La pathologie et la neuroinflammation liées à TDP-43
dans des différents modéles d'ischémie cérébrale: le
rôle thérapeutique potential d'un analogue de la
Withaferin A (IMS-088)
Thèse
Saisampath Thammisetty
Doctorat en sciences pharmaceutiques
Philosophiæ doctor (Ph. D.)
La pathologie et la neuroinflammation liées à
TDP-43 dans des différents modéles d’ischémie
cérébrale: Le rôle thérapeutique potential
d’un analogue de la Withaferin A (IMS-088)
Thèse
Sai Sampath Thammisetty
Sous la direction de :
Jasna Kriz, directeur de recherche
Frédéric Calon, codirecteur de recherche
Résumé
Les accidents vasculaires cérébraux (AVC) résultent d’une insuffisance d’apport sanguin au cerveau et constituent la troisième cause de décès mondial. Expérimentalement et cliniquement, un AVC est suivi de réactions inflammatoires aiguës et prolongées et environ un tiers des survivants souffrent de démence post-AVC. Bien que plusieurs mécanismes aient été expliqués comme étant impliqués dans la pathologie de l’AVC, une thérapie efficace visant à minimiser les conséquences post-AVC n’a toujours pas été établie. Dans cette thèse, nous avons exploré la pathologie de la protéine TDP-43 et l’association de la neuroinflammation dans le modèle d’occlusion de l’artère cérébrale moyenne dans le vieillissement (chapitre 2), un modèle d’hypoperfusion cérébrale chronique (HCC) : modèle pour la démence vasculaires et les effets thérapeutiques d’IMS-088 dans le modèle d’hypoperfusion cérébrale chronique (chapitre 3). Dans le premier chapitre, nous observons qu’une accumulation cytoplasmique de TDP-43 ubiquitinée dépendante de l’âge peut moduler les réponses inflammatoires post-ischémiques comme l’activation microgliale et de grands infarctus chez les souris âgées post-accident vasculaire cérébral (AVC). Nous avons également observé qu’une augmentation/surexpression de TDP-43 potentialise les réponses NF-κB et les lésions ischémiques post-AVC. Finalement, nous observons la présence dans le cytoplasme de structures immunoréactives de TDP-43 dans des tissus humains post-mortem-AVC.
Dans le chapitre 2, nous montrons que HCC induit une accumulation cytoplasmique en plus de la formation d’agrégats insolubles de phospho-TDP-43 dans les neurones corticaux. La présence de TDP-43 cytoplasmique a également été détectée dans les maladies humaines (démence vasculaire). La dérégulation de TDP-43 a été associée avec une activation microgliale et de la voie NF-κB en plus du développement de déficits moteurs et cognitifs après 2 mois de HCC. Finalement, dans le chapitre 3, nous avons étudié l’efficacité du nouveau médicament IMS-088 (analogue semi-synthétique de withaferin A) qui vise à moduler l’inflammation post-ischémique en ciblant la voie NF-κB chez les souris HCC. Après 2 mois de traitement oral de IMS-088, les souris HCC ont montré une amélioration importante des résultats cognitifs et moteurs en plus d’une restauration de la balance nucléaire et cytoplasmique de TDP-43 ainsi qu’une diminution de l’activation microgliale,
de la mort cellulaire et une amélioration de l’autophagie. Selon ces résultats, IMS-088 devrait être considéré comme un candidat potentiel pour des interventions thérapeutiques futures pour HCC et pour d’autres protéinopathies avec TDP-43.
Abstract
Stroke results from insufficient blood supply to the brain and is the third leading cause of death in the world. Experimentally and clinically, stroke is followed by acute and prolonged inflammatory responses, and about a third of stroke survivors suffer from post-stroke dementia. Although numerous mechanisms have been explained to be involved in stroke pathology, an effective therapy to minimize post stroke consequences is still to be established. In this thesis, we have explored TDP-43 protein pathology and associated neuroinflammation in middle cerebral artery occlusion model (MCAO) in aging (chapter 2) and in chronic cerebral hypo-perfusion model(CCH): model for vascular dementia and therapeutic effects of IMS-088 in the chronic cerebral hypo-perfusion (CCH) model (chapter 3). In the chapter 2, we observed age-dependant cytoplasmic accumulation of ubiquitinated TDP-43 can modulate the post-ischemic inflammatory responses like microglial activation and large infarctions after acute stroke. We also observed that an increase / over expression of TDP-43, potentiates NF-κB response and ischemic injury after stroke. Finally, we observed the presence of cytoplasmic TDP-43 immunoreactive structures in post-mortem-stroke human brain tissues.
In the chapter 3, we reported that CCH induces cytoplasmic accumulation and formation of insoluble phospho-TDP-43 aggregates in cortical neurons. Presence of cytoplasmic TDP-43 was also detected in human disease (vascular dementia). Further, mislocalization of TDP-43 was associated with microglial and NF-κB activation as well as development of cognitive and motor impairments after 2 months of CCH. Finally, in chapter 3 we studied the efficacy of a novel drug IMS-088 (semi-synthetic analog of withaferin A) which is aimed to modulate post ischemic inflammation by targeting NF-κB pathway in the CCH mice. After 2 months of oral IMS-088 treatment, CCH mice showed marked improvement in cognitive and motor outcome along with restoration of nuclear vs cytoplasmic TDP-43 balance and decreased microglial activation, cell death and improved autophagy. Basing on these evidences, IMS-088 should be considered as potential candidate for therapeutic interventions in CCH and other TDP-43 proteinopathies.
