de ces derniers par des protéines spécifiques, l tudeàde leurs rôles dans les systèmes biologiques et leur analyse complète par différentes techniques.2 Dans ce contexte, la glycochimie est la base essentielle de la glycobiologie et a fortiori des glycosciences. Le développement de nouvelles méthodes stéréosélectives et efficaces pour la formation de liaisons glycosidiques etàl o te tio àdeàgl a es complexes est donc un enjeu crucial.3
1 R.V. Stick, S. Williams, Carbohydrates: The Essentials Molecules of Life, Oxford, Elsevier.
2 Transforming Glycoscience: A Roadmap for the Future, 2012, Washington DC, National Academies Press. 3 T.J. Boltje, T. Buskas, G.J. Boons, Nat. Chem., 2009, 1, 611.
Santé
Etude des sucres en biologie Développement pharmaceutique
Énergies
Développement de nouvelles énergies renouvelablesMatériaux
Conception de nouveaux matériauxGLYCOSCIENCES
2 La glycosylation
2.1 Généralités
Les glycanes, des molécules organiques composées de sucres constituent avec les acides nucléiques et les protéines, une des grandes familles de macromolécules biologiques. Ces macromolécules se construisent à partir de monomères : acides aminés pour les protéines, nucléotides pour les acides nucléiques et monosaccharides pour les glycanes (Figure 2).
Figure 2 : Trois types de macromolécules biologiques (droite) obtenues par polymérisation des monomères correspondants (gauche)
La polymérisation de ces monomères s effe tueàvia la création de liaisons covalentes présentant une certaine orientation. La nature des liaisons et la structure des unités qui composent la macromolécule sont porteuses d i fo atio à aisà toutes les molécules biologiques ne sont pas porteuses du eàpote tielàd i fo atio .àUn polysaccharide tel que la cellulose, o pos àd u eà unique unité répétitive -D-glucopyranose est pauvre en information. Par contre, la multitude de
monosaccharides présents dans la nature, liés de façon régio- et stéréosélective à d aut es o osa ha idesàouàd aut esà ol ules peut conduire à des macromolécules riches en information.4 Les glycanes englobent les polysaccharides (cellulose, chitine, amidon, glycogène…) et les glycoconjugués complexes (glycopeptides, glycolipides, glycoprotéines) dans lesquels des unités sucres sont jointes par des liaisons covalentes appelées liaisons glycosidiques.5 Cette liaison est le résultat de la réaction clé en glycochimie : la réaction de glycosylation.
Dans cette réaction, le donneur de glycosyle possède un substituant anomérique qui est transformé e à u à o à g oupeà pa ta tà pa à l a tio à d u à promoteur. Un accepteur de glycosyle (nucléophile) attaque ensuite le centre anomérique du donneur (électrophile) pour donner naissance à la liaison
4 R.H. Garret, C.M. Grisham, Biochimie 2ème édition, 1995, Bruxelles, De Boeck Université.
covalente qui caractérise le produit formé. Le plus souvent deux glycosides diastéréoisomères sont formés. Ils ne diffèrent que par la stéréochimie du carbone hemi-acétalique et sont nommés respectivement αà età β.à áà tit eà d e e ple,à lo sà d u eà a tio à deà gl cosylation avec du D -glu op a ose,à l a o e αà està a a t is à pa à u eà liaiso à a o i ueà a iale.à Da sà leà asà o à la liaiso àaàu eào ie tatio à uato iale,àilàs agitàdeàl a o eàβà(Schéma 1).
Schéma 1 : Principe de la réaction de glucosylation
E fi ,à laà atu eà duà g oupeà u l ophileà deà l a epteu à lors de la glycosylation conduit à une classification des glycosides. Les glycosides sont de type O-glycoside, N-glycoside, S-glycoside et C-glycoside. Selon la nature du glycoside cible, il existe de nombreuses méthodes de glycosylation à disposition et notamment pour effectuer des réactions de O-glycosylation.
2.2 Les différentes méthodes de O-glycosylation
2.2.1 Les pionniers
La première glycosylation a été réalisée par Mickaël6 en 1879 avec la synthèse de glycosides arylés à pa ti àd u à hlo u e de glycosyle. A la fin du XIXème siècle, Fischer7 a développé une méthode basée su à l utilisatio à d al ool comme accepteur e à p se eà d a ideà pe ettant d o te i à un mélange d a talsàdeàt peàfu a oseàetàp a ose. En 1901, Koenigs et Knorr8 ont mis au point une nouvelle méthode pour la formation de liaisons glycosidiques en utilisant un donneur de glycosyle de type halogénure a e à duà a o ateà d a ge tà o eà p o oteu .à E suite,à )emplen et Gerac9 puis Heilferich et Wedermeyer10 ont amélioré cette méthode en employant une catalyse hétérogène à base de sels de e u e.àL i o ie tà deà etteà thode,à ie à u effi a e,à està laà g atio à deà déchets toxiques. Enfin, la contribution de Lemieux11 a pe isàd ta li que la réactivité du donneur et la stéréosélectivité de la glycosylation corrèlent directement avec la nature des groupements protecteurs et plus particulièrement celui p ot gea tàl al ool en position 2.
6 A. Mickael, J. Am. Chem. Soc., 1879, 1, 305. 7 E. Fischer, Chem. Ber., 1893, 26, 2400.
8 W. Koenigs, E. Knorr, Chem. Ber., 1901, 34, 957. 9 G. Zemplen, A. Gerecs, Chem. Ber., 1930, 63, 2720.
10 B. Heilferich, K.F. Wedermeyer, Liebigs Ann. Chem., 1949, 563, 139. 11 R.U. Lemieux, Ad. Carbohydr. Chem. Biochem., 1954, 9, 1.
