Effet de la cristallinité sur la perméabilité aux gaz
de films à base d’acide polylactique
Mémoire
Amir Ghassemi
Maîtrise en génie chimique
Maître ès sciences (M.Sc.)
Québec, Canada
© Amir Ghassemi, 2016
Effet de la cristallinité sur la perméabilité aux gaz
de films à base d’acide polylactique
Mémoire
Amir Ghassemi
Sous la direction de :
Denis Rodrigue, directeur de recherche
Carl Duchesne, codirecteur de recherche
iii
Résumé
Le but principal de ce travail est de déterminer l’effet de la cristallinité sur la perméation aux gaz pour des films polymères. En particulier, on utilise l’acide polylactique (PLA) comme matrice et différents gaz (azote, dioxyde de carbone, hydrogène, méthane et oxygène) pour les propriétés de transfert. À cet effet, le travail a été divisé en trois parties.
Dans la première partie, on utilise le talc comme agent de nucléation afin de modifier la cristallinité du PLA. Dans ce cas, on remarque que la perméabilité au gaz et le coefficient de diffusion sont réduits en augmentant la teneur en talc (0-3% poids). On constate aussi que l’augmentation de la cristallinité due à la nucléation hétérogène ne modifie pas significativement les propriétés mécaniques, sauf pour la déformation à la rupture. Néanmoins, les propriétés de transfert sont diminuées.
Dans la deuxième partie, l’effet du temps de recuit pour modifier la cristallinité du PLA seul et avec 3% de talc a été étudié. Dans ce cas, on constate que la perméabilité aux gaz et le coefficient de diffusion sont réduits en augmentant le temps de recuit (jusqu'à 40 minutes). On constate également que l'augmentation de la cristallinité ne modifie pas les propriétés mécaniques, à l'exception du module de Young.
Enfin, la température de recuit pour améliorer la cristallinité du PLA seul ou avec 3% de talc a été étudiée. Bien que la perméabilité aux gaz et le coefficient de diffusion aient diminué en augmentant la température de traitement (de 60 à 120°C), l'augmentation de la cristallinité finale n’a pas changé de manière significative les propriétés mécaniques, à l'exception du module de Young et la déformation à la rupture pour le composite ayant 3% de talc.
iv
Abstract
The main purpose of this work is to determine the effect of crystallinity on the gas permeation of polymer films. In particular, polylactic acid (PLA) was used as the matrix and various gases were selected (nitrogen, carbon dioxide, hydrogen, methane and oxygen) for the gas transport properties. To this end, the work was divided into three parts.
In the first part, talc was used as a nucleating agent to modify the crystallinity of PLA. In this case, it was noted that the gas permeability and the diffusion coefficient were reduced with increasing talc content (0-3% by weight). It was also observed that increased crystallinity was related to heterogeneous nucleation, but had limited effect on mechanical properties, with the exception of strain at break. However, the transport properties were decreased. In the second part, annealing time was used to modify PLA crystallinity with 0% and 3% talc. In this case, it was found that gas permeability and diffusion coefficient were lowered with higher annealing time (up to 40 min). It was also observed that higher crystallinity did not change the mechanical properties except for the Young’s modulus.
Finally, annealing temperature was modified to improve the crystallinity of neat PLA and 3% talc composite. While gas permeability and diffusion coefficient both decreased with increasing annealing temperature (from 60 to 120°C), crystallinity changes did not significantly modify the mechanical properties, except for the Young’s modulus and strain at break of the 3% talc composite.
v
Tables des matières
Résumé _________________________________________________________________ iii Abstract ________________________________________________________________ iv Tables des matières ________________________________________________________ v Liste des Tableaux _______________________________________________________ vii Nomenclature _____________________________________________________________ x Liste des symboles ______________________________________________________ xi Remerciements __________________________________________________________ xii Avant-propos ___________________________________________________________ xiii Chapter 1 ________________________________________________________________ 1 1.1 Introduction ______________________________________________________ 1 1.2 L’acide polylactique _______________________________________________ 2 1.2.1 La production de PLA ____________________________________________ 3 1.2.2 Composition structurelle __________________________________________ 5 1.2.3 Propriétés du PLA _______________________________________________ 6 1.2.4 Applications du PLA ____________________________________________ 15 1.3 Le talc _________________________________________________________ 20 1.4 Fabrication de pellicules par extrusion-soufflage ________________________ 23 1.5 Le recuit (traitement thermique) _____________________________________ 26 1.5.1 Effet du recuit sur les polymères ___________________________________ 27 1.6 Objectifs et structure du mémoire ____________________________________ 33 Chapter 2 _______________________________________________________________ 36 2.1 Résumé _________________________________________________________ 36 2.2 Abstract ________________________________________________________ 37 2.3 Introduction _____________________________________________________ 38
vi 2.4 Methodology ____________________________________________________ 40 2.4.1 Materials _____________________________________________________ 40 2.4.2 Sample Preparation _____________________________________________ 41 2.4.3 Thermal Properties _____________________________________________ 42 2.4.4 Gas Permeability _______________________________________________ 42 2.4.5 Scanning Electron Microscope (SEM) ______________________________ 43 2.4.6 Mechanical Properties ___________________________________________ 43 2.5 Results and Discussion ____________________________________________ 44 2.5.1 Morphology ___________________________________________________ 44 2.5.2 Effect of Talc Content ___________________________________________ 44 2.5.3 Effect of Annealing Time ________________________________________ 52 2.5.4 Effect of Annealing Temperature __________________________________ 59 2.6 Conclusion ______________________________________________________ 65 2.7 Acknowledgements _______________________________________________ 66 Chapter 3 _______________________________________________________________ 67 3.1 Conclusion générale ______________________________________________ 67 3.2 Recommandations pour travaux futurs ________________________________ 69 Bibliographie ____________________________________________________________ 71 Annexe A _______________________________________________________________ 80 Annexe B _______________________________________________________________ 82
vii
Liste des Tableaux
Tableau 1.1. Comparaison des propriétés des polymères biodégradables typiques avec le
LDPE, le PS et le PET (Clarinval et al., 2005). ... 8
Tableau 1.2. Températures de transition d’une série de copolymères PLA (Bigg, 1996). ... 13
Tableau 1.3. Secteurs d’activité pour les produits à base de PLA Ingeo et NatureWorks. .. 17
Tableau 1.4. Quelques produits commercialisés à base de PLA (Platt, 2006). ... 18
Tableau 1.5. Propriétés fonctionnelles du PLA pour l’emballage (Jamshidian et al., 2010). ... 19
Tableau 1.6. Principales applications du PLA en 2003 et estimation pour 2020 (Jamshidian et al., 2010). ... 20
Tableau 1.7. Propriétés du talc (Wypych, 1999). ... 22
Table 2.1. Temperature profile for compounding (extruded with L/D=40). ... 41
viii
Liste des Figures
Figure 1.1. Différentes voies de polymérisation pour l’acide polylactique (Drumright et al.,
2000). ... 3
Figure 1.2. Schéma de la production de PLA via le prépolymère et le lactide (Drumright et al., 2000). ... 4
Figure 1.3. Schéma de la production de PLA via le prépolymère et le lactide (Drumright et al., 2000). ... 5
Figure 1.4. Structure schématique des stéréo-isomères L et D (Jamshidian et al., 2010). ... 6
Figure 1.5. Comparaison de températures de transition vitreuse et de fusion du PLA avec d’autres thermoplastiques (Lim et al., 2008). ... 12
Figure 1.6. Les températures de transition vitreuse pour des PLA ayant différents ratios L:D en fonction du poids moléculaire (Dorgan et al., 2005). ... 13
Figure 1.7. Température du pic de fusion du PLA en fonction du contenu de méso-lactide pour des données tirées de: (○) Witzke (1997) et (●) Hartmann (1998). La ligne solide est calculée sur la base de l’équation 1-3. (Witzke, 1997) (Hartmann, 1998; Lim et al., 2008). ... 15
Figure 1.8. Structure moléculaire du talc (Wypych, 1999). ... 21