Table of contents
Résumé ... iii
Abstract ... v
Table of contents ...vii
List of Tables ... xi
List of Figures ... xiii
List of Abbreviations ... xv
Acknowledgement ... xvii
Foreword ... xix
1. Introduction ... 1
1.1 Trans active response DNA binding protein 43 ... 2
1.2 TDP-43 Pathophysiology in Disease ... 5
1.3 TDP-43 in Neurodegenerative disorders ... 9
1.3.1 Amyotrophic lateral sclerosis and frontotemporal dementia ... 9
1.3.2 Alzheimer’s disease ... 10
1.3.3 Parkinson’s disease and Parkinson-dementia complex ... 11
1.3.4 Huntington’s disease... 12
1.4 Cerebral ischemia ... 13
1.4.1 Pathophysiology of stroke ... 13
1.4.2 Molecular mediators of neuroinflammation in cerebral ischemia ... 15
1.4.3 Cellular mediators of inflammation ... 18
1.4.4 Post stroke dementia ... 24
1.4.5 Animal models of stroke... 25
1.5 Withania somnifera or its derivatives as therapeutic candidates for cerebral ischemia ... 27
1.6 Hypothesis and Aims of the thesis ... 41
1.7 References ... 44
2. Chapter - Age-related cytoplasmic mislocalization of TDP-43 enhances NF-kB mediated inflammation and exacerbates ischemic injury... 65
2.1 Preamble ... 66
2.2 Résumé ... 66
2.3 Abstract ... 67
2.5 Materials and methods ... 70
2.6 Results ... 74
2.6.1 Increase in cytoplasmic TDP-43 and pathological TDP-35, TDP-25 fragments in the 12 months old mice after MCAO ... 74
2.6.2 Formation of TDP-43-Ubiquitin aggregates in neuronal cells and TDP-43 mislocalization in microglial cells after MCAO in 12 months old mice ... 77
2.6.3 Enhanced inflammatory response in 12 months old mice 72 hrs after MCAO ... 80
2.6.4 Larger ischemic lesions and increased neuronal death are observed in 12 months old mice after MCAO ... 83
2.6.5 TDP-43 cytoplasmic mislocalisation exacerbates ischemic injury and neuroinflammation after stroke ... 85
2.6.6 Increase in the cytoplasmic TDP-43 immunoreactivity in human stroke ... 89
2.7 Discussion ... 90
2.8 Conclusion ... 94
2.9 Acknowledgements ... 94
2.10 Funding ... 94
2.11 References ... 95
3. Chapter - IMS-088 reverses TDP-43 pathology and improves cognitive and motor deficits in a model of vascular dementia ... 99
3.1 Preamble ... 100
3.2 Résumé ... 100
3.3 Abstract ... 101
3.4 Introduction ... 102
3.5 Materials and Methods ... 104
3.6 Results ... 109
3.6.1 Chronic cerebral hypoperfusion causes deregulation of TDP-43 in mouse and human disease ... 109
3.6.2 After chronic cerebral hypoperfusion, microglial cells display inflammatory response and TDP-43 deregulation... 112
3.6.3 Chronic hypoperfusion increases NF-κB mediated neuroinflammation and neuronal death... 115
3.6.4 CCH leads to an early onset of cognitive deficits followed by motor impairment ... 116
3.6.5 IMS-088 restores the nuclear TDP-43 and abrogates the formation of TDP-35 and TDP-25 pathological fragments... 118
3.6.6 IMS-088 treatment decreased microglial activation by reducing NF-B activation
pathway ... 121
3.6.7 IMS-088 treatment alleviates cognitive and motor performance in CCH mice 124 3.6.8 IMS-088 promotes clearance of the insoluble phospho-TDP-43 aggregates by promoting autophagy: helps in minimizing cell death ... 126
3.7 Discussion ... 129
3.8 Acknowledgements ... 135
3.9 References ... 135
4. General discussion ... 143
4.1 General discussion ... 144
4.2 Inflammation in stroke / neurodegeneration ... 145
4.2.1 Microglia in cerebral ischemia/neuroinflammation ... 145
4.2.2 Nuclear factor kappa B (NF-κB) in post stroke inflammatory ... 148
4.3 Neuroinflammation in cognitive impairment ... 149
4.4 Role of autophagy in cerebral ischemia ... 151
4.5 Post stroke dementia (PSD) and therapeutic interventions ... 152
4.5.1 Interventions ... 153
4.6 Conclusion ... 154
List of Tables
Table 1.1. TDP-43 RNA targets involved in neural development and function (Sephton et al.,
2012) ... 5
Table 1.2 Animal models of stroke (Krafft et al., 2012) ... 26
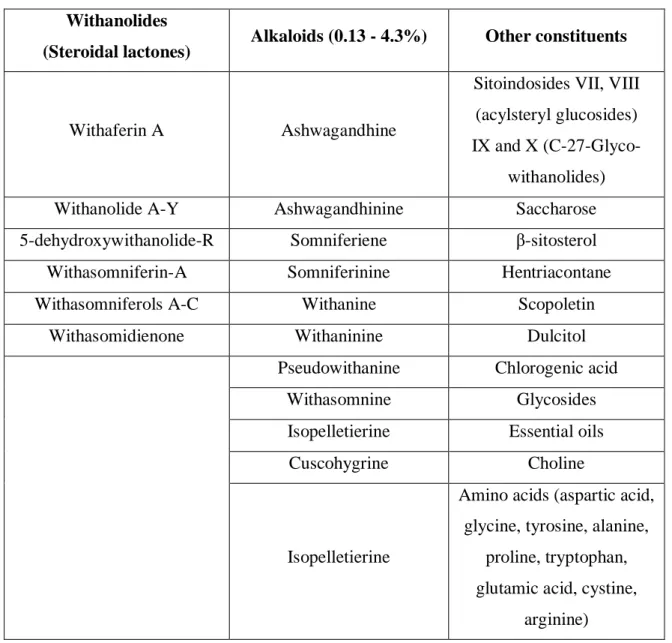
Table 1.3 Major constituents of the Withania somnifera plant extracts (adapted from Dutta et al., 2017) ... 28
Table 1.4 Summary of clinical trials with WS extracts depicting Dosage and Duration updated and modified from (Dutta et al., 2017) ... 31
Table 1.5 IC50 values of WA in cell culture models ... 33
Table 1.6. IC50 for NF-κB activation inhibition by novel withanolides ... 40
Table 2.1. Details about the stroke human subjects used in the project ... 74
List of Figures