2.2.2 Nouvelles méthodes de O-glycosylation
A partir des années 1960, de nombreuses méthodes utilisant des donneurs de type bromure, chlorure, acétate ou hémiacétal sont développées pour accéder à de simples glycosides ou à des oligosaccharides complexes (Figure 3a).12
A partir des années 1970, le développement de nouvelles conditions réactionnelles exploitant de nouveaux substituants anomériques a permis la mise au point de glycosylations plus efficaces et stéréosélectives.
Figure 3 : Différents donneurs de glycosyle développés (a) de 1900 à 1970 (b) de 1970 à 1990 (c) de 1990 à 2015
Durant les années 1970 et 1980, de nouveaux donneurs sont utilisés. On peut citer les thioglycosides développés par Ferrier13 et Garreg14 ,les cyanoéthylidènes15 et les dérivésàd o thoeste sàexploités par Kochetkov, les O-imidates utilisés par Sinaÿ16, et Mukaiyama17 ou encore les glycosides fluorés popularisés par Schmidt18 (Figure 3b).
A partir des années 1990, une nouvelle vague de méthodes de glycosylation voit le jour permettant ainsi l a sà àdeàsi plesàgl osides ou à des glycanes de complexité croissante plus efficacement et de façon stéréosélective (Figure 3c).
Bie à u ilàe isteàu eà ultitude de méthodes de glycosylation et que cette réaction soit très répandue et appliquée, son mécanisme intime reste étonnamment un des moins bien maîtrisés dans le
12 A.V. Demchenko, Handbook of Chemical Glycosylation, 2008, John Wiley & Sons. 13 R.J. Ferrier, R.W. Hay, N. Vethaviyasar, Carbohydr. Res., 1973, 27, 55.
14 P.J. Garreg, Adv. Carbohydr. Chem. Biochem., 1997, 52, 179.
15 N.K. Kochetkov, L.V. Backinowsky, Y.E. Tsvetkov, Tetrahedron Lett., 1977, 41, 3681.
16 J.R. Pougny, J.C. Jacquinet, M. Nasser, D. Duchet, M.L. Milat, P. Sinaÿ, J. Am. Chem. Soc., 1977, 99, 6762. 17 T. Mukaiyama, T. Nakatsuka, S. Shoda, Chem. Lett., 1979, 8, 487.
domaine de la chimie organique selon Crich.19Deà eàfait,àl lu idatio àdesàd tailsàduà a is eàdeàlaà gl os latio à età l ide tifi atio à desà i te diai esà a tio elsà i pli u sà esteà u à th eà deà
e he heàd a tualit .
2.3 Le mécanisme de la glycosylation
2.3.1 Différents types de mécanismes
Il est communément accepté que la réaction de glycosylation peut faire appel à deux types de mécanismes dit associatif (SN2) ou dissociatif (SN1). Laà o de satio àd un donneur et d un accepteur de glycosyle débuteà pa àl a ti atio àduàg oupe e tàa o i ueà duàdo eu I par un électrophile adapté (E+). L esp eàa ti eàII peut ensuite subir une substitution de type SN2 par un nucléophile approprié, nommé accepteur de glycosyle (Schéma 2a).
Schéma 2 : (a) Mécanisme général de la réaction de glycosylation (b) Profils énergétiques pour les réactions de substitution bimoléculaire et unimoléculaire
áp sà leà d pa tà duà g oupe e tà pa ta t,à l io à II peut également conduire à la formation de l io à oxocarbénium III de type pai eàd io sàl hesà SSIP) ou pai eàd io sài ti esà CIP . Ce dernier peut alors être intercepté par le contre-io àdeàl le t ophileà(X-) pou àg e àl i te diai eàIV, dans lequel le groupe X est lié de façon covalente au carbone anomérique du donneur. L atta ueà u l ophileàsu à les intermédiaires réactionnels II et IV s effe tue alors via un mécanisme de type SN2 alors que celle su à l i te diai eà III (SSIP) est plutôt de type SN1.20 Dans ce dernier cas, la conformation de l i te diai eà a tio elàestàp o heàdeà elleàdeàl tatàdeàt a sitio ET2 (Schéma 2b).
La structure du donneur de glycosyle et plus particulièrement sa stéréochimie et la nature des groupements protecteurs jouent aussi un rôle crucial sur le mécanisme de la glycosylation. En effet, on distingue deux voies bien distinctes dites respectivement « participante » et « non-participante ».
19 L. Bohé, D. Crich, C. R. Chimie, 2011, 14, 3.
20 M.T.C. Walvoort, J. Dinkelaar, L.J. van den Bos, G. Lodder, H.S. Overkleeft, J.D.C. Codée, G.A. van der Marel, Carbohydr. Res., 2010, 345, 1252. ET2 ET1 ET ΔG Coordonnées de réaction (a) (b)
2.3.2 Deux voies réactionnelles
2.3.2.1 Voie dite non-pa ti ipa te
Si la réaction de gl os latio àsuitàu eà oieàditeà o -pa ti ipa te , le promoteur facilite le départ du groupement partant en position anomérique formant un cation glycosyle. Celui-ci est stabilisé par réso a eà d lo alisa tà laà ha geà positi eà su à l o g eà e do li ueà pou à fo e à l io à glycosyl oxocarbenium (Schéma 3a). Le carbone anomérique de cet ion est alors hybridé sp2 ce qui antérise théoriquement l atta ueàduà ucléophile sur les deux faces du cycle pyranose de façon équiprobable. Par ailleurs, le produit thermodynamique d a o ieà αà està fa o is à de a tà leà p oduità i ti ueà d a o ieàβàintroduisant laà otio àd effet anomérique qui est explicité dans le paragraphe 2.4.1.