Figure 1.9. Talc de qualité Mistron (Wypych, 1999). ... 22
Figure 1.10. Schéma du procédé d’extrusion-soufflage (McKeen, 2012). ... 25
Figure 1.11. Micrographies optiques polarisées d’une pellicule de PET d’une épaisseur de 100 µm: a) cristallisé par fonte à 1 K/min, b) cristallisé par fonte à 1 K/min, puis recuit à 473 K pour 60 min, c) trempé dans de l’eau glacée, puis recuit à 473 K pour 60 min. La barre d’échelle est de 20 µm (Zia et al., 2010). ... 29
Figure 1.12. Courbes de contrainte-déformation d’échantillons recuits pendant 1 min à 100, 160 et 220°C pour l’échantillon de contrôle, FA (non contraint) et TA (contraint) (Gupta et al., 1981b). ... 31
Figure 2.1. SEM images of (a) neat PLA ×1000, (b) neat PLA ×2000, (c) PLA/0.75% talc ×1000, (d) PLA/0.75% talc ×2000, (e) PLA/1.5% talc ×1000, (f) PLA/1.5% talc ×2000, (g) PLA/3% talc ×1000, (h) PLA/3% talc ×2000. ... 45
Figure 2.2. Effect of talc content on the crystallinity and crystallization temperature of neat PLA and PLA/talc composites. ... 46
ix
Figure 2.3. Effect of talc content on gas permeability of PLA/talc composites. ... 49 Figure 2.4. Effect of talc content on gas diffusion coefficient and solubility of PLA/talc
composites. ... 50 Figure 2.5. Effect of crystallinity on hydrogen permeability for PLA/talc composites. ... 51 Figure 2.6. Effect of talc content on the strain at break and Young’s modulus of neat PLA
and PLA/talc composites. ... 52 Figure 2.7. Effect of annealing time on the crystallinity of neat PLA and PLA + 3% talc
composite. ... 53 Figure 2.8. Effect of annealing time on gas permeability of neat PLA and PLA + 3% talc
composite. ... 55 Figure 2.9. Effect of annealing time on gas diffusion and solubility of neat PLA and PLA +
3% talc composite. ... 57 Figure 2.10. Effect of crystallinity on hydrogen gas permeability of neat PLA and PLA +
3% talc composite. ... 57 Figure 2.11. Effect of annealing time on the strain at break and Young’s modulus of neat
PLA and PLA + 3% talc composite. ... 58 Figure 2.12. Effect of annealing temperature on the crystallinity of neat PLA and PLA +
3% talc composite. ... 59 Figure 2.13. Effect of annealing temperature on the gas permeability of neat PLA and PLA
+ 3% talc composite. ... 61 Figure 2.14. Effect of annealing temperature on the gas diffusion coefficient and solubility
of neat PLA and PLA + 3% talc composite. ... 63 Figure 2.15. Effect of crystallinity on hydrogen permeability of neat PLA and PLA + 3%
talc composite. ... 64 Figure 2.16. Effect of annealing temperature on the strain at break and Young’s modulus of
neat PLA and PLA + 3%talc composite. ... 65 Figure 4.1. Configuration de la vis dans l’extrudeuse. ... 80 Figure 4.2. Design des éléments de la vis utilisée pour l’extrusion. ... 81
x
Nomenclature
ASTM American Society for Testing and Materials
DMA Dynamic mechanical analysis
DSC Differential scanning calorimetry
HDPE High density polyethylene
LDPE Low density polyethylene
OPS Oriented polystyrene
PE Polyethylene
PET Polyethylene terephthalate
PLA Polylactic acid
PP Polypropylene
PS Polystyrene
PVC Polyvinyl chloride
PVDF Polyvinylidene fluoride
SEM Scanning electron microscopy
Tc Crystallization temperature
Tg Glass transition temperature
xi
Liste des symboles
A Area (cm2)
D Diffusion coefficient (cm2/s)
ΔH0
m Melting enthalpy for the 100% crystalline polymer matrix (J/g)
ΔHm Melting enthalpy for polymer matrix (J/g)
l Film thickness (cm)
L/D Screw length/barrel diameter (-)
P Permeability coefficient (Barrer = 10−10cm3(STP) cm cm⁄ 2 s cmHg)
p Gas pressure (psi)
S Solubility coefficient (cm3(STP)/cm3.cmHg)
T Absolute temperature (K)
V Volume (cm3)
xii
Remerciements
J’aimerais profiter de cette occasion pour exprimer ma gratitude à l’égard de tous ceux qui m’ont soutenu tout au long de mon parcours pour l’obtention d’une maîtrise en génie chimique.
Je voudrais tout d’abord exprimer ma gratitude pour mon superviseur, le professeur Denis Rodrigue. Il a été une grande source de soutien et un réel guide tout au long de mes activités de recherche à l’Université Laval.
Je tiens à remercier sincèrement mon codirecteur, le professeur Carl Duchesne, pour son aide, le support de son encadrement et pour ses connaissances scientifiques.
Je tiens également à remercier tous les gens qui m’ont aidé pour l’utilisation des installations nécessaires pour accomplir mon travail au département de génie chimique de l’Université Laval. En particulier M. Yann Giroux, le technicien du groupe. Je remercie également mes collègues pour leur bonne compagnie.
Je reconnais aussi le soutien financier du Conseil de Recherches en Sciences Naturelles et en Génie du Canada (CRSNG), Centre de Recherche sur des systèmes Polymères et Composites à haute performance (CREPEC), et le soutien technique du Centre de Recherche sur les Matériaux Avancés (CERMA).
xiii
Avant-propos
Ce mémoire est composé de trois chapitres, dont un article. Le premier chapitre contient une introduction générale sur l’acide polylactique ainsi que ses propriétés et ses applications. De plus, on y présente le procédé de production de pellicules, le talc, et les composites à base de PLA. Finalement, on présente l’effet du traitement thermique sur les polymères. Dans chaque cas, la littérature récente liée au sujet est présentée et discutée.
Dans le second chapitre, les effets de la teneur en talc (0-3% poids), de la durée (0-60 min) et de la température (60-120°C) du traitement thermique sur la cristallinité du PLA est présentée. Celle-ci est ensuite reliée à d’autres propriétés comme la perméabilité aux gaz et les propriétés mécaniques en tension. De plus, ce chapitre décrit la manière dont la cristallinité influence la perméabilité aux gaz et les propriétés mécaniques. Enfin, ce chapitre a été soumis pour publication et la référence est fournie ci-dessous. Ma contribution consiste en l’exécution des travaux expérimentaux, la collecte et l’analyse des données, ainsi que la rédaction de l’article qui a été corrigé par mes directeurs.
Amir Ghassemi, Siavosh Moghaddamzadeh, Carl Duchesne, Denis Rodrigue, “Effect of Annealing on Gas Permeability and Mechanical Properties of Polylactic Acid/Talc Composite Films”, Journal of Plastic Film and Sheeting, soumis, août 2016.
Le dernier chapitre comprend une conclusion générale sur le travail effectué et des recommandations pour les travaux futurs.
1
Chapter 1
1.1 Introduction
De nos jours, les polymères sont de plus en plus utilisés dans l’industrie de l’emballage alimentaire. Le type d’emballage est principalement basé sur l’application et peut être une combinaison de polymère, métal, verre, papier et carton. La durabilité et la dégradabilité des matériaux d’emballage sont deux aspects importants: le premier est important parce que l’emballage doit être stable pour protéger le contenu et le deuxième est nécessaire pour une dégradation rapide dans l’environnement (Bohlmann, 2005).
Les avantages des polymères à base de produits pétrochimiques sont: (a) un faible coût et une grande vitesse de production, (b) de hautes performances mécaniques dépendant du type de polymère, (c) de bonnes propriétés barrière et (d) une bonne étanchéité et résistance à la chaleur. D’un autre côté, ces polymères comportent de nombreux inconvénients tels que: (a) la diminution des réserves de ressources pétrolières et gazières, (b) l’augmentation des prix du pétrole et du gaz au cours des dernières décennies, (c) les préoccupations environnementales relativement à leur dégradation ou leur incinération au niveau du réchauffement climatique, (d) des coûts importants et la contamination croisée dans leur recyclage, ainsi que (e) des risques de toxicité pour les consommateurs relativement à la migration de monomères, d’oligomères ou d’additifs vers les substances comestibles (Amass et al., 1998; Chandra et al., 1998; Mohanty et al., 2000; Siracusa et al., 2008).
L’augmentation des besoins en matériaux polymériques dans les industries de l’emballage, de l’automobile et de la construction, couplée aux sources limitées de polymères à base de pétrole, a entraîné un besoin pour les polymères biosourcés. Ces polymères possèdent des sources naturellement abondantes et les polymères étant biodégradables, peuvent se décomposer dans la nature. Ceci donne plusieurs avantages par rapport aux polymères d’origine pétrochimique. Une grande quantité de matières premières fait désormais l’objet d’études en vue de produire de nouveaux polymères biodégradables (connus sous le nom de biopolymères) (Wool, 2013). Les biopolymères sont divisés en deux catégories, à savoir naturels et synthétiques (Yu et al., 2006). Parmi les polymères naturels, on trouve les
2
polysaccharides, les protéines, le caoutchouc naturel et les hydrates de carbone, tandis que du côté des polymères synthétiques on trouve les polyamides, les polyandydrides et les différents types de polyester (acide polyglycolique et acide polylactique).