Figure 1.1. The TDP-43 architecture __________________________________________ 3 Figure 1.2 RNA processing pathways controlled by TDP-43 (Buratti and Baralle, 2012) _ 3 Figure 1.3 Cell death and activation of pattern recognition receptors (Iadecola and Anrather, 2011) __________________________________________________________________ 21 Figure 1.4. Phenotypes and activation of microglia after ischemic stroke _____________ 22 Figure 1.5 Resolution of inflammation and tissue repair (Iadecola and Anrather, 2011) _ 23 Figure 1.6. Structure of Withaferin A or 5,6-epoxy-4,22,27-trihydroxy-1-oxoergosta-2,24-dienoic acid delta-lactone. Molecular formula C28H38O6. Molecular weight 470.606 g/mol
______________________________________________________________________ 32 Figure 1.7 Activation of nuclear factor-κB: Canonical and non-canonical pathways (Rosebeck et al., 2016) ____________________________________________________ 35 Figure 1.8. Structural modifications made in the WA core structure where for Withaferin A: R = R = H, for 27-O-methylwithaferin A: R1 = CH3, R = H; for 4-O-methylwithaferin A: R1
= H, R2 = CH3; for 4.27-O-dimethylwithaferin A: R1 = R2 = CH3__________________ 39
Figure 2.1. Increase in cytoplasmic TDP-43 and pathological TDP-35, TDP-25 fragments in the 12 months old mice after MCAO _________________________________________ 76 Figure 2.2. Formation of TDP-43-Ubiquitin aggregates in neuronal cells and TDP-43 mislocalization in microglial cells after MCAO in 12 months old mice ______________ 79 Figure 2.3. Enhanced inflammatory response in 12 months old mice 72 hrs after MCAO 82 Figure 2.4. Larger ischemic lesions and increased neuronal death are observed in 12 months old mice after MCAO _____________________________________________________ 84 Figure 2.5. TDP-43 mislocalisation exacerbates ischemic injury and neuroinflammation after stroke _________________________________________________________________ 87 Figure 2.6. TDP-43 mislocalization was observed in human subjects died from stroke __ 90 Figure 3.1. Chronic cerebral hypoperfusion causes deregulation of TDP-43 in mouse and human disease __________________________________________________________ 111 Figure 3.2. After chronic cerebral hypoperfusion, microglial cells display inflammatory response and TDP-43 deregulation. _________________________________________ 114 Figure 3.3. Chronic hypoperfusion increases NF-κB mediated neuroinflammation, neuronal death and early onset of cognitive deficits followed by motor impairment ___________ 117 Figure 3.4. IMS-088 restores the nuclear TDP-43 and abrogates the formation of TDP-35 and TDP-25 pathological fragments ____________________________________________ 120 Figure 3.5. IMS-088 treatment decreased microglial activation by reducing NF-B activation pathway. ______________________________________________________________ 123 Figure 3.6. IMS-088 treatment alleviates cognitive and motor performance in CCH mice _____________________________________________________________________ 125
Figure 3.7. IMS-088 promotes clearance of the insoluble phospho-TDP-43 aggregates by promoting autophagy: helps in minimizing cell death ___________________________ 128
List of Abbreviations
Aβ : Amyloid beta
AD : Alzheimer’s’ disease ADP : Adenosine di phosphate AICD : Amyloid intra cellular domain AIF : Apoptosis inducing factor APP : Amyloid precursor protein ALS : Amyotrophic lateral sclerosis
BALB/c : Albino laboratory bred strain of the house mouse BCL3 : B cell lymphoma encoded protein 3
BSA : Bovine serum albumin b.w : Body weight
C90rf72 : Chromosome 9 open reading frame 72 CCL2 : Chemokine(c-c) motif ligand 2
CCL5 : Chemokine(c-c) motif ligand 5 CCD : Charge coupled device
CCH : Chronic cerebral hypo-perfusion
CD11b : Cluster of differentiation 11b (macrophage marker) Cox-2 : Cyclooxygenase 2
DMSO : Dimethyl sulfoxide
DNA : De-oxy ribose nucleic acid
EDTA : Ethylene di-amine tetra acetic acid FTLD : Fronto-temporal lobar degeneration FUS : Fused in sarcoma
GFAP : Glial fibrillary acidic protein GFP : Green fluorescent protein
GM-CSF : Granulocyte monocyte colony stimulating factor HEK293 : Human embryonic kidney cells 293
HEPES : Hydroxyethyl piperazine ethane sulfonic acid HIV : Human immune deficiency virus
HnRNP : Heterogeneous ribonucleoprotein HO1 : Hemeoxygenase 1
HSP90 : Heat shock protein 90 HTT : Huntingtin gene
Iba-1 : Ionized calcium binding adapter molecule 1 IC50 : Half maximal inhibitory concentration
ICAM1 : Intercellular adhesion molecule 1 IGF-1 : Insulin growth factor 1
INOS : Inducible Nitric oxide synthase IL-1β : Interleukin-1 beta
IL-4 : Interleukin-4 IL-6 : Interleukin-6 IL-10 : Interleukin-10 IL-17 : Interleukin-17
IKB : Inhibitor of kappa B IKBβ : Inhibitor of kappa B β
IKB : Inhibitor of kappa B IKKB : Inhibitory kappa kinase B i.p : Intraperitoneal
LD50 : Median lethal dose
LMN : Lower motor neuron LPS : Lipopolysaccharide MCA : Middle cerebral artery
MCAO : Middle cerebral artery occlusion MMP : Matrix metallo proteinase m-RNA : Messenger ribo nucleic acid m-TOR : Mammalian target of rapamycin