2.3.2.2 Voie dite pa ti ipa te
Dans ce second cas, le cation glycosyle peut être stabilisé de façon covalente par le groupement protecteur en position 2 de type ester, acétamide ou thioester. Da sàleà asàd u àeste , l atta ueàduà doublet non-lia tà deà l o g eà du groupe carbonyle forme un intermédiaire bicyclique appelé ion dioxalénium (Schéma 3b). Avec cette assista eà a hi i ue,à l atta ue du nucléophile est alors privilégiée sur la face oppos eà à l io à dioxalénium générant le glycoside 1,2-trans comme produit final.21Ilà està i t essa tà deà ote à ueà laà fo atio à deà l io à dio al iu à peutà gale e tà p o e i à di e te e tà d u e attaque intramoléculaire de type SN2 concomitante au départ du groupement partant, sans générer un cation glycosyle.
Schéma 3 : Mécanismes possibles pour la réaction de glycosylation
Bien que ces mécanismes soient généralement admis, ces deux voies ne sont pas suffisantes pour expliquer la majorité des résultats de glycosylation. Cestà pou uoi laà e he heà deà l e iste eà deà l io glycosyl oxocarbénium et son étude détaillée sont essentielles en glycochimie etàfo tàl o jetàdeà la seconde partie de cette introduction générale. Par ailleurs, les nombreuses études sur le mécanisme de glycosylation soulignent gale e tàlesà ultiplesàfa teu sài flue ça tàl effi a it àetàlaà stéréosélectivité de cette réaction.
2.4
Fa teu s i flue ça t l effi a it et la st os le ti it de la gl os latio
2.4.1 Leffet a o ère
Le comportement conformationnel est une des principales différences entre les cyclohexanes substitués et les sucres. En effet, sur un carbocycle à six chaînons, les conformations présentant les substituants en position équatoriale sont énergétiquement plus favorables que celles présentant les substituants en position axiale (Schéma 4a). La p se eàd u àh t oato eàe do li ueà odifieàlesà caractéristiques du cycle (longueur de liaison, angle de valence) et crée des effets stéréoélectroniques ayant un effet prononcé sur la conformation et donc sur la réactivité de ces composés (Schéma 4b).
Edward a longuement étudié la conformation et la réactivité des sucres et a rapporté en 1955 que les sucres possédant des substituants de type alkoxy en position anomère adoptaient en majorité une conformation où le substituant anomérique était en position axiale (Schéma 4c).22 Ce phénomène a également été observé par Lemieux23 qui a présenté ces résultats lo sà d u eà o f e eà deà l American Chemical Society à San Francisco en le nommant pour la première fois effet anomère.24
Schéma 4 : Comparaison de la stéréochimie prédominante da s les la ges a o es sous l effet anomère
Le D-glucopyranose à l tatà atu elàe isteà ajo itai e e tàsousàsaàfo eà , -trans (Schéma 4d) or dès que la position anomère est méthylée,à leà atioà α/βà està i e s à pou à do e à leà d i à αà majoritaire (Schéma 4e). Cetà effetà està d auta tà plusà p o o à ueà leà groupement en position anomère est électronégatif (Schéma 4f).25 Généralement, leffet anomère s e p i eà o eà leà sultatà d i te a tio sà dipôle-dipôle des liaisons polaires au niveau du carbone anomérique. áut e e tà dit,à lesà dou letsà d le t o sà présents sur leà su stitua tà d a o ie βà i t agisse t de
22 J.T. Edward, Chem. Ind., 1955, 1102.
23 R.U. Lemieux, P. Chu, Abstracts of papers, 133rd National Meeting of the American Chemical Society, 1958,
San Francisco.
24 E. Juaristi, G. Cuevas, The Anomeric Effect, 1995, CRC Press. 25 R.U. Lemieux, Pure Appl. Chem., 1971, 25, 527.
manière répulsive avec les doublets non-liants deà l o g eàduà leà défavorisant ainsi la position équatoriale de ce substituant (Figure 4a).
Figure 4 : Rationalisation de l'effet anomère (a) Interactions dipôle-dipôle (b) Interactions orbitalaires De plus, lorsque le substituant anomère est en position axiale, il est stabilisé par hyperconjugaison en
aiso à deà l o ie tatio à p ipla ai e d une orbitale occupée de type p (non-lia te à deà l o g e endocyclique età l o italeà a ti-lia teà σ*à duà a o eà a o i ue.26 áà l i e se, dans le cas où le substituant anomère est en position équatoriale, les orbitales non-lia tesàdeàl o g e endocyclique eàpeu e tài te agi àa e àl o italeàσ* du C1, ces orbitales étant orientées dans des plans différents (Figure 4b).E à l a se eà deà g oupesà pa ti ipa ts,à l effetà a o e est un des facteurs majeurs qui influence la stéréosélectivité de la glycosylation. N a oi s,à d aut esà pa a t es peuvent prédominer devant cet effet.
2.4.2 La structure du donneur de glycosyle
La stéréosélectivité de la glycosylation peut être profondément influencée par la structure du donneur de glycosyle et la nature des groupements protecteurs.