1.2 L’acide polylactique
Carothers (chez DuPont) a découvert le PLA, ou polylactide, en 1932 (Mehta et al., 2005). Au début, il était seulement possible de produire un PLA de bas poids moléculaire en chauffant l'acide lactique sous vide tout en éliminant l'eau de condensation. Le problème à cette époque était donc d’augmenter le poids moléculaire. Le PLA de poids moléculaire élevé fut synthétisé en utilisant la polymérisation par ouverture de cycle du lactide. À l’origine, le PLA a été utilisé en combinaison avec de l'acide polyglycolique (PGA) comme matériau de suture et vendu sous le nom Vicryl aux U.S.A. (Jamshidian et al., 2010).
Comparé à d’autres biopolymères, les nombreux avantages de la production de PLA sont: (a) la production du monomère de lactide à partir d’acide lactique, lequel est produit par la fermentation d’un maïs ou d’une source agricole renouvelable telle que l'amidon et les racines de Tapioca, (b) la fixation de quantités significatives de dioxyde de carbone par les plants de maïs, (c) les économies d’énergie significatives, (d) la possibilité de le recycler en acide lactique par hydrolyse ou alcoolyse, (e) la capacité de produire des emballages hybrides compostables en papier-plastique, (f) la réduction du volume des sites d’enfouissement, (g) l’amélioration de l’économie agricole et (h) la capacité de modifier les propriétés physiques par le biais de modifications matérielles (Dorgan et al., 2000).
En bref, le PLA touche les sciences et les technologies liées à l’agriculture (croissance des cultures), à la biologie (fermentation) et à la chimie (polymérisation). Il est classifié comme
Generally Recognized As Safe (GRAS) par la United States Food and Drug Administration
(FDA) et est sans danger pour toutes les applications liées à l’emballage alimentaire (Conn et al., 1995; FDA, 2002).
3
1.2.1 La production de PLA
Le PLA peut être préparé à la fois par condensation directe de l’acide lactique et par la polymérisation par ouverture de cycle du lactide dimère cyclique (Figure 1.1). Parce que la voie directe de condensation est une réaction d’équilibre, les difficultés à retirer l’infime quantité d’eau dans les dernières étapes de la polymérisation limitent généralement le poids moléculaire final réalisable par cette approche. La plupart des travaux ont porté sur la polymérisation par ouverture de cycle, bien que Mitsui Toatsu Chemicals ait breveté un procédé de distillation azéotropique en utilisant un solvant à point d’ébullition élevé pour entraîner le retrait de l’eau dans le procédé d’estérification directe afin d’obtenir un PLA de poids moléculaire élevé (Enomoto et al., 1994; Kashima et al., 1995).
Figure 1.1. Différentes voies de polymérisation pour l’acide polylactique (Drumright et al., 2000).
4
Figure 1.2. Schéma de la production de PLA via le prépolymère et le lactide (Drumright et al., 2000).
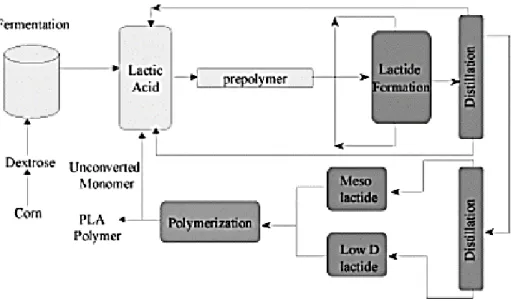
Cargill Dow LLC a breveté un processus continu et à faible coût pour la production de polymères à base d’acide lactique (Gruber et al., 1992; Gruber et al., 1993). Le processus combine les avantages environnementaux et économiques considérables de synthétiser à la fois le lactide et le PLA fondu plutôt qu’en solution et, pour la première fois, fournit un polymère biodégradable commercialement viable à partir de ressources renouvelables. Le processus débute par une réaction de condensation continue de l’acide lactique aqueux pour produire un prépolymère de PLA de faible poids moléculaire (Figure 1.2). Ensuite, le prépolymère est transformé en un mélange de lactide stéréo-isomère en utilisant des catalyseurs d’étain pour améliorer la vitesse et la sélectivité de la réaction de cyclisation intramoléculaire. Le mélange de lactide fondu est ensuite purifié par distillation sous vide. Enfin, un polymère PLA fondu de poids moléculaire élevé est produit en utilisant une polymérisation par ouverture de cycle du lactide au moyen de catalyseurs d’étain, ce qui élimine l’utilisation de solvants coûteux et nocifs pour l’environnement. Une fois la polymérisation terminée, tout monomère restant est éliminé sous vide et recyclé au début du procédé (Figure 1.3) (Westervelt, 2000).
5
Figure 1.3. Schéma de la production de PLA via le prépolymère et le lactide (Drumright et al., 2000).
1.2.2 Composition structurelle
L’acide lactique (acide 2-hydroxypropionique), le seul monomère du PLA, est produit par fermentation ou par synthèse chimique. Ses deux configurations optiquement actives, les stéréo-isomères L(+) et D(-), sont produites par la fermentation bactérienne (homo-fermentation et hétéro-(homo-fermentation) d’hydrates de carbone (Figure 1.4). La production industrielle d’acide lactique utilise le procédé de fermentation lactique au lieu de la synthèse, car les voies de synthèse présentent de nombreuses et importantes limites, incluant une capacité limitée en raison de la dépendance à l’égard d’un sous-produit d’un autre processus, l’incapacité à ne créer que le stéréo-isomère d’acide L lactique souhaité, et les coûts de fabrication élevés (Datta et al., 2006).
6
Figure 1.4. Structure schématique des stéréo-isomères L et D (Jamshidian et al., 2010).
1.2.3 Propriétés du PLA
Le PLA possède des propriétés uniques telles qu’une bonne apparence, une résistance mécanique élevée et une faible toxicité, mais ses bonnes propriétés barrière ont élargi ses applications. De nombreux chercheurs ont étudié les différentes propriétés du PLA utilisé seul ou en combinaison avec d’autres polymères sous forme de mélange ou de copolymère. Certains d’entre eux seront introduits ici.
Le groupe de Auras a étudié les propriétés mécaniques, physiques et barrière de deux types de pellicules de PLA désignés par les noms de 4030-D, lequel est nominalement constitué à 98% de L-lactide, et de 4040-D, nominalement constitué à 94% de résines L-lactide (Auras et al., 2003). Les résultats de ces deux échantillons ont été comparés à du polystyrène (PS) et du polyéthylène téréphtalate (PET). Les pellicules de PLA ont montré une bonne résistance à la traction avec des valeurs plus élevées que le PS, mais inférieures au PET. Le 4030-D et le 4040-D possédaient tous deux une température de fusion (Tm) et une température de
transition (Tg) plus faible que le PET et le PS, ce qui rend le PLA mieux adapté au scellage
à chaud et au traitement thermique. En termes de propriétés barrière, les coefficients de perméabilité du CO2 et du O2 étaient inférieurs à ceux du PS et du PET (Auras et al., 2003).
Pour les modules de traction et de flexion, le PLA possède des valeurs plus élevées par rapport au PS (polystyrène), au PP (polypropylène) et au HDPE (polyéthylène de haute densité). Pour la résistance au choc Izod avec entaille, le PLA a la plus faible valeur comparée
7
au PS, au PP et au HDPE. L’allongement à la rupture est faible et près de 4%, ce qui est tout juste supérieur à celui du PS (Dorgan et al., 2000).
La faible température de transition vitreuse du PLA limite ses usages dans les emballages traités thermiquement. En raison de sa déformation et de sa faible température de fusion, il est préférable de l’utiliser pour des applications de thermo-scellage et de thermoformage. Cinq grandes propriétés des polymères biodégradables typiques sont comparées dans le Tableau 1.1 avec le LDPE (polyéthylène basse densité), le PS et le PET. Il est donc possible de prédire les champs d’application d’un polymère par ces propriétés physiques, mécaniques et barrière.
Une autre propriété importante des polymères est leur taux de cristallinité. La cristallinité indique la quantité de régions cristallines dans le polymère par rapport au contenu amorphe. La cristallinité influence de nombreuses propriétés du polymère telles que la dureté, le module, la résistance à la traction, la rigidité, etc. Lors de la sélection d’un polymère en vue d’une application, la cristallinité joue un rôle de premier plan.
Les cristaux de PLA se présentent selon trois positions structurelles appelées les formes α, ß et γ. Ils sont caractérisés par différentes configurations d’hélices et de symétries cellulaires, lesquelles se développent selon différents traitements thermiques et/ou mécaniques. La forme α croît par fusion ou cristallisation à froid, la forme ß se développe par étirement mécanique de la forme α, qui est plus stable. La forme γ se développe sur un substrat de hexaméthylbenzène tel que rapporté récemment (Di Lorenzo, 2005).