Myd88 : Myeloid differentiation primary response gene 88 NEMO : Nuclear factor kappa B essential modulator NeuN : Neuronal nuclear marker
NF-κB : Nuclear factor kappa B
NIK : Nuclear factor kappa B inducing kinase NRP-1 : Receptor neurophilin-1
OPTN : Optineurin protein
PARP1 : Poly ADP-Ribose Polymerase-1 PBS : Phosphate buffer saline
PD : Parkinson disease P.O : Per oral
PSD : Post stroke dementia PVDF : Polyvinyl difluoride
RRM1 : Ribonucleotide-diphosphate reductase 1 RRM2 : Ribonucleotide-diphosphate reductase 2 SDS : Sodium do decyl sulphate
SiRNA : Small interference ribo nucleic acid SOD1 : Superoxide dismutase
TDP-43 : Trans active response DNA binding protein-43 TG : Thioguanine
TGF- : Transforming growth factor beta TLR2 : Toll like receptor 2
TLR2KO : Toll like receptor 2 knock out TNFα : Tumor necrosis factor α
UCCAO : Unilateral carotid artery occlusion UMN : upper motor neuron
UTR : Untranslated region VaD : Vascular dementia
VEGF : Vascular endothelial growth factor WA : Withaferin A
Acknowledgement
The work described in this thesis would not have been possible without the invaluable guidance, instruction, assistance and support that I received from many people during my years as a graduate student in University Laval. I am using this opportunity to express my gratitude to everyone who supported me throughout my graduation period.
First and foremost, I would like to express my deepest sense of gratitude to my supervisor Dr. Jasna Kriz, for giving me the opportunity to do my doctoral training in her laboratory and for being an exceptional mentor over the years. I could not have imagined a better supervisor for my PhD. Her strong passion for science, adherence to excellence, integral and simplistic view on scientific matters and continual guidance in every aspect of my training had significant influence on me as a researcher, professional, and individual. I am forever grateful to her for all of the time, care, and effort that she has put into my training. On the personal level she has always amazed me by her patience and humility. Without her encouragement, patient way of guidance and sharing knowledge, this work would have been impossible. Words cannot describe how grateful I am for having come across Dr. Jasna Kriz in my life.
I would also like to sincerely thank Dr. Frédéric Calon for all his guidance over the years. He has always been there to give sound advice and careful instructions which have helped to shape the work presented in this thesis. He has been patient with me to guide through the difficulties and troubles during the lab work.
I am really grateful to Dr. Jean-Pierre Julien, Professor, Faculty of Medicine, University Laval and chief scientific officer IMSTAR therapeutics. Part of the work presented in this thesis could not have been possible without his generous support in providing the novel drug to test in the experimental model I used.
I express heartful thanks to my brother Dr. Kallol Dutta who patiently explained to me details about scientific and technical aspects of protocols that I used in the thesis and has always been kind enough to share his knowledge related to science field.
I specially thank a wonderful human being Dr. Hejer Boutej who constantly motivated me during my odds and taught me techniques related to molecular biology.
All this work would have not been as successful without the nice atmosphere among colleagues in the lab. I owe special thanks to Yuan Cheng Weng without his hand in inducing stroke this work would not be possible. I would like to thank our lab manager Melanie Lalancette-Hebert for her valuable suggestions in shaping the manuscripts for publication. I would like to thank Reza Rahimian for always sharing his scientific knowledge and for his kindly help in giving me valuable comments in writing thesis. I owe special thanks to Priyanka Patel, Manasa and Mena Fatma for sisterly love and affection. I would like to thank my dear friend Dr. Vincent Picher-Martel for his fun-filled friendly support outside and inside the lab. I would like to thank Dr. Anna Planeus and Dr. Saikali for their generous support in providing the post-mortem human brain sections for carrying out experiments. Also, I would like to thank the members of the lab, past and present, who laid the groundwork for, or contributed to, the work presented here: Dr. Daniel Phaneuf, Dr. Yasuyuki Ohta, Dr. Senthil Krishansamy, Dr. Prakash Kumar Mishra, Dr. Pierre Cordeau Jr, Dr. Silvia Pozzi, Dr. Yohei Iguchi, Dr. Karine Plouard, Genevieve Soucy, Christine Bareil, Louis Charles Beland, Tereza Iljutic, Laurence Renaud, Romina Barreto, Sunny and Bunny.