Comme précisé dans le paragraphe 2.3,àl assistance anchimérique influence la stéréosélectivité de la glycosylation. Notamment,àdesà tudesà o t e tàl e iste eàdeàpa ti ipatio àavec des groupements acyles en position 2 de type O-acétate27, N-acétate28 ou S-acétate29 1 favorisant laàfo atio àd une liaison glycosidique 1,2-trans.
En revanche, Boons30 a montré u à pa ti à d u à do eu à protégé en position 2 par un éther S-(phénylthiométhyle)benzyl, la glycosylation produit préférentiellement un α-glucoside. Dans ce cas, il y a participation de l the àet fo atio àd u àio àsulfo iu àa o eà2, le nucléophile attaquant par laàfa eài f ieu eàduàdo eu àdeàgl os leà àl oppos àduàsulfo deà li ue (Schéma 5a).
En plus deà etteà pa ti ipatio à oisi e ,à u eà pa ti ipatio à plusà loi tai e à des groupements en position C3 ou C6 peut également influencer la stéréosélectivité de la réaction de glycosylation. Kim31 aà o t à u u à g oupe e tà a tateà e à positio à C (Composé 3) ou C6 (Composé 4) affecte la
26 C.M. Filloux, Angew. Chem. Int. Ed., 2015, 54, 8880.
27 Y. Zeng, Z. Wang, D. Whitfield, X. Huang, J. Org. Chem., 2008, 73, 7952.
28 F.W. Ballardie, B. Capon, W.M. Dearie, R.L. Foster, Carbohydr. Res., 1976, 49, 79. 29 S. Knapp, B.A. Kirk, Tetrahedron Lett., 2003, 44, 7601.
30 T.J. Boltje, J-H. Kim, J. Park, G-J. Boons, Org. Lett., 2011, 13, 284.
stéréoséle ti it à deà laà a tio à deà a os latio à o duisa tà ajo itai e e tà à u à α-mannoside suiteà àl atta ueàdeàl a epteu nucléophile de façon oppos eà àl io àdio al iu . (Schéma 5b) Enfin, la participation du groupe en C4 a aussi été observée. Elle favorise laàβ-glucosylation par la formation du 1,2,4-orthoacétate 5 et l atta ueà u l ophileà à l oppos à deà età o thoa tate.32 A l i e se,à etteà pa ti ipatio à e à C à p i il gieà l a o eà αà da sà leà asà d u eà gala tos latio à o à l atta ueàs effe tue sur la face inférieure du cycle 6 (Schéma 5c).33
Schéma 5 : Exemples d'assistance anchimérique (a) Participation en 2 (b) Participation en 3 et 6 (c) Participation en 4
Ces quelques exemples de participations proches ou lointaines qui contrôlent la stéréochimie anomérique du produit formé illustrent le rôle joué par le groupe protecteur. Les conditions réactionnelles et la structure de l a epteu de glycosyle peuvent également influencer la stéréosélectivité de la réaction de glycosylation.
2.4.3 L effet de sol a t
Amplement étudié, le solvant influence la sélectivité de la glycosylation. En effet, un solvant dit « polaire »à fa o iseà laà β-glycosylation.34 Cependant, la simple solvatation ne doit pas être prise seulement en considération. Il est montré que les solvants éthérés p i il gie tàu eàα-glycosylation suite à la stabilisation possible du cation glycosyle (Schéma 6a).35
32 Y. Ma, G. Lian, Y. Li, B. Yu, Chem. Commun., 2011, 47, 7515.
33 A.V. Demchenko, E. Rousson, G-J. Boons, Tetrahedron Lett., 1999, 40, 6523.
34 H. Satoh, H.S. Hansen, S. Manabe, W.F. van Gunsteren, P.H. Hünenberger, J. Chem. Theory. Comput., 2010, 6,
1783.
35 R. Eby, C. Schuerch, Carbohydr. Res., 1974, 34, 79.
Schéma 6 : Rationalisation de l'effet du solvant (a) avec l'éther diéthylique (b) avec l'acétonitrile D aut eàpa t,àlaàfo atio àdeàβ-glycosides via des intermédiaires réactionnels axiaux est privilégiée e à utilisa tà l a to it ile.à Da s ce cas, le piégeage du cation glycosyle par le solvant conduit à la fo atio àd un ion nitrilium. Ce dernier formé in situ adopte exclusivement une orientation axiale fa o isa tàai siàl atta ueàduà u l ophileàsu àlaàfa eàopposée au cycle pyranose (Schéma 6b).
Récemment, Mong36 a révisé ce mécanisme en démontrant queàl o ie tatio àa ialeàdeàl io à it iliu à est due à l effetà a o i ueà ais est gale e tà e fo eà pa à laà pa ti ipatio à deà l o g eà e à position 2 formant ainsi un intermédiaire de type oxazolinium (Schéma 6c). Cet intermédiaire a été prouvé indirectement par la fo atio àdeàl o azoli eà o espo da te après hydrolyse.
La stéréosélectivité de la réaction de glycosylation dépend également de la structure du donneur et plus particulièrement de sa conformation et a fortiori de elleàdeàl i te diai eà a tio elàfo .àà D sàlo s,àl a al seàdesài te diai esà a tio elsàdeàt peà atio àgl os leàetàe àpa ti ulie àl i pa tà de leur conformation sur la stéréosélectivité de la glycosylation a été largement étudié.
3
Etat de l’a t su l’e iste e d’u atio glycosyle
La connaissance des intermédiaires réactionnels est essentielle pour le développement de méthodologies de synthèse et les intermédiaires de type cationique ont été fortement étudiés.