Di Lorenzo et al. (2005) ont mesuré les taux de cristallisation du PLA sur une large plage de température, utilisant à la fois les méthodes isothermes et non-isothermes. Ils ont déterminé que le taux de cristallisation du PLA à des températures comprises entre 100 et 118°C est très élevé. Ils ont conclu que le taux de cristallisation élevé du PLA en dessous de 120°C doit être attribué au taux élevé de croissance radiale des sphérulites qui sont des régions sphériques semi-cristallines situées à l’intérieur des polymères linéaires non ramifiés (Di Lorenzo, 2005).
8
Tableau 1.1. Comparaison des propriétés des polymères biodégradables typiques avec le LDPE, le PS et le PET (Clarinval et al., 2005).
Polymère Tg (°C) Tm (°C) Résistance à la rupture (MPa) Young (MPa) Module de Élongation à la rupture (%)
LDPE (low density
polyethylene) -100 98 to 115 8 to 20 300 to 500 100 to 1000 PCL (polycaprolactone) -60 59 to 64 4 to 28 390 to 470 700 to 1000 Amidon - 110 to 115 35 to 80 600 to 850 580 to 820 PBAT (polybutyrate adipate terephthalate) -30 110 to 115 34 to 40 - 500 to 800 PTMAT (polytetramethylene adipate terephthalate) -30 108 to 110 22 100 700 PS (polystyrene) 70 to 115 100 34 to 50 2300 to 3300 1.2 to 2.5 Cellulose - - 55 to 120 3000 to 5000 18 to 55 PLA (polylactic acid) 40 to 70 130 to 180 48 to 53 3500 30 to 240 PHB (polyhydroxybutyrate) 0 140 to 180 25 to 40 3500 5 to 8 PHA (polyhydroxyalkanoate) -30 to 10 70 to 170 18 to 24 700 to 1800 3 to 25 PHB-PHV (polyhydroxybutyrate-hydroxyvalerate) 0 to 30 100 to 190 25 to 30 600 to 1000 7 to 15 PVA (polyhydroxyvalerate) 58 to 85 180 to 230 28 to 46 380 to 530 - Cellulose acetate - 115 10 460 13 to 15 PET (polyethylene terephthalate) 73 to 80 245 to 265 48 to 72 200 to 4100 30 to 300 PGA (polyglycolic acid) 35 to 40 225 to 230 890 7000 to 8400 30 PEA (polyester amide) -20 125 to 190 25 180 to 220 400
Di Lorenzo et al. (2005) ont également démontré qu’en modifiant l’architecture des liaisons par l’introduction de ramifications, différentes propriétés relatives à la fluidité du fondu étaient obtenues. Les propriétés thermiques et rhéologiques de deux types commerciaux de PLA, linéaire et ramifié, ont été étudiées (Di Lorenzo, 2005). La cinétique de cristallisation du polymère ramifié était plus rapide que celle du polymère linéaire. De plus longs temps de relaxation dans la région terminale du matériau ramifié introduisent une viscosité supérieure à faible taux de cisaillement. Ils ont conclu qu’en utilisant la modification de structure au
9
moyen de polymères ramifiés, la capacité d’utilisation du PLA dans de nombreuses opérations de traitement serait élargie.
Les propriétés optiques du PLA sont importantes dans les opérations de teinture pour les textiles et dans diverses applications d’emballage où un certain degré de clarté est souhaitable. Hutchinson et al. (2006) ont déterminé les propriétés optiques du PLA par des mesures ellipsométriques en fonction de différentes proportions de stéréo-isomères. Ils ont développé une équation pour l’indice de réfraction du PLA avec une large gamme de proportions stéréo-isomères (série L) dans les longueurs d’onde allant de 300 à 1300 nm en utilisant les coefficients de Cauchy (Hutchinson et al., 2006).
1.2.3.1 Propriétés barrière
L’un des facteurs les plus importants pour les polymères utilisés à des fins d’emballage alimentaire est leur barrière ou performance de perméabilité contre le transfert de gaz, de vapeur d’eau et de molécules aromatiques. Les propriétés du PLA relativement à sa perméation au gaz (L:D ratio 96:4) ont été étudiées (Lehermeier et al., 2001) et les valeurs suivantes ont été rapportées: à 30°C, la perméation du PLA au N2 était de 1,3
(10−10𝑐𝑚3𝑐𝑚 𝑐𝑚⁄ 2𝑠 𝑐𝑚𝐻𝑔), et l’énergie d’activation de 11,2 kJ/mole. Pour l’oxygène, les
valeurs correspondantes étaient de 3,3 (10−10𝑐𝑚3𝑐𝑚 𝑐𝑚⁄ 2𝑠 𝑐𝑚𝐻𝑔) et 11,1 kJ/mol. Les
valeurs pour la perméation du dioxyde de carbone étaient de 1,2 (10−10𝑐𝑚3𝑐𝑚 𝑐𝑚⁄ 2𝑠 𝑐𝑚𝐻𝑔) et 6,1 kJ/mole. Pour le méthane, une valeur de 1,0
(10−10𝑐𝑚3𝑐𝑚 𝑐𝑚⁄ 2𝑠 𝑐𝑚𝐻𝑔) et une énergie d’activation de 13,0 kJ/mole ont été trouvées.
Il a été conclu que la ramification de la chaîne de polymère et les changements mineurs au contenu stéréochimique L:D n’ont aucun effet sur les propriétés de perméation, mais que la cristallinité avait un impact important sur la perméation des gaz mentionnés. Par exemple, en raison de la cristallinité plus élevée de la pellicule de PLA orientée biaxialement, la perméation au CH4 était 4,5 fois inférieure à celle des autres pellicules. Les propriétés de
perméabilité du PLA pour tous les gaz étudiés étaient très semblables à celles du polystyrène (Lehermeier et al., 2001).
10
Dans une recherche effectuée par Bao et al. (2006), des résultats différents par rapport aux travaux antérieurs ont été obtenus pour la perméation du PLA à certains gaz purs. Ils ont utilisé une méthode de temporisation pour la détermination de la perméation du PLA aux gaz purs et ont déterminé la diffusivité et la solubilité du N2, CO2 et O2 dans des pellicules de
PLA. Par exemple, à 30°C, la perméabilité au N2, sa diffusivité et sa solubilité dans le PLA
(98,7% L, 1,3% D) étaient de 0,05 (10−10𝑐𝑚3𝑐𝑚 𝑐𝑚⁄ 2𝑠 𝑐𝑚𝐻𝑔), 2,4×10-8 cm2/s, et
2,2×10−4𝑐𝑚3⁄𝑐𝑚3𝑐𝑚𝐻𝑔, respectivement. L’énergie d’activation mesurée pour la
perméation au N2 était de 34,6 kJ/mole (Bao et al., 2006).
Shogren (1997) a rapporté que le taux de transmission de la vapeur d’eau du PLA à 6, 25 et 49°C, était de 27, 82 et 333 g/m2 par jour pour la forme cristalline et de 54, 172 et 1100 g/m2
par jour pour la forme amorphe, respectivement (Shogren, 1997). Il a rapporté des énergies d’activation de l’ordre de 5 et 0,1 kJ/mole pour les PLA amorphes et cristallins, respectivement.
Siparsky et al. (1997) ont utilisé un modèle “solution-diffusion” pour déterminer les paramètres de perméabilité à la vapeur d’eau de différentes pellicules de PLA, de copolymères PLA avec caprolactone, et de mélanges avec du polyéthylène glycol (Siparsky et al., 1997b). Ces paramètres comprenaient le coefficient de solubilité S (𝑐𝑚3(𝑆𝑇𝑃) 𝑐𝑚⁄ 3. 𝑐𝑚𝐻𝑔), qui est une mesure de la concentration d’eau à l’équilibre
disponible pour l’hydrolyse, ainsi que le coefficient de diffusion D (cm2/s), qui caractérise la
vitesse de diffusion de la vapeur d’eau dans la pellicule sous des conditions spécifiques. Ils ont calculé le coefficient de perméabilité P (Barrer = 10−10cm3(STP) cm cm⁄ 2 s cmHg)
au moyen de l’équation suivante:
𝑷 = 𝑺 𝑫 (1-1)
Ils ont étudié les valeurs de S et D pour des pellicules de PLA avec différents pourcentages de lactides L et D et ont conclu que le degré de cristallinité avait peu d’influence sur les paramètres de perméabilité mesurés.