I would like to thank Mr. Kapil Sehgal for his unconditional help with confocal microscopy. I would like to express my thanks to my friends in Quebec Mr. M. Siddhartha, Dr. Ramesh Muddala, Mrs. Shrabani Hazra Dutta, Dr. Ranjan Maity, Dr. Jayesh Patel, Dr. Hemanta Adhikary, Dr. Arojit Mitra, Dr. Minty Thomas, Dr. Pallavi Jain, Jaya Mishra, Mrs. Kanchan Bisht, Mrs. Anandita Parasor, Mrs. Richa and Mrs. Praveena Uppari for many fun-filled memories. To all the little angels Martha, Kairav, Alice, Kenza, Camilla and Aaria, I would like to say thank you for all the joy and serenity whenever you played with me.
The chain of my gratitude would be definitely incomplete if I would forget to express my deepest regards and sincere thanks to my parents and my family for encouraging me in every decision I made in my life, always believing in my choices that led me to this place. Special thanks to my mother, without her continuous support and encouragement, I never would have been able to achieve my goals. Thank you.
Foreword
Current thesis covers the literature review that brief the information regarding general introduction about TDP-43 protein and its role in various neurodegenerative diseases in first part of the first chapter. In the second part of the first chapter provides general information regarding cerebral ischemia and pathological events involved. This part also covers the various neuroinflammatory mediators and animal models involved in cerebral ischemia. The final part of the first chapter covers the information regarding various Withania somnifera or its derivatives as therapeutic candidates for cerebral ischemia.
The second chapter of this thesis is presented in the form of a research manuscript where I am the principal author. The manuscript was accepted for publication in the journal of neuroinflammation on October 26-2018 and the manuscript number is JNEU-D-18-00533R1
Age-related cytoplasmic mislocalization of TDP-43 enhances NF-kB mediated inflammation and exacerbates ischemic injury. Sai Sampath Thammisetty; Jordi
Pedragosa; Yuan-Cheng Weng; Frederic Calon; Anna Planas; Jasna Kriz.
The third chapter of this thesis is presented in the form of a research manuscript where I am the principal author. The manuscript will be submitted soon.
IMS-088 reverses TDP-43 pathology and improves cognitive and motor deficits in a model of vascular dementia. Sai Sampath Thammisetty; Laurence Renaud; Vincent
Picher-Martel; Yuan Cheng Weng; Frédéric Calon; Stephan Saikali; Jean-Pierre Julien; Jasna Kriz.
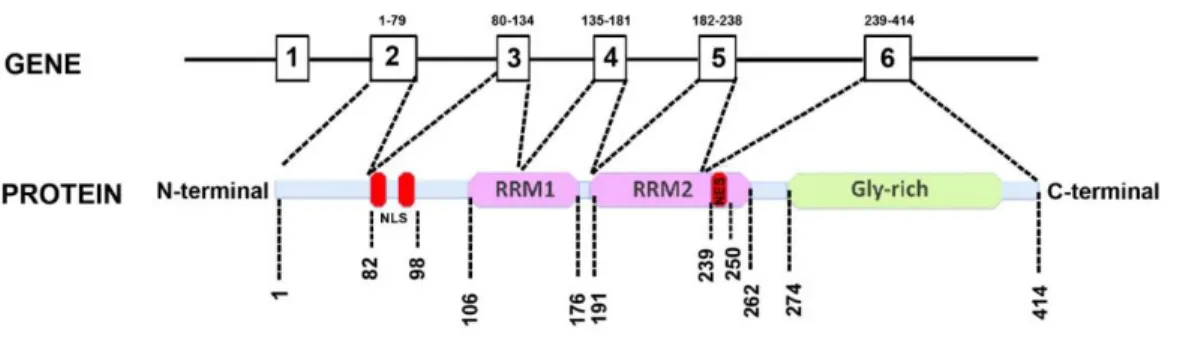
1.1 Trans active response DNA binding protein 43
The transactive response DNA binding protein of molecular weight 43 or, TDP-43 in short, was originally described as a putative inhibitor of human immune-deficiency virus (HIV) transcription which mediated its action either by altering or blocking the assembly of transcription complexes that are capable of responding to viral Tat protein (Ou et al., 1995). The eukaryotic TDP-43 is highly conserved, ubiquitously expressed nuclear protein that is capable of shuttling between the nucleus and cytoplasm in a transcription-dependent manner (Ayala et al., 2008). In humans it is encoded by the TARDBP gene in chromosome 1 which is made of six exons that can be alternatively spliced to yield 11 different isoforms, with the mRNA encoding TDP-43 being the major species (Wang et al., 2004). The full-length protein contains a N-terminal domain, two highly conserved RNA recognition motifs (RRM1 and RRM2), as well as a glycine-rich C-terminal sequence (Figure 1.1). Both the RRM domains are involved in multiple aspects of RNA metabolism, including transcription, splicing, RNA transport, RNA stability and turnover as well as microRNA biogenesis (Buratti and Baralle, 2010; Freibaum et al., 2010) (Figure 1.2). The RRM1 domain reportedly binds to single-stranded UG or TG repeat motifs with very high affinity (Ayala et al., 2005) whereas the RRM2 also contains a leucine-rich nuclear export signal (Winton et al., 2008). Incidentally, the RRM1 and C-terminal domain are also necessary for autoregulation of TDP-43 that requires RNA‐binding activity through the association to specific sequences in the 3′ UTR of the TDP‐43 transcript as well as the presence of the 321–366 amino acid region (Ayala et
al., 2011; Avendano-Vazquez et al., 2012).