3.1 Etude des intermédiaires cationiques
3.1.1 Historique
C est en étudiant le réarrangement de Wagner du camphène chloré en isobornyl chloré en 1922 que Meerwein37 a proposé ueàl iso isatio observée est pas due à une migration du chlore mais due àu à a a ge e tàd u ài te diai eà atio i ue.àC est ai sià u està e laà otio àd u eàtelleà espèce transitoire. Plus tard, Ingold et Hugues38 ont étudié en détail les cinétiques et la stéréochimie de substitutions nucléophiles nommées a posteriori SN1 et ont confirmé l i pli atio à pote tielleà d i te diai esà atio i uesàen chimie organique. Ensuite Whitmore39 a généralisé cette notion pour
36 C.S. Chao, C.Y. Lin, S. Malani, W.C. Hung, K.K. Mong, Chem. Eur. J., 2011, 17, 12193. 37 H. Meerwein, K. van Emster, Chem. Ber., 1922, 55, 2500.
38 L.C. Bateman, M.G. Church, E.D. Hughes, C.K. Ingold, N.A. Taher., J. Chem. Soc., 1940, 979. 39 F.C. Whitmore, J. Am. Chem. Soc., 1932, 54, 3274.
de nombreuses autres réactions. Cette notion est néanmoins restée controversée. De manière anecdotique, áda sà uià doutaità deà l e iste eà d u eà telleà esp eà fu ti eà empêcha Whitmore de publier dans un journal prestigieux (Journal of the American Chemical Society) duàfaitàdeàl utilisatio à de la notation R+ pour dénoter une espèce cationique.40
Bien que Meerwein, Ingold, Hughes, Whitmore, Barlett, Cram età d aut esà ont contribué fondamentalement au développement de la chimie des intermédiaires déficients en électron(s), l o se atio à di e teà d espèces cationiques stables resta longtemps un but inaccessible. Il a fallu attendre le début des années 1960 pou à u Olah41 caractérise pour la première fois par Résonance Magnétique Nucléaire (RMN) in situ un carbocation. L utilisatio d un milieu très acide dit superacide, défini dans le paragraphe 4, a permis de générer un carbocation stable. De plus, la faible nucléophilie des contre-ions présents et formés lors de la réaction contribue à la bonne stabilité des sels obtenus et a permis pa foisàd isoler le carbocation. Le premier carbocation 11 a été généré à partir du chlorure de t-butyle 10 et est caractérisé par un déplacement 13C très déblindé à 273 ppm (Schéma 7).
Schéma 7 : Première mise en évidence par RMN d'un carbocation généré en milieu superacide Le déblindage significatif du signal deà l io à 11 par rapport à celui du précurseur covalent 10 met clairement en évide eà leà a a t eà atio i ueà deà l espèce générée en solution. Ce déblindage indique que le carbone subit u à ha ge e tàd h idatio sp3 en sp2. Cetteàte h i ueàd a al seàe à milieu superacide a ensuite été appliquée à d autres fonctions mettant en évidence des intermédiaires de type azonium42, oxonium43, sulfonium44 ou encore carboxonium45 et notamment les ions alkoxycarbénium.46 L e se leà deà esà tudesà a o duità à l att i utio à duà Prix Nobel de
Chimie au professeur Olah en 1994 pour sa contribution à la chimie des carbocations.
40 G.K.S. Prakash, P.v.R. Schleyer, Stable carbocation chemistry, 1997, Wiley-Interscience, New-york.
41 G.A. Olah, E.B. Baker, J.C. Evans, W.S. Tolgyesi, J.S. McIntyre, I.J. Bastien, J. Am. Chem. Soc., 1964, 86, 1360. 42 G.A. Olah, K.K. Laali, Q. Wang, G.K.S. Prakash, Onium Ions, 1998, Wiley-Interscience, New-york.
43G.á.àOlah,àD.H.àO B ie ,àC.U.àPitt a ,àJ. Am. Chem. Soc., 1967, 89, 2996. 44 G.A. Olah, P.L. Szilagyi, J. Org. Chem., 1971, 36, 1121.
45 G.A. Olah, Chem. Eng. News., 1967, 45, 76.
3.1.2 Les ions alkoxycarbénium en milieu superacide
Des ions alkoxycarbénium tertiaires préparés dans un premier temps par Meerwein47 ont été caractérisés par Olah48 par RMN.àLeà a o eàdeàl ion oxocarbénium présente un signal déblindé entre 220 et 250 ppm selon laà atu eàp i ai eàouàte tiai eàdeàl io à(Figure 5).49
Figure 5 : Caractérisation par RMN d'ions alkoxycarbénium (a) tertiaires et (b) primaires (déplacements chimiques en ppm)
3.1.3 Gé atio d io s alko a iu par oxydation électrochimique
Plus récemment, Yoshida a étudié ces ions alkoxycarbénium et plus particulièrement ceux dérivés de sucre par une méthode électrochimique. áàpa ti àd the sàα-silylés50ouàd h i-thioacétals51, Yoshida
a développé une méthode permettant laàg atio àd io sàalko a ium à longue durée de vie
en solution par oxydation électrochimique à basse température. Ainsi, il a été possi leàd a al se àpa à RMN les ions N-acyliminium et alkoxycarbénium générés dans le dichlorométhane deutéré en utilisant le tétrafluoroborate de tétrabutylammonium (Bu4NBF4) comme électrolyte. (Schéma 8)
Schéma 8 : Génération d'ions alkoxycarbénium à partir d'hémithioacétals
Cependant, bien que de nombreux ions oxocarbénium aient pu être générés et analysés par cette méthode électrochimique, cette technique aàpasàpa is l a al seàpa à‘MNàduà atio àglycosyle.51 Yoshida a ensuite amélioré son procédé en développant deux méthodes indirectes séquentielles en
o eàpot àouàe àflu à o ti u.52
47 H. Meerwein, K. Bodenbenner, P. Borner, F. Kunert, K. Wunderlich, Ann. Chem., 1960, 38, 632. 48 G.A. Olah, D.G. Parker, N. Yoneda, F. Pelizza, J. Am. Chem. Soc., 1976, 98, 2245.