Tsuji et al. (2006) ont étudié les effets du contenu en D-lactide, du degré de cristallinité (Xc)
11
d’eau (WVTR) (Tsuji et al., 2006). Ils ont observé que le WVTR des pellicules de PLA diminuait de façon monotone avec l’augmentation du Xc de 0 à 20%, tandis qu’il se stabilisait
pour un Xc supérieur à 30%. Ils ont donc suggéré que ce changement avait lieu en raison de
la résistance supérieure à la perméation de la vapeur d’eau de régions amorphes limitées par rapport à celle des régions amorphes libres. Ils ont également conclu que les changements de Mn des pellicules de PLA dans l’intervalle de 9×104 à 5×105 g/mole et de la teneur en unités
de D-lactide des pellicules de PLA dans la fourchette de 0 à 50% avaient des effets négligeables sur les valeurs de WVTR.
En outre, l’orientation modifie les propriétés barrière. Le PLA orienté (OPLA) a été étudié avec le PET et le polystyrène orienté (OPS) à l’égard de leurs propriétés physiques, mécaniques et barrière (Auras et al., 2005). Il a été conclu, en termes de barrière à la vapeur d’eau, que le PET donnait la meilleure performance, suivi par l’OPS et l’OPLA. Dans le cas des propriétés barrière à l’oxygène, le PET a montré les plus faibles coefficients de perméabilité à l’oxygène, suivi par l’OPLA et l’OPS, qui ont montré de très mauvaises performances barrière à l’oxygène.
1.2.3.2 Propriétés thermiques
Comme pour de nombreux polymères thermoplastiques, le PLA semi-cristallin présente une Tg et une Tm. Au-dessus de la Tg (~ 58°C), le PLA est caoutchouteux, tandis qu’au-dessous
de la Tg, il devient un verre encore capable de se mouvoir jusqu’à ce qu’il soit refroidi à sa
température de transition β, à environ -45°C, en dessous de laquelle il se comporte comme un polymère cassant (Henton et al., 2005). La Figure 1.5 compare les valeurs de Tg et Tm du
PLA avec d’autres polymères. Comme on le voit, le PLA a une Tg relativement élevée et une
12
Figure 1.5. Comparaison de températures de transition vitreuse et de fusion du PLA avec d’autres thermoplastiques (Lim et al., 2008).
La Tg du PLA dépend à la fois du poids moléculaire et la pureté optique du polymère (Figure
1.6). La Tg augmente avec le poids moléculaire jusqu’à des valeurs maximales à un poids
moléculaire infini de 60,2, 56,4 et 54,6°C pour un PLA constitué à 100, 80, et 50% de stéréo-isomère L, respectivement. En outre, le PLA avec une teneur plus élevée en L-lactide, possède des valeurs de Tg plus élevées que la Tg du même polymère avec la même quantité
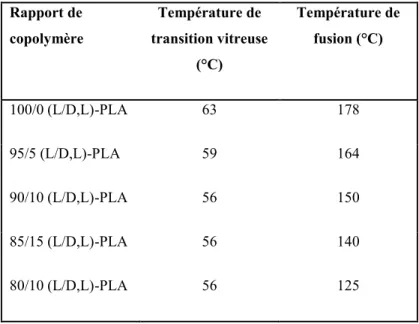
de D-lactide (Dorgan et al., 2005). Des relations similaires ont été rapportées par Tsuji (Tsuji et al., 1996). Le Tableau 1.2 montre la transition vitreuse et les températures de fusion que produisent différents PLA avec différents ratios de copolymère (Lim et al., 2008).
13
Figure 1.6. Les températures de transition vitreuse pour des PLA ayant différents ratios L:D en fonction du poids moléculaire (Dorgan et al., 2005).
Tableau 1.2. Températures de transition d’une série de copolymères PLA (Bigg, 1996). Rapport de copolymère Température de transition vitreuse (°C) Température de fusion (°C) 100/0 (L/D,L)-PLA 63 178 95/5 (L/D,L)-PLA 59 164 90/10 (L/D,L)-PLA 56 150 85/15 (L/D,L)-PLA 56 140 80/10 (L/D,L)-PLA 56 125
14
En général, la relation entre la Tg et le poids moléculaire peut être représentée par l’équation
de Flory-Fox:
𝑻𝒈= 𝑻𝒈 ∞−𝑲
𝑴̅𝒏 (1-2)
où 𝑇𝑔∞ est la 𝑇
𝑔 au poids moléculaire infini, K est une constante qui représente le volume
libre excédentaire des groupes terminaux pour les chaînes de polymère et 𝑀̅𝑛 est la masse molaire moyenne en nombre. Les valeurs de 𝑇𝑔∞ et K sont de l’ordre de 57 à 58°C et (5,5 à
7,3)×104 tel que rapporté dans la documentation écrite sur le sujet pour le PLLA (acide
L-polylactique) et de PDLLA (acide D,L-L-polylactique), respectivement (Jamshidi et al., 1988). Le comportement de la transition vitreuse du PLA dépend également de l’histoire thermique du polymère. La trempe du polymère à partir de sa masse fondue à une vitesse de refroidissement élevée (>500°C/min, comme lors du moulage par injection par exemple) se traduira par un polymère hautement amorphe. Les polymères de PLA à cristallinité faible ont tendance à vieillir rapidement, en quelques jours, à des conditions ambiantes (Celli et al., 1992; Cai et al., 1996). Ce phénomène est un contributeur important à la fragilisation du PLA.
La Tm du PLA est également une fonction de sa pureté optique. La Tm maximale pouvant
être obtenue de façon pratique pour un PLA (L ou D) stéréo-chimiquement pur est d’environ 180°C avec une enthalpie de 40 à 50 J/g. La présence de méso-lactide dans la structure du PLA peut abaisser la Tm de jusqu’à 50°C, en fonction de la quantité de D-lactide incorporée
au polymère. La Figure 1.7 montre la variation de la Tm en fonction du contenu en
méso-lactide introduit dans le PLA selon les données recueillies par Witzke (Witzke, 1997) et Hartmann (Hartmann, 1998). La relation entre la Tm et le contenu en méso-lactide peut être
assez bien estimée par l’expression suivante (Witzke, 1997):
𝑻𝒎(°𝐂) ≈ 𝟏𝟕𝟓°𝐂 − 𝟑𝟎𝟎𝑾𝒎 (1-3)
où Wm est la fraction de méso-lactide sous le niveau de 0,18 et 175°C est la température de
fusion d’un PLA constitué à 100% de L-lactide. Les valeurs typiques de la Tm pour le PLA
15
conséquences importantes, car elle contribue à élargir les fenêtres de transformation, à réduire la dégradation thermique et hydrolytique, et à diminuer la formation de lactide.
Figure 1.7. Température du pic de fusion du PLA en fonction du contenu de méso-lactide pour des données tirées de: (○) Witzke (1997) et (●) Hartmann (1998). La ligne solide est calculée sur la base de l’équation 1-3. (Witzke, 1997) (Hartmann, 1998; Lim
et al., 2008).
Pyda et al. (2004) ont déterminé la capacité thermique du PLA dans les états liquides et solides allant de 5 à 600 K (Pyda et al., 2004). La capacité thermique (Cp-liquide, J K-1 mole-1)
peut être représentée sous une forme simple:
𝑪𝐩−𝐥𝐢𝐪𝐮𝐢𝐝𝐞= 𝟏𝟐𝟎. 𝟏𝟕 + 𝟎. 𝟎𝟕𝟔𝟕 𝑻 (1-4)
où T est en Kelvin (K).
1.2.4 Applications du PLA
Le PLA a un potentiel d’utilisation dans une vaste gamme d’applications. Le Tableau 1.3 présente une vue d’ensemble du PLA de NatureWorksTM et IngeoTM avec des exemples de
16
concerne l’emballage alimentaire sont idéales pour conserver la fraîcheur des produits frais et pour les produits dont la qualité n’est pas menacée par la perméabilité du PLA à l’oxygène. Le PLA connaît une croissance comme une alternative "verte" pour les polymères utilisés dans l’emballage alimentaire. De nouvelles applications ont été revendiquées dans le domaine des produits frais, où des contenants de PLA thermoformés sont utilisés dans les marchés de détail pour les fruits, les légumes et les salades. Le potentiel d’utilisation de ces produits emballés dans le PLA est illimité.
La principale application du PLA à ce jour est dans l’emballage (près de 70%). Le Tableau 1.6 présente une estimation pour 2020. Il montre l’augmentation pour d’autres applications, en particulier dans les fibres et les tissus (Jamshidian et al., 2010).
17
Tableau 1.3. Secteurs d’activité pour les produits à base de PLA Ingeo et NatureWorks.