The glycine-rich C-terminus, a region that mediates protein-protein interactions, is required for TDP-43 to participate in alternative splicing, and also enables TDP-43 to bind to several proteins of diverse nature and function including the heterogeneous nuclear ribonucleoprotein (hnRNP) family, a class of proteins involved in the biogenesis of mRNA (Figure 1.2) (Wang et al., 2004; Buratti et al., 2005). Thus, given the role of TDP-43 in alternative splicing, it is not surprising that in cell culture models and human post mortem brain tissue,
Figure 1.1. The TDP-43 architecture
TAR DNA-binding protein 43 (TDP-43) protein contains two RNA-recognition motifs (RNA-recognition motif 1 (RRM1) and RRM2), a carboxy-terminal glycine-rich domain, a bipartite nuclear localization signal (NLS) and a nuclear export signal (NES) (Warraich et
al., 2010; Vanden Broeck et al., 2014).
Figure 1.2 RNA processing pathways controlled by TDP-43 (Buratti and Baralle, 2012)
The major nuclear and cytoplasmic processes in which TDP-43 has been shown to play a role under physiological conditions are shown. Key protein–protein interactions with hnRNPs and FUS/TLS are shown together with the pathways that are most affected by these interactions. Ovals represent TDP-43 (green), FUS-TLS (yellow), hnRNP of the A/B family (orange), other hnRNPs proteins or factors (grey). The red line represents RNA and the continuous black line represents DNA.
full-length TDP-43 has been localized predominately to the nuclear compartment. However, as first reported by Buratti and Baralle as unpublished observations, later studies have verified the presence of small amounts of cytosolic TDP-43 under normal, physiologic conditions (Buratti and Baralle, 2008; Winton et al., 2008).
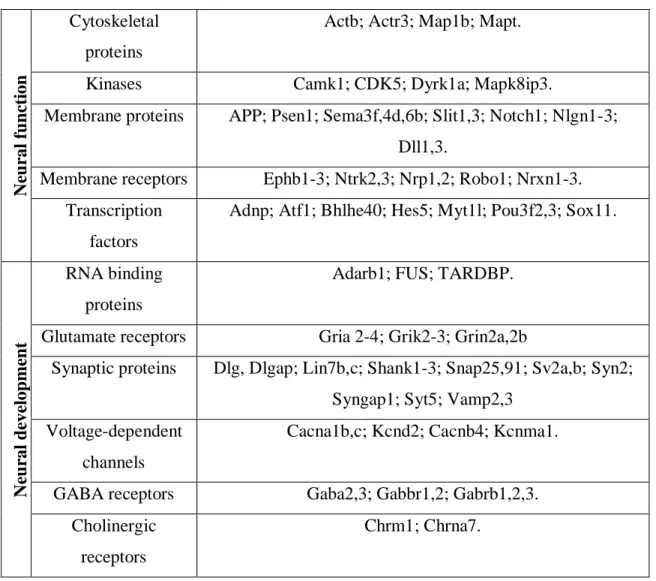
Within the nervous system, TDP-43 binds to >6,000 pre-mRNAs and affects the levels of ∼600 mRNAs and the splicing patterns of another 950 (Polymenidou et al., 2011). TDP-43 is also involved in neural development and function through its interactions with other RNA binding proteins (RNP) and its regulation of specific RNA targets as shown in Table 1.1.
In addition to full-length TDP-43 (observed at 43 KDa), other isoforms of the protein have also been detected in human brain and spinal cord tissues. A 35 KDa isoform is controversial, with reports claiming it to be either a C-terminally cleaved fragment (Che et
al., 2011; Kitamura et al., 2016) or N-terminally truncated spliced variant of the full length
protein (Xiao et al., 2015). Aggregates of this isoform reportedly cause formation of cytoplasmic inclusions and results in alteration of RNA processing and neuronal death (Che
et al., 2015). The C-terminally cleaved TDP-35 has been shown to contain the 2 intact RRM
domains as well as the glycine rich region (Kitamura et al., 2016). A 25 KDa C-terminally cleaved shorter fragment (CTF25) has also been reported by several investigators which lacks RRM1 and a portion of RRM-2 domain. Rodent models expressing this CTF25fragment has been reported to show cognitive impairment similar to that observed in Frontotemporal lobar degeneration (FTLD) models and its effects could be further altered by aging or stress (Caccamo et al., 2012; Caccamo et al., 2013). The cleavage site that generates CTF25 has been reported to be at Asp174. TDP-43 is cleaved initially after Asp174 by the endoplasmic reticular membrane-bound enzyme caspase-4, which further results in activation of caspases-3/7 to accelerate TDP-43 fragmentation. Experimental blockage of this cleavage resulted in a severe delay in TDP-43 clearance and prolonged necrotic cell death (Li et al., 2015).
N eu ral fu n cti on Cytoskeletal proteins
Actb; Actr3; Map1b; Mapt.
Kinases Camk1; CDK5; Dyrk1a; Mapk8ip3.
Membrane proteins APP; Psen1; Sema3f,4d,6b; Slit1,3; Notch1; Nlgn1-3; Dll1,3.
Membrane receptors Ephb1-3; Ntrk2,3; Nrp1,2; Robo1; Nrxn1-3. Transcription
factors
Adnp; Atf1; Bhlhe40; Hes5; Myt1l; Pou3f2,3; Sox11.
N eu ral d eve lop me n t RNA binding proteins
Adarb1; FUS; TARDBP.