49 G.A. Olah, J.M. Bollinger, J. Am. Chem. Soc., 1967, 89, 2993. 50 S. Suga, S. Suzuki, J-I. Yoshida, Org. Lett., 2005, 7, 4717.
51 S. Suzuki, K. Matsumoto, K. Kawamura, S. Suga, J-I. Yoshida, Org. Lett., 2004, 6, 3717. 52 S. Suga, K. Matsumoto, K. Ueoka, J-I. Yoshida, J. Am. Chem. Soc., 2006, 128, 7710.
3.1.3.1 M thode one pot
Dans cette approche53,à leà te psà d a cumulation est fortement réduit. Après oxydation électrochimique du diphényl-disulfure (ArSSAr) à -78 °C dans le dichorométhane en présence du Bu4NBF4,àl io à21 formé réagit avec un hémithioacétal 22 o duisa tà àl io ào o a iu 16.àL ajout d u à u l ophileà o duità auà p oduità d additio après neutralisation avec de bons rendements. (Schéma 9)
Schéma 9: Génération et caractérisation de l io pa la thode « one pot »
3.1.3.2 Méthode en flux continu
A partir de l oxydation électrochimique one-pot , Yoshida54 a dévéloppé une technique en flux continu pour générer des cations alkoxycarbénium en solution. Afin de générer les ions visés, un réacteur microfluidique composé de trois microréacteurs liés par deux microtubes a été utilisé. Ce dispositif permet dans un premier temps dajoute simultanément le cation 21, issu deàl o datio àduà diphényl-disulfure, avec le thioacétal 22 dans le microréacteur M1 afin de générer l io à oxocarbénium 16 (Schéma 10a). Ce dernier réagit avec le nucléophile 23 ajouté dans le microréacteur M2 (Schéma 10b) formant ainsi le produit désiré 24 de façon quantitative après la neutralisation du milieu dans le microréacteur M3 par ajout de triéthylamine (Schéma 10c).
Schéma 10 : Génération et réaction d'ion alkoxycarbénium dans un système en flux continu
Cette méthodologie électrochimique a ensuite été appliquée à la réaction de glycosylation. A partir du thioglucoside perbenzylé 25, la réaction de gl os latio às effe tueàdans le dichlorométhane en présence de tétrakis(pentafluorophényl)borate de tétrabutylammonium (Bu4NB(C6F5)4) au lieu de
53 K. Matsumoto, K. Ueoka, S. Suzuki, S. Suga, J-I. Yoshida, Tetrahedron, 2009, 65, 10901.
Bu4NBF4 comme électrolyte afi àd ite àlaàfo atio àduàsucre fluoré. áp sàl additio àd u àa epteu à de glycosyle 26, un mélange de glucosides 27 est obtenu avec un rendeme tàdeà à%àetàu à atioàα/βà de 32/68 (Figure 6).
Figure 6 : Réaction de glycosylation réalisée dans un réacteur microfluidique
Alors que la formation du cation glycosyle 28 est admise dans ce processus, aucunes données de caractérisation du cation glycosyle o tà puà t eà o te ues. Cela a conduit Yoshida à proposer l h poth seàselo àla uelle cet ion formé e isteàpasàsousàfo eàli e mais est plutôt stabilisé par coordination avec le disulfure formé in situ démontrant une durée de vie très courte pour cet intermédiaire.
L io à gl os là o o a iu à aà gale e tà t à p opos à o eà i te diai eà lo sà deà a tio sà deà glycosylation par voie électrochimique utilisant un thioglycoside comme donneur. Sinaÿ55 et Lubineau56 ont montré que le thioglycoside de départ 17 pe dàu à le t o àlo sàdeàl o datio àpou à donner le radical cation 18. Le clivage de la liaison C1-S de ce dernier libère alors le radical thioaryl et le cation glycosidique 19. Celui-ci est ensuite piégé par le nucléophile présent (alcool) pour former le β-glycoside 20 ajo itai e e tà ua dàlaà a tio àestà alis eàda sàl a to it ileà Schéma 11).
Schéma 11: Existence de l'ion glycosyl oxocarbénium dans la glycosylation par la voie électrochimique
55 C. Amatore, A. Jutand, J-M. Mallet, G. Meyer, P. Sinaÿ, J. Chem. Soc., Chem. Commun., 1990, 718. 56 G. Balavoine, A. Gref, J-C. Fischer, A. Lubineau, Tetrahedron Lett., 1990, 31, 5761.
3.1.4 La du e de ie de l io o o a iu
Alors que les ions alkoxycarbénium sont stables et parfaitement caractérisés, Jencks et Amyes57 ont montré que la durée de vie de certains ions dioxocarbénium et oxocarbénium estàdeàl o d eàdeàlaà nanoseconde en utilisant une méthode basée sur l i hi itio àdeàla solvolyse. (Figure 7)
Figure 7 : Durée de vie de différents ions oxocarbéniums
Steeken58 a généré des ions similaires stabilisés via des cations radicaux et a mesuré par conductance que leur durée de vie da sàl eau était gale e tàdeàl o d eàdeàla nanoseconde. Afin de déterminer si l io àgl os le nu (sans groupe protecteur) existe réellement en milieu aqueux, Jencks57 et Bennet59 ont estimé indépendamment sa durée de vie et arrivent aux mêmes conclusions. Par extrapolation des durées de vie obtenues pour les ions oxocarbénium acycliques, Jencks a obtenu une valeur de 5.10-12 secondes pour la durée de vie de l ion glycosyle.* En étudiant l h d ol seàduàselàdeàp idi iu à du 2-déoxy-β-D-glucopyranose, Bennet a calculé un temps de vie de 2,5.10-12 secondes pour le cation glycosyle en solution aqueuse.
Ces valeurs démontrent que l o servation de cette espèce ionique, bien que très convoitée, reste un objectif ambitieux. Dans cette optique, son étude a été effectuée de manière indirecte en exploitant principalement trois méthodes distinctes : les effets isotopiques cinétiques, les calculs théoriques d i te diai esà h poth ti uesà età lesà tudesà pa à ‘MNà basse température où le donneur est préalablement activé.
3.2 Etude du mécanisme de la glycosylation par mesure des effets
isotopiques cinétiques
L tudeàdeàl Effet Isotopique Cinétique (EIC ou KIE : Kinetic Isotope Effect) est un outil puissant pour examiner le mécanisme et plus particulièrement évaluer la nature des intermédiaires réactionnels mis en jeu lo sàd u eà a tio . En effet, la molécule substituée pa àl isotopeàd u à l e tàp se teà les mêmes propriétés du point de vue de la réactivité que la molécule non marquée mais produit des effets perceptibles d u àpoi tàdeà ue thermodynamique et cinétique.60
3.2.1 La otio d effet isotopi ue i tique
Seà asa tàsu àl tudeàduàdeut iu àd Urey et Rittenberg61, Bigeleisen et Mayer62 ont publié en 1947, les premiers travaux su à etteà otio à d EIC. Le principe était d tudie à laà pe tu atio à i duiteà pa à
57 T.L. Amyes, W.P. Jencks, J. Am. Chem. Soc., 1989, 111, 7888.
58 S. Steenken, J. Buschek, R.A. McClelland, J. Am. Chem. Soc., 1986, 108, 2808; S. Steenken, R.A. McClelland, J. Am. Chem. Soc., 1989, 111, 4967.
59 X. Huang, C. Surry, T. Hiebert, A.J. Bennet, J. Am. Chem. Soc., 1995, 117, 10614.
*Ceà uià o espo dà àu eàdu eàd u à illia dàdeàfoisàplusà ou teà u u àflashàd appa eilàphoto. 60 E.M. Simmons, J.F. Hartwig, Angew. Chem. Int. Ed., 2012, 51, 3066.
Prix Nobel de Chimie en 1934 pour la d ouve te de l’h d og e lou d. 61 H.C. Urey, D. Rittenberg, J. Chem. Phys., 1933, 1, 137.
l isotopeàe àa o da eà atu elleàouàa plifi eàsu le système. Ce phénomène est observable avec les éléments les plus courants : H, C, O, N.... Cependant, les effets isotopiques pour les isotopes de l hydrogène sont plus intenses que pour les autres atomes car ils sont directement liés à la variation de la masse atomique (plus grande différence entre l h d og e et le deutérium).
Il y a EIC lorsque la liaison molécule-isotopeàestàaffe t eàlo sàdeàl tapeà i ti ue e tàd te i a te.à Da sà eà as,à laà su stitutio à isotopi ueà d u à e t eà a tio elà aà u eà i fluence sur la vitesse de l tapeàle teàdeàla a tio .àL EIC s e p i eàdo à o eàu à appo tàdeà o sta teàdeà itesse,ào àleà u ateu à està laà o sta teà deà itesseà pou à laà a tio à a e à l isotopeà ajeu à e à a o da eà naturelle et le dénominateur est la consta teàdeà itesseàpou àlaà a tio àa e àl isotopeàdeàplusàfai leà abondance naturelle. Il existe deux principaux t pesà d effetà isotopi ue respectivement nommés primaire et secondaire.
L effet isotopique primaire implique la rupture de la liaison molécule-isotopeà auà ou sà deà l tapeà cinétiquement déterminante. Dans le cas de la réaction de glycosylation par exemple, il y a un effet cinétique isotopique primaire impliquant le carbone du centre anomérique. Les vitesses respectives des réactions impliquant les liaisons covalentes avec le 12C1 et 13C1 et le groupe partant sont donc fortement influencées (Schéma 12).
Leffet isotopique est dit secondaire lorsque la liaison molécule-isotope est seulement perturbée (allongement,à a ou isse e t… .àL i te sit àdeà esàeffetsàse o dai esàestàplusàfai leà ueà elleàdesà effets primaires ce qui exige une plus grande précision expérimentale.
Schéma 12 : Illustration des effets isotopiques primaire et secondaire dans la réaction de glycosylation Singleton63 a mesuré de faibles effets isotopiques cinétiques du 2H et du 13C par RMN en abondance naturelle avec une grande précision lo sà deà l tudeà de la réaction de Diels-Alder entre un dérivé d isop eàetàl a h d ideà al i ue (Figure 8a).àLo sàdeà l a a e e tàdeà laà a tio ,àleà p oduità 29 esta tà s e i hità e à isotopeà lou d. De cet enrichissement détecté et mesuré par RMN, le pou e tageàd isoprène a a tàpasàréagi et récupéré est obtenu. Appliquant la formule ci-dessous (Équation 1), Singleton accède aux effets isotopiques du 2Het du 13C (Figure 8b).