Secteur Entreprises Applications disponibles commercialement
1. Applications « plastique » de Ingeo
Thermoformé rigide Emballage pour fruits et légumes frais Plateaux à viande
Contenants opaque pour produits laitiers Boulangerie, herbes fraîches et bonbons Emballage électronique
Articles jetables (verres, tasses et ustensiles) Films orientés biaxialement Film de plastification
Suremballage Pellicules diverses Étiquettes
Sachets rigides
Bouteilles Lait courte durée de vie Les huiles comestibles Eau en bouteille
2. Applications « fibres » de Ingeo
Vêtements Sous-vêtements et article de mode
Non-tissés Lingettes, produits d'hygiène, couches, doublures Renfort pour le papier
Ameublement Couvertures et panneau, tissus d'ameublement Produits décoratifs
Tapis industriels Produits agricoles et géotextiles Moquette résidentiel / commercial Remplissage Oreillers, couettes, matelas, meubles
18
Tableau 1.4. Quelques produits commercialisés à base de PLA (Platt, 2006).
Produit Nom de la compagnie
Emballage
Films et plateaux pour biscuits, fruits, légumes et de la viande
Treophan, Natura, IPER, Sainsburys, Sulzer, Ecoproducts, RPC
Yogourt Crystalline/Cargill Dow
Emballage transparent rigide avec un film imprimé amovible sur le côté
Panasonic
Plateaux et bols pour la restauration rapide McDonalds Enveloppe avec fenêtre transparente, sac en papier
pour le pain avec fenêtre transparente
Mitsui, Ecocard
Agriculture et horticulture
Films pour emballage Novamont, Cargill Dow Longue durée de vie et bien de consommation
Vêtements (T-shirt, chaussettes) FILA/Cargill Dow, Kanebo Gosen
Couverture Ingeo
Boîtier du Walkman Sony
CD (compact disk) Sanyo Marvic Media/Lacea Clés informatiques Fujitsu
Petit élément pour ordinateur portable Fujitsu/Lacea Couverture de roue de secours Toyota
19
Tableau 1.5. Propriétés fonctionnelles du PLA pour l’emballage (Jamshidian et al., 2010).
Pourcentage de la production totale (2003)
Pourcentage de la production totale estimée (2020)
Secteur Cargill Dow Hycail Cargill Dow Hycail
Emballage 70 70 20 55 Bâtiment -- -- -- -- Agriculture 1 12 -- 6 Transport -- -- 20 2 Meubles -- -- -- -- Appareils électrique et électronique 1 1 10 10 Articles ménagers -- 12 -- 6
Autres (fibres et tissus) 28 3 to 5 50 21
Autres (analyse) -- -- -- --
Total 100 100 100 100
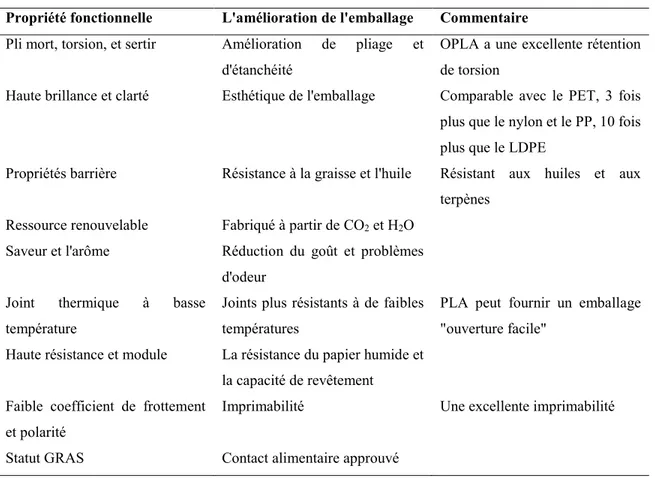
Dans le domaine de l’emballage, deux produits spécifiques ont reçu une attention particulière, à savoir les pellicules fonctionnelles et les contenants rigides thermoformés. Le PLA apporte une nouvelle combinaison d’attributs à l’emballage, comprenant la rigidité, la clarté, la thermo-soudabilité à basse température, ainsi qu’une combinaison intéressante de propriétés barrière incluant certaines caractéristiques relatives à la rétention de la saveur et des arômes. Les propriétés fonctionnelles et les avantages du PLA dans ces domaines sont présentés dans le Tableau 1.5.
Les produits de PLA commercialisés démontrent que le PLA n’est pas utilisé uniquement en raison de sa dégradabilité ou qu’il soit fabriqué à partir de ressources renouvelables. Il est utilisé parce qu’il fonctionne très bien et parce qu’il présente d’excellentes propriétés à un prix compétitif. De nombreux produits à base de PLA sont commercialisés sur le marché aujourd’hui et leur variété, ainsi que leur consommation, augmentent rapidement (Tableau 1.6).
20
Tableau 1.6. Principales applications du PLA en 2003 et estimation pour 2020 (Jamshidian et al., 2010).
Propriété fonctionnelle L'amélioration de l'emballage Commentaire Pli mort, torsion, et sertir Amélioration de pliage et
d'étanchéité
OPLA a une excellente rétention de torsion
Haute brillance et clarté Esthétique de l'emballage Comparable avec le PET, 3 fois plus que le nylon et le PP, 10 fois plus que le LDPE
Propriétés barrière Résistance à la graisse et l'huile Résistant aux huiles et aux terpènes
Ressource renouvelable Fabriqué à partir de CO2 et H2O
Saveur et l'arôme Réduction du goût et problèmes d'odeur
Joint thermique à basse température
Joints plus résistants à de faibles températures
PLA peut fournir un emballage "ouverture facile"
Haute résistance et module La résistance du papier humide et la capacité de revêtement Faible coefficient de frottement
et polarité
Imprimabilité Une excellente imprimabilité Statut GRAS Contact alimentaire approuvé
1.3 Le talc
Le talc est le constituant majeur des roches connues sous le nom de pierre à savon ou stéatite. Sa paragenèse est associée au métamorphisme hydrothermal de dolomies siliceuses et, par conséquent, il peut être accompagné de trémolite, laquelle peut être une source de préoccupation pour de nombreuses applications potentielles. Il faut donc une demande spéciale pour le talc (Wypych, 1999).
La composition du talc varie en fonction de sa source. Le facteur le plus important est la quantité de trémolite présente (Wypych, 1999). Aux États-Unis, par exemple, les talcs du Montana sont considérés comme étant sans amiante ni trémolite. Les talcs californiens, en forme de plaques, contiennent des quantités mineures de trémolite (moins de 3%), alors que les talcs durs contiennent entre 5 et 25% de trémolite. Certains talcs industriels extraits dans
21
l’état de New York contiennent de 25 à 50% de trémolite (Wypych, 1999). L’autre composant important est l’eau, laquelle se combine chimiquement dans la couche d’oxyde de magnésium ou de brucite. La Figure 1.8 présente la structure moléculaire du talc. Le talc ne peut pas perdre son eau à moins d’être chauffé à plus de 800°C mais (Wypych, 1999), dans ce cas, la structure en forme de plaques est complètement perdue et les propriétés du talc se trouvent modifiées. Les surfaces planes de la structure en forme de plaque sont maintenues ensemble par de très faibles forces de van der Waals, et par conséquent, le talc peut être délaminé à des forces de cisaillement relativement faibles, ce qui crée la texture glissante du talc et le rend facile à disperser (Wypych, 1999).
Figure 1.8. Structure moléculaire du talc (Wypych, 1999).
La structure en forme de plaque fournie aux matériaux à base de talc des propriétés importantes telles qu’une résistivité élevée et une faible perméabilité aux gaz. Cela vient du fait que la voie de diffusion est très complexe. Plusieurs autres propriétés uniques du talc sont liées à sa structure même et comprennent son effet lubrifiant, causé par son délaminage facile et sa faible abrasion parce que le talc est le plus doux minéral dans l’échelle de dureté de Mohs et des propriétés hydrophobes de sa surface. Son hydrophobie peut encore être augmentée par un revêtement de surface constitué de stéarate de zinc (Wypych, 1999). La Figure 1.9 montre la structure en forme de plaque du talc. Certaines propriétés du talc sont présentées dans le Tableau 1.7.
22
Figure 1.9. Talc de qualité Mistron (Wypych, 1999).
Tableau 1.7. Propriétés du talc (Wypych, 1999).
Noms: talc, magnesium silicate hydroxide, phyllosilicate CAS#: 14807-96-6
Formule chimique: Mg3Si4O10(OH)2 Fonctionnalité: OH or silane modifié
Composition chimique: SiO2 – 46.4-63.4%, MgO – 24.3-31.9%, CaO – 0.4-13%, Al2O3 – 0.3-0.8%, Fe2O3
– 0.1-1.8%
Les oligo-éléments: Pb, As, Cd, Zn, Ba, Sb
PROPRIÉTÉS PHYSIQUES
Densité, g/cm3: 2.7-2.85 Dureté Mohs: 1-1.5 Perte au feu, %: 4.8-17
Conductivité thermique, W/K.m: 0.02 Température maximale d'utilisation, °C: 900
Coefficient de dilatation thermique, 1/K: 8 Chaleur spécifique, kJ/kg.K: 0.82
PROPRIÉTÉS CHIMIQUES
Teneur en humidité, %: 0.1-0.6 pH de la suspension dans l'eau: 8.7-10.6
23
PROPRIETES OPTIQUES & ELECTRIQUES
Indice de réfraction: 1.57-1.59 Luminosité: 78-93 Blancheur: 70-93
Couleur: blanc Constante diélectrique: 7.5
MORPHOLOGIE
Forme des particules: lamellaire Structure en crystal: monoclinique
Clivage: basal
Taille des particules, μm: 1.4-19 Absorption d'huile, g/100 g: 22-57
Finesse Hegman: 0-7
Ratio d'aspect: 5-20 Épaisseur de la particule, μm: 0.2-6
Analyse granulométrique: résidus sur un tamis de 325 mesh: 0.1-2%
Aire de surface spécifique, m2/g: 2.6-35
PRINCIPALES APPLICATIONS: papier, peinture, toiture, plastiques, céramiques, aliments pour animaux, cosmétiques, calfeutrage, amortissement sonore, mastics, antiagglomérants, produits d'étanchéité, isolation électrique, plâtre, lubrifiants, tuiles, appareils, meubles de jardin, emballage alimentaire, film agricole.
APPLICATIONS MAJEURS POUR LES POLYMÈRES: PP, PE, PC, ABS, PPS, PS, caoutchouc.
1.4 Fabrication de pellicules par extrusion-soufflage
Dans le procédé d’extrusion-soufflage de pellicules, le PLA fondu est extrudé pour former un tube en utilisant une filière annulaire. En soufflant de l’air à travers la tête de la filière, le tube est gonflé en une mince bulle tubulaire avant d’être refroidi. Le tube est ensuite aplati entre les rouleaux de pincement et repris par l’enrouleur (Figure 1.10). Le rapport entre le diamètre de la bulle et le diamètre de la filière est appelé le rapport de soufflage (Blow-up
ratio ou BUR). Des rapports BUR de 2:1-4:1 avec une température de filière de 190-200°C
24
Tweed et al., 2006). En faisant varier le BUR, le TUR (take-up ratio), la vitesse de la vis, la pression de l’air et la vitesse de l'enrouleur, des pellicules de différentes épaisseurs (10-150 μm) et de différents degrés d’orientation peuvent être obtenus.
Le PLA a une densité spécifique d’environ 1,24 g/cm3, ce qui est beaucoup plus élevé que
les polyoléfines (0,91 à 0,96 g/cm3). Alors que le PLA peut être traité dans des extrudeuses
conçues pour les polyoléfines, si l’extrudeuse fonctionne déjà aux alentours de la puissance maximale de l’entraînement de la vis, l’extrudeuse peut ne pas avoir assez de puissance pour traiter le PLA en raison de la densité substantiellement plus élevée du PLA (Natureworks., 2003). Par rapport aux polyoléfines, le PLA possède la plus faible la résistance à l'état fondu, et donc, la formation d’une bulle stable lors de l’extrusion-soufflage est plus difficile. En conséquence, l’extrusion-soufflage de pellicules de PLA nécessite souvent l’utilisation d’additifs tels que des agents augmentant la viscosité pour renforcer sa la résistance à l'état fondu. Ces additifs protègent le polymère contre la dégradation et/ou couple les chaînes de polymères afin d’atténuer la perte globale de poids moléculaire et de viscosité de la masse fondue du polymère. Néanmoins, la formulation de ces agents de couplage est souvent un secret industriel. L’un de ces agents de couplage pour le PLA disponible sur le marché est constitué d’un copolymère de styrène, de méthyle méthacrylate et le glycidyle méthacrylate (Tweed et al., 2006). Sodergard et al. (2003) ont décrit un procédé pour stabiliser le PLA et améliorer sa la résistance à l'état fondu par l’addition d’un composé de peroxyde organique (par exemple, le butylperoxybenzoate, le peroxyde de dibenzoyle, ou le tert-butylperoxyacétate) au cours du traitement en fondu, lors duquel le peroxyde est ajouté à environ 0,01 à 3% en poids de PLA (Sodergard et al., 2003).
25
Figure 1.10. Schéma du procédé d’extrusion-soufflage (McKeen, 2012).
Puisque les pellicules de PLA sont assez rigides et ont une élongation beaucoup plus faible que les polyoléfines, l’écrasement de la bulle dans les rouleaux de pincement tend à produire des rides, lesquelles ont tendance à demeurer en permanence sur la pellicule en raison des propriétés de pli permanent élevées du PLA. Ce problème peut être surmonté en incorporant des matières de remplissage dans le PLA lors de l’extrusion. Pour réduire l’adhérence entre les pellicules, Hiltunen et al. (2000) ont mélangé un PLA avec un plastifiant de triacétine (triacétate de glycérol), avec divers agents anti-adhésion tels que le talc, TiO2 et CaCO3. Ils
ont affirmé que les pellicules soufflées résultant de ce mélange possédaient des forces de rupture meilleures que celle du polyéthylène typique et des pellicules de PP (Hiltunen et al., 2000). Des additifs de glissement (par exemple l’oléamide, le stéaramide, le N,N’-éthylène-bis-stéaramide et l’oléyle palmitamide) ont également été ajoutés pour réduire le coefficient de friction résultant du chevauchement des pellicules (Tweed et al., 2006). Typiquement, un additif de glissement de moins de 0,5-1,0% par poids du polymère est utilisé, des quantités excessives pouvant compromettre la capacité des encres d’impression et des autocollants à adhérer à la surface de la pellicule. Afin d’éviter d’avoir recours à des techniques de copolymérisation, de mélange ou à des plastifiants, Tweed et al. (2006) ont mis au point une
26
méthode pour obtenir des pellicules soufflées de PLA en augmentant la viscosité du PLA par étapes successives dans une unité de refroidissement de polymère ou encore par refroidissement interne du mandrin de la filière au moyen d’air ou de fluide pour réguler la température de la filière (Tweed et al., 2006).
1.5 Le recuit (traitement thermique)
La production de pellicule n’est généralement pas la fin du processus de production. Il existe de nombreux procédés de traitement après leur production. Les plus simples sont les processus de découpage et de finition et les processus liés à l’emballage. Il est souvent souhaitable de combiner différentes pellicules dans des structures multicouches qui ne pouvaient être produites directement par les méthodes de production de pellicules dont nous venons de parler.
Des structures multicouches peuvent être construites en laminant deux ou plusieurs pellicules ensemble. Lorsque cela est fait, un adhésif doit parfois être appliqué entre les couches afin de s’assurer qu’elles colleront l’une à l’autre. L’adhésif est souvent appliqué comme revêtement. Le revêtement continu d’une pellicule est appelé un « revêtement de bande ». D’autres revêtements sont appliqués pour modifier certaines propriétés telles que la couleur, l’aspect et la perméabilité. Les revêtements sont habituellement des liquides qui peuvent nécessiter un séchage. Certains revêtements sont appliqués au moyen de procédés de dépôt en phase vapeur ou à vide. De minces couches de métal peuvent être déposées par un processus appelé métallisation (McKeen, 2012). Les propriétés physiques de la pellicule peuvent également être affectées par son étirement, un processus appelé orientation. Tous ces processus doivent être effectués à une vitesse élevée et un coût minimal. D’autres procédés utilisés dans le traitement des pellicules sont le recuit, l’oxydation (méthode de décharge corona), le revêtement, la métallisation, l’étirage (orientation), le laminage, et le parage (McKeen, 2012).
Le recuit (traitement thermique) de polymères peut être défini comme un processus secondaire lors duquel le polymère est porté à une certaine température, maintenu pendant un certain temps, puis refroidit à la température ambiante. Le recuit des polymères semi-cristallins peut changer la structure cristalline, le degré de cristallinité, la perfection des
27
cristaux, l’orientation à la fois des phases cristalline et amorphe, la morphologie structurelle contiguë et le nombre de chaînes de liaison entre les cristallites (LeGrand, 2002). Des changements morphologiques directs sont également observés sur le recuit d’échantillons massivement cristallisés en dessous de leur température d’équilibre. Le recuit de polymères cristallins peut entraîner un certain polymorphisme puisqu’une forme cristalline se transforme en une autre. Le processus de recuit dépend du temps et de la température. Le recuit de polymères cristallins liquides entre la température de transition vitreuse et la température de fusion conduit à l’augmentation du niveau de cristallinité et à une plus grande perfection cristalline, ce qui les rapprochent du comportement des polymères semi-cristallins conventionnels. L’effet du recuit sur la microstructure du polymère est discuté à la section suivante.