Glutamate receptors Gria 2-4; Grik2-3; Grin2a,2b
Synaptic proteins Dlg, Dlgap; Lin7b,c; Shank1-3; Snap25,91; Sv2a,b; Syn2; Syngap1; Syt5; Vamp2,3
Voltage-dependent channels
Cacna1b,c; Kcnd2; Cacnb4; Kcnma1.
GABA receptors Gaba2,3; Gabbr1,2; Gabrb1,2,3. Cholinergic
receptors
Chrm1; Chrna7.
Table 1.1. TDP-43 RNA targets involved in neural development and function (Sephton
et al., 2012)
1.2 TDP-43 Pathophysiology in Disease
The role of TDP-43 in neurodegenerative conditions has primarily focused on two conditions-amyotrophic lateral sclerosis (ALS) and FTLD, even though now it’s proteinopathy has been reported in multiple other conditions as briefly discussed in the next section. Based on data from multiple studies it is now generally accepted that TDP-43-associated proteinopathy is in response to “loss of normal function” of the protein in neuronal nucleus rather than “gain of toxic function” by its mis-localized or aggregated form in the cytoplasm (Vanden Broeck et al., 2014). In majority of ALS-FTLD cases, the full length, wild type TDP-43 is found aggregated in neuronal cytoplasm. The entrapment of TDP-43 in
aggregated form also reduces the availability of the protein for normal functioning which in turn affects turnover and functioning of multiple other proteins that may contribute to the overall disease pathology (Prpar Mihevc et al., 2016). TDP-43 autoregulates its synthesis, in part by directly binding and enhancing splicing of an intron in the 3’untranslated region of its own transcript, thereby triggering non-sense mediated RNA degradation. Interestingly, if the autoregulatory mechanism of TDP-43 is perturbed, a gain of novel functions ensues which has been shown to exacerbate disease pathology (White et al., 2018).
Studies have showed that the full-length protein is not particularly prone to aggregation by itself unless it is purified in vitro or highly overexpressed both in vitro and in
vivo, which by itself constitutes non-physiological conditions (Romano et al., 2015).
However, when exposed to conditions mimicking aging such as chronic, mild oxidative stress or inflammation, both wild type and mutant variants of TDP-43 have an increased aggregation propensity (Wilson et al., 2011; Vaccaro et al., 2012; Correia et al., 2015; Cragnaz et al., 2015). Particularly mutant TDP-43 has been shown to be actively form cytoplasmic accumulation and aggregation. However, in this case, mere cytoplasmic mislocalization without formation of inclusion bodies, was reported to be toxic for cells (Barmada et al., 2010). TDP-43 trapped in aggregates are known undergo post-translational modifications such as hyperphosphorylation, ubiquitination and SUMOylation (Neumann et
al., 2006b; Hasegawa et al., 2008; Seyfried et al., 2010). Hyperphosphorylation is believed
to be a compensatory defense mechanism to stop or prevent pathogenic TDP-43 from aggregation (Li et al., 2011) whereas ubiquitination is necessary for the clearance of the soluble form of the protein. However, cellular macroaggregates of oligomeric TDP-43 have been demonstrated to be effectively cleared only by autophagy. Thus, a concerted role of the ubiquitin-proteasome system and autophagy seems to be necessary to maintain cellular TDP-43 homeostasis (Scotter et al., 2014).
Shorter isoforms of TDP-43, as mentioned above, are predominantly found in patient samples rather than in non-disease controls (Neumann et al., 2006b). Thus, they may represent the toxic species, an observation that is corroborated in transgenic animal models expressing a C-terminal shorter isoform (Walker et al., 2015). These shorter isoforms are also believed to form oligomers that may contribute to cell-to-cell propagation of the disease
via a prion-like mechanism (Nonaka et al., 2013; Feiler et al., 2015; Smethurst et al., 2016). TDP-43 possesses the potential to propagate from cell to cell based on seeded aggregation whereby mis-folded proteins recruit and initiate template-directed mis-folding of the native protein to form new aggregates and this could, in part, be by an exosome-dependent pathway (Iguchi et al., 2016; Iguchi et al., 2017).
Although TDP-43 proteinopathy-associated neuronal dysfunction has been studied in detail in experimental models, post-mortem human tissues (brain and spinal cord) have also revealed that cytologic pathology, almost universally, involves glial proliferation and activation as well. Reports also show that glial cells in post-mortem ALS-FTLD brain are strongly immunoreactive for TDP-43 (Zhang et al., 2008). Recent findings, particularly in ALS models, have shown that glial pathology may play a critical role in disease progression via secondary astrocyte and microglial activation (Cassina et al., 2005). Introduction of human wild type TDP-43 via gene transfer using lentiviral vectors into rat primary motor cortex resulted in marked increase in the expressions of glial fibrillary acidic protein (GFAP) and ionized calcium binding adaptor molecule 1 (iba-1). A concomitant increase in levels of pro-inflammatory cytokines, including interleukin (IL)-6, tumor necrosis factor (TNF-α) was also observed (Herman et al., 2012). However, a study conducted with patient iPSCs showed that cytoplasmic TDP-43 accumulation can cause cell-autonomous astroglial pathology and may directly contribute to the initiation of glial activation, rather than this glial activation being a secondary effect of a primary neuronal pathology (Serio et al., 2013). This could mean that astrocyte death is associated with subsequent death of neurons by the loss of a critical astrocyte-secreted neurotrophins. Gliosis could conceivably represent replacement of one neurotrophic population of astrocytes by another astrocyte population that lacks this neurotrophic capacity (Díaz-Amarilla et al., 2011).