=
� [ 1 − �/�
� 1 −
0
]
Équation 1 : Formule pour calculer l'EIC
63 D.A. Singleton, A.A. Thomas, J. Am. Chem. Soc., 1995, 117, 9357.
R :àI t g atio àpou àl isotopeà i eu àda sàleàp oduitàfi al R0:àI t g atio àpou àl isotopeà i eur dans le produit de départ F : Conversio àdeàl isop eà1H)
Figure 8 : (a) Réaction de Diels-Alder entre l'isoprène et l'anhydride maléique ; (b) Effets isotopiques cinétiques du 2H et du 13C du composé 29 en appliquant l'Equation 1
Lesà aleu sà p o hesà deà l u it à o eà celles obtenues pour les centres C2 et C3 traduisent des centres non réactionnels. Au contraire, les valeurs plus élevées obtenues pour les centres C1 et C4 respectivement de 1,022 et 1,017 prouvent que ces sites so tài pli u sàda sàl tapeà i tiquement déterminante de la réaction.à Pa à ailleu s,à à oi sà d u e erreur expérimentale ou de la présence d i pu et s, la différence entre les effets observés pour C1 et C4 indique que la formation des liaisons impliquant ces atomes estàpasàe a te e tàs h o e.
La mesure de leffetà isotopi ueà i ti ueà p i ai eà duà13C est une méthode pour distinguer les mécanismes monomoléculaire et bimoléculaire. En effet, les valeurs comprises entre 1,02 et 1,06 traduisent un mécanisme associatif (SN2) alors que les valeurs inférieures ou égales à 1,01 suggèrent un mécanisme dissociatif (SN1).64Plusieurs groupes dans le monde appliquent la méthode de Singleton pour des études mécanistiques de la réaction de glycosylation.
3.2.2 Les effets isotopiques cinétiques et la réaction de glycosylation
En 2002, Berti65 a étudié la rupture chimique et enzymatique de la liaison glycosidique des α- etàβ-glucopyranosides de méthyle. Lh d ol seàa ideà HClO4 2M) de la liaison glycosidique est effectuée à
à°Càe àp se eàd u à talo ài te eà su i iteàdeàsodiu àpuisàlaà a tio àestàneutralisée après une o e sio àd e i o à %. Les EIC sont déterminés pa àl i t g atio àdesàspe t esà‘MNàetàl utilisatio à de la formule décrite précédemment. Lesàα- etàβ-glucopyranosides de méthyle 32 et 33 conduisent à u à la geàd a o es 34 (Schéma 13a). Selon la nature de la catalyse, les valeurs diffèrent. Les faibles valeurs observées pou àl h d ol seàacide des dérivés αà32 , àetàβà33 (1,01) etàl h d ol seà enzymatique du dérivé 32 (1,010) prédisent un mécanisme de type SN1 avec un intermédiaire de t peà o o a iu .à E à e a he,à l h d ol seà e z ati ueà duà d i à βà 33 semble privilégier un mécanisme à fort caractère SN2 (1,032) (Schéma 13b).
64 J.K. Lee, A.D. Bain, P.J. Berti, J. Am. Chem. Soc., 2004, 126, 3769. 65 P.J. Berti, K.S.E. Tanaka, Adv. Phys. Org. Chem., 2002, 37, 239.
Schéma 13 : Effets isotopiques cinétiques primaires du 13C observés lors de l'hydrolyse des α- et β-D-glucopyranosides de méthyle par (a) catalyse acide et (b) catalyse enzymatique
Il est également important de mentionner que lesàEICàse o dai esàdeàl α-glucoside de méthyle en catalyse enzymatique sont différents de ceux obtenus en catalyse acide.
Récemment, Crich a analysé en détail le mécanisme de la glycosylation impliquant des αàetàβ-manno et glucopyranosides en utilisant les EIC.66áfi àd ite àlaàs th se de substrats de départ marqués, toutes les mesures ont été réalisées en abondance naturelle en utilisant un appareil RMN de 800 MHz. Le triflate de mannosyle généré à partir du phénylsulfoxyde de 4,6-O-benzylidène-2,3-di-O-méthyl-α-D-mannopyranose 35 dans le dichlorométhane par addition de Tf2O en présence de 2,4,6-tri-tert-butylpyrimidine, a été utilisé comme donneur de glycosyle dans la réaction de glycosylation. L isopropanol ajouté en défaut assure une conversion partielle du triflate 36 e àα- etàβ-mannosides d isop op le 37 et 38 (Schéma 14). Cette réaction est ensuite neutralisée à -72 °C par une solution aqueuse saturée de NaHCO3.à Lesà tau à deà o e sio à e à α- età β-mannosides sont déterminés par analyse RMN 1H quantitative du brut réactionnel.
Schéma 14 : Réaction de mannosylation étudiée en utilisant les EIC d'abondance naturelle
Après isolation des deux mannosides 37 et 38, les EIC ont été déterminés sur le spectre RMN du 13C en prenant comme étalon interne le carbone du benzylidène considéré comme éloigné du centre réactionnel et donc sans EIC notable (valeur égale à 1). Ainsi, pour chaque anomère isolé, l i t g atio à duà C à vs le car o eà duà e z lid eà pe età d obtenir le paramètre Rp.à L i t g atio à similaire du dérivé sulfoxyde de départ donne la valeur du paramètre RO L i t g atio à des