Photodynamic therapy of cancer: an update.
Texte intégral
Figure
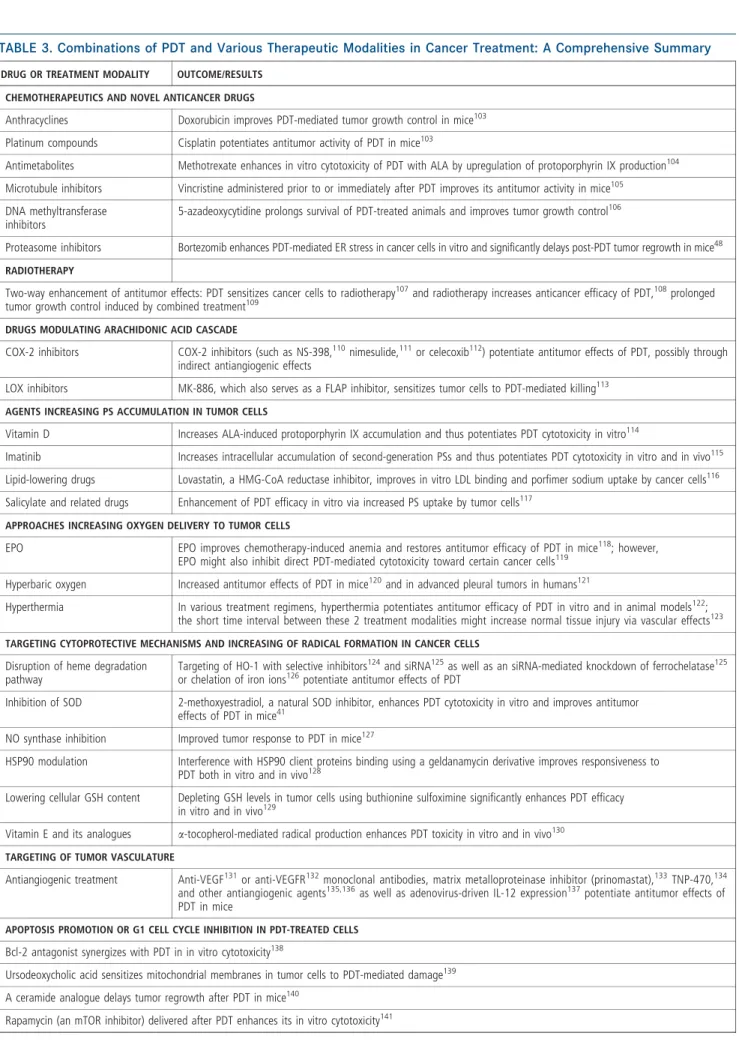
Documents relatifs
Somatic mutations of EGFR at tyrosine kinase domain have been associated with clinical response to tyrosine kinase inhibitors (TKIs) in lung cancer patients.. In this study,
The parameter D can be set to a higher value to achieve the required entropy rate at the output of the TRNG and at the same time, all the output bits of the generator can be used
Top-down proteomics consists in the analysis of intact proteins using liquid chromatography coupled to mass spectrometry (LC-MS), followed by their identification by tandem mass
L’accès à ce site Web et l’utilisation de son contenu sont assujettis aux conditions présentées dans le site LISEZ CES CONDITIONS ATTENTIVEMENT AVANT D’UTILISER CE SITE
A cet égard, nous pourrions faire l’hypothèse que le chant de l’Oiseau est emprunt de signifiants énigmatiques au sens où Jean Laplanche les a conceptualisés dans sa théorie de
Mais dans les textes de Freud, comme souvent, il y a des torsions qui peuvent faire comprendre une chose et son contraire – preuve d’une pensée dialectique sans cesse en
30 Department of Clinical Immunology and Infectious Diseases, National Research Institute of Tuberculosis and Lung Diseases, Shahid Beheshti University of Medical Sciences,..
(ed.): 2011, Ethical Dilemmas in Assisted Reproductive Technologies.. Bioconstitu- tionalism in the