1.5.1 Effet du recuit sur les polymères
1.5.1.1 Effet du recuit sur les structures conventionnelles
En général, la cristallinité pendant le recuit, dépendamment du procédé utilisé, augmente lorsque le polymère est chauffé entre les températures de transition vitreuse et de fusion (Joijode, 2011). Le chauffage augmente la mobilité des chaînes et conduit à une recristallisation. Par conséquent, les propriétés mécaniques (et spécialement les modules) sont améliorées (Joijode, 2011).
Les effets du recuit sur des fibres de Nylon 6 mènent à une augmentation de la cristallinité globale ainsi que de la taille des cristaux jusqu’à ce qu’un arrangement parfait des cristaux soit obtenu. Par conséquent, la densité de la fibre augmente de manière significative (Murthy et al., 1995). Le procédé de recuit est différent d’un procédé comme l'étirage pour les fibres et les fils. Cependant, la cristallinité est augmentée par le procédé de recuit en raison de la cristallisation des parties amorphes orientées dans la fibre, et non de l’augmentation du nombre de parties orientées par le procédé d'étirage (Murthy et al., 1995).
La taille des cristaux de polyéthylène et de fluorure de polyvinylidène a été augmentée en utilisant un recuit thermique à la température de transition (Tg) vitreuse ou au-dessus de
28
amélioré la cristallinité du polymère par nucléation et par certains mécanismes de croissance des sphérolites pour le polyéthylène téréphtalate (Zia et al., 2010). En outre, l'épaisseur des lamelles du polyéthylène a été augmentée et les grands repliements de la chaîne de molécules de polymère conduisirent à la perfection cristalline (Statton et al., 1960). Cette augmentation de l’épaisseur est due à la fusion partielle de petits cristaux et à la recristallisation au moyen de lamelles plus épaisses pendant le recuit thermique (Kawai, 1965).
Dans le procédé de recuit de l'oxyde de polyéthylène, l'état initial est la présence de cristaux non intégrés et repliés pour la cristallisation. Cependant, l’augmentation de l’épaisseur lamellaire est causée par l’augmentation de l’épaisseur des cristaux et de la chaîne repliée des polymères (Cheng et al., 1991; Cheng et al., 1990). Selon la croissance des sphérolites de l’oxyde de polyéthylène, des températures distinctes telles que la température de transition vitreuse ou la température de fusion dépendent de la taille des sphérolites et des taux de refroidissement après recuit (Cheng et al., 1990; Price et al., 1962). L'étude du recuit près de la température de fusion a montré des chaînes repliées sur les surfaces lamellaires et l'épaississement des bords lamellaires des cristaux. En outre, la recristallisation des côtés des lamelles est une source de sites de nucléation pour la cristallisation (Chen et al., 2003). La destruction de la morphologie sphérolitique par la fusion des régions cristallisées au cours du recuit à haute température leur laisse plus de possibilité pour relaxer leur morphologie cristalline initiale (Massa et al., 2003).
Un modèle du processus de recuit à haute température pour le polyéthylène téréphtalate a été présenté dans des études précédentes (Gupta et al., 1981a; Gupta et al., 1981b). Cependant, la vitesse de cristallisation augmente en même temps que la température de recuit; c’est le stade de “cristallisation primaire” où les cristaux se développent rapidement dans les noyaux existants pour les fibres de PET. Au fil du temps, de petits cristaux s’unissent et cette réduction du nombre de petits cristaux crée une parfaite formation de cristal uniforme. En poursuivant le processus de recuit, la perfection des cristaux s’améliore alors que les défauts cristallins disparaissent durant la phase de cristallisation secondaire (Gupta et al., 1981a). La Figure 1.11 présente des micrographies optiques polarisées de pellicules de PET pour (a) une vitesse de refroidissement linéaire de 1 K/min, (b) un refroidissement à 1 K/min, puis recuit à 473 K pour 60 minutes et (c) une trempe dans un bain de glace, puis recuit à 473 K
29
pendant 60 minutes (Zia et al., 2010). On voit que le refroidissement linéaire permet de former une superstructure sphérolitique et qu’un recuit post-refroidissement conduit à une augmentation de la taille des sphérolites. De petites sphérolites sont nucléées dans le matériau amorphe d’origine au cours du recuit. Également, les mécanismes de nucléation et de croissance se produisent tous les deux au cours du processus de recuit, lorsque les lamelles ont la forme cristalline dominante dans les sphérolites (Zia et al., 2010).
Figure 1.11. Micrographies optiques polarisées d’une pellicule de PET d’une épaisseur de 100 µm: a) cristallisé par fonte à 1 K/min, b) cristallisé par fonte à 1 K/min, puis recuit à 473 K pour 60 min, c) trempé dans de l’eau glacée, puis recuit à
473 K pour 60 min. La barre d’échelle est de 20 µm (Zia et al., 2010).
L’augmentation des propriétés mécaniques survient en raison de la progression de la cristallisation au cours du recuit. Le module et la résistance des pellicules sont augmentés par le procédé de recuit en raison de la cristallisation. Pendant ce temps, des chaînes de polymères rayonnent à partir du site de nucléation pour former des sphérolites. Cependant, une simple chaîne de polymère peut se trouver dans plus d’une lamelle ou sphérolites. Par conséquent, les molécules de liaison (parties inter-cristallines moléculaires) précisent la force de la pellicule polymère et les polymères de poids moléculaires élevés ont des chaînes polymères
(a)
(c)
30
plus longues. Ainsi, le nombre de molécules de liaison est supérieur à la normale et permet au polymère d’atteindre une plus grande force.
L’effet du recuit sur les propriétés mécaniques dépend de la température de recuit, du temps de recuit, du mode de recuit (par exemple, contraint ou non contraint) et de l’histoire de l’échantillon (par exemple, un pré-étirage).
D’après des études antérieures, la température joue un rôle important dans le processus de recuit pour des propriétés mécaniques telles que le module et la résistance à la traction. Près de la température de fusion pour des polymères semi-cristallins et au-delà à la température de transition vitreuse pour les polymères amorphes, sont des choix appropriés pour la température de recuit (Choi et al., 2004; Mikos et al., 1993; You et al., 2006). Par exemple, des études sur la température de recuit entre 60 et 200°C pour le Nylon 6-6 ont montré que le module du polymère se trouvait amélioré de manière significative jusqu’à 150°C en raison du mouvement des chaînes jusqu’à cette température, alors qu’un recuit à 200°C, au contraire, diminuait les propriétés mécaniques (Babatope et al., 1992). En comparant le recuit à 60°C (près de la Tg) et le recuit à 100°C, les propriétés mécaniques s’amélioraient
davantage à 60°C à cause de la rupture des liens d’hydrogène près de la température de transition vitreuse, mais les chaînes de polymère ne disposaient pas de suffisamment de mobilité, alors qu’un recuit à 100°C entraîne la rupture des liaisons hydrogène puis la création de nouvelles liaisons avec des chaînes mobiles. En outre, les résultats démontrent de moins bonnes propriétés mécaniques pour un recuit à 200°C en raison de la dégradation thermique des molécules de liaison (Babatope et al., 1992).
Un autre facteur ayant un impact sur la microstructure du polymère est le mode de recuit (contraint ou non contraint). Toutefois, Gupta et Kumar (1981) ont rapporté l'effet du type de recuit sur les propriétés mécaniques des filaments de PET. La Figure 1.12 montre que les échantillons contraints ont une contrainte à la rupture plus élevée et un allongement inférieur à celui de l’échantillon témoin, mais les échantillons sans contrainte sont fragiles et non ductiles (Gupta et al., 1981b).
31
Figure 1.12. Courbes de contrainte-déformation d’échantillons recuits pendant 1 min à 100, 160 et 220°C pour l’échantillon de contrôle, FA (non contraint) et TA
(contraint) (Gupta et al., 1981b).
Pour l’échantillon non contraint, les molécules de liaison orientées sont en petit nombre. Une concentration élevée de tension peut donc aisément déformer le polymère. La température de déformation augmente avec le recuit et la contrainte diminue du fait des orientations réductrices des chaînes amorphes du polymère. Au contraire, dans le recuit contraint, l’élongation des chaînes de polymère se produit de façon uniforme. Donc, la distribution des déformations est uniforme et la phase cristalline résiste à la déformation. Une orientation supérieur de la zone amorphe est donc plus résistante jusqu’au point de rupture (Gupta et al., 1981b).