CRISPR-Cas : un nouvel outil pour l'étude des génomes de bactériophages
Texte intégral
Figure
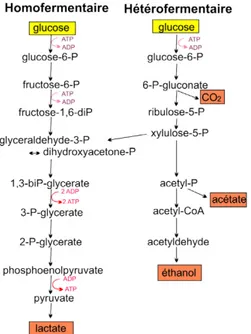

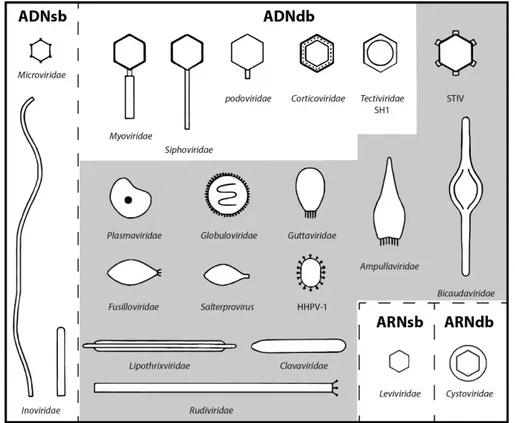
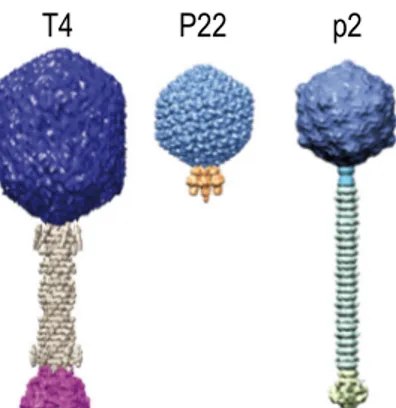

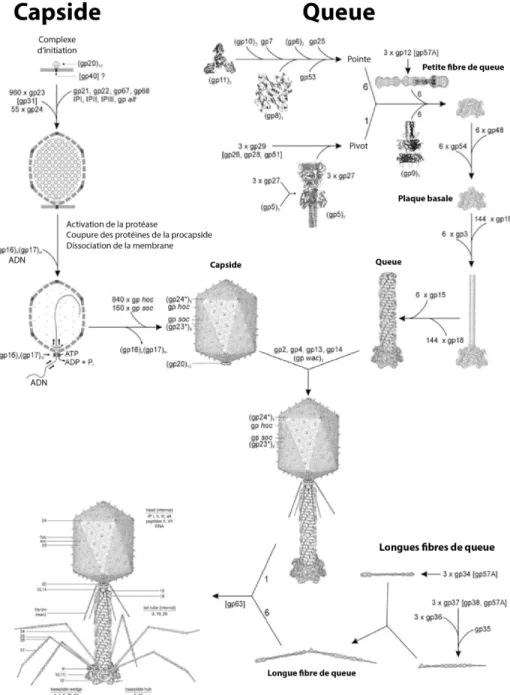


Outline
Documents relatifs
Étendre chaque paire de 12-mers dans les deux directions (sans gaps), jusqu’à ce que le score chute en dessous d’un certain seuil.. Si l’alignement (sans gaps) trouvé dépasse
Étant donné deux génomes A et B, un MUM est un facteur commun de A et B de longueur dépassant un certain seuil d (par défaut d=20) tel que.. - Il est
SDU 5HF$ &KH] ( FROL 5HF$ SHUPHW OD UHFRPELQDLVRQ LQWUDFKURPRVRPLTXH HQWUH V«TXHQFHV UKV SRXUUHFRPELQDWLRQKRWVSRWHQDQJODLVD\DQW¢GHGLYHUJHQFH 3HWLW HW DO &KH] %DFLOOXV
tRNAscan-SE (Lowe and Eddy, Nucleic Acids Res.,25, 955-64 (1997)) qui s’appuie sur deux méthodes existantes (tRNAscan et EufindtRNA ( Pavesi al., Nucleic Acids Res., 22, 1247-56
Pour certains k mers rares même avec un grand jeu d’apprentissage comme un génome entier, il peut être difficile d’obtenir des estimations précises et inversement certains k
MAUVE évite ce problème en utilisant des « Multiple Maximal Unique Matches » (multi-MUMs) de longueur minimum k comme ancres, c’est-à-dire des régions qui sont trouvées
• Utilisation d’une heuristique pour fournir l’alignement final entre les deux séquences (alignement local qui va renvoyer les deux sous-régions les plus conservées entre
• les listes ordonnées sont ensuite parcourues pour identifier les k-mers qui sont trouvés dans deux ou plusieurs séquences mais qui apparaissent au plus une fois dans chacun