Microglia surrounding neurons containing TDP-43 aggregates are involved both in controlling inflammation as well as TDP-43 clearance. It has been reported that microglia are involved in uptake of extracellular TDP-43 and responds by secreting IL-1β and activating nucleotide binding and oligomerization domain like receptor protein 3 -dependent and noncanonical IL-18 processing. These microglia induce neurodegeneration by a non-cell autonomous mechanism in which the TDP-43 aggregates bind to and colocalize with
MAPK/MAK/MRK overlapping kinase to trigger caspase-3/IL-18 signaling (Leal-Lasarte et
al., 2017). In contrast, if microglia are depleted of TDP-43, they have been reported to initiate
unregulated synaptic pruning (Paolicelli et al., 2017). A novel neuroprotective role of microglia was reported from mice expressing a truncated variety of TDP-43 that lacked the nuclear localization signal but whose expression was under a tetracycline suppressor. On discontinuing tetracycline administration there was a massive increase in TDP-43 production in motor neurons but without any significant response from surrounding microglia. However, if tetracycline was re-continued, thereby halting further TDP-43 production, the microglia became hyper-reactive, their processes contacted the TDP-43 laden motor neuron and phagocytosed TDP-43 (Spiller et al., 2018). However, the significance of this microglia-mediated TDP-43 clearance is hard to correlate with disease pathology as the TDP-43 variant used in the study is not naturally occurring.
The involvement of inflammation in neurodegenerative diseases is now a well-established fact, albeit whose exact role and regulation needs more elucidation. A critical factor involved in this inflammatory process is the nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB). NF-κB is ubiquitously expressed in neurons and the constitutive κB activation is associated with processing of neuronal information. On one hand NF-κB can regulate the transcription of genes expressing chemokines, cytokines, proinflammatory enzymes, adhesion molecules, proinflammatory transcription factors, while on the other hand it modulates factors to affect neuronal survival. In glial cells, NF-κB is directly responsible for regulating their reactive state and chemical messenger secretion (Shih
et al., 2015). Thus, unsurprisingly, NF-κB is found to be elevated in neurodegenerative
conditions. But surprisingly, TDP-43 has been reported to be a co-activator of NF-κB. In both human samples from ALS patients as well as transgenic animal model of ALS overexpressing human wild type TDP-43, NF-κB was found to be associated with TDP-43 by interaction between its P65 subunit and TDP-43’s RRM-1 domain (Swarup et al., 2011b). This provided evidence of a direct interplay between TDP-43 and neuroinflammation and is not only limited to ALS pathology; in clinically diagnosed cases of mild cognitive impairment TDP-43 was found to interact with NF-κB (Ohta et al., 2014). Hence it is clear that TDP-43’s involvement in neurodegenerative disorders are multifaceted and complex and whose better understanding is a necessity in order to manage these conditions.
1.3 TDP-43 in Neurodegenerative disorders
Irrespective of the large body of data accumulated over the years regarding the functions as well as potential dysfunctions of TDP-43, it has not been possible yet to clearly point out the exact modality its involvement in neurodegenerative diseases. However, as laid out in the following sections, the molecular signature of TDP-43 pathology is present in almost all neurodegenerative diseases processes encompassing motor dysfunctions to cognitive impairment and dementia.
1.3.1 Amyotrophic lateral sclerosis and frontotemporal dementia
ALS is a “relentlessly progressive” neurodegenerative disorder primarily affecting motor neurons which supply to voluntary muscles, including lower motor neurons (LMN) in the medulla and anterior horn of the spinal cord as well as upper motor neurons (UMN) in the cerebral cortex (de Carvalho and Swash, 2016). A hallmark of ALS is the presence of various inclusion bodies in degenerating lower motor neurons of the brainstem, the spinal cord and in corticospinal UMN (Sasaki and Maruyama, 1994) and occasionally in surrounding reactive glial cells (Miller et al., 2004). These inclusions are classified as ‘Lewy body-like’ or ‘Skein-like’ or Bunina bodies, which are cystatin C-containing eosinophilic intraneuronal inclusions, and are found in the cell bodies of motor neurons in ALS (Kawashima et al., 1998; He and Hays, 2004; Okamoto et al., 2008). These inclusions mostly comprise of mis-folded and/or ubiquinated, hyperphosphorylated or SUMOylated aggregates of different proteins such as SOD1, TDP-43 or FUS, OPTN, UBQLN2 and the translational product of intronic repeats in the gene C9ORF72 (Neumann et al., 2006a; Deng et al., 2010; Blokhuis et al., 2013; Dangoumau et al., 2013).
frontotemporal lobar degeneration (FTLD) and alterations in cognition are also associated with ALS (Wilson et al., 2001). An estimated 50% of ALS cases show some degree of cognitive decline and about 15% also manifest symptoms of FTD (Ringholz et al., 2005). Non-mutated TDP-43 is found in aggregates in spinal cord motor neurons, hippocampal and frontal cortex neurons and glial cells in all sporadic ALS patients and the vast majority of SOD-1-negative familial ALS patients (Mackenzie et al., 2007; Tan et al.,
2007) . In FTLD, TDP-43 aggregates are present in the most common subtype of the disease, FTLD with ubiquitinated inclusions, now referred to as FTLD-TDP (Neumann et al., 2006a). ALS and FTD show a remarkable overlap at the genetic, symptomatic and pathological level and they may reflect two ends of a disease spectrum (Chen-Plotkin et al., 2010; Geser et al., 2011).
1.3.2 Alzheimer’s disease
Alzheimer’s disease (AD) is the most common form of dementia (60-70%) and is characterized by presence of amyloid plaques, hyperphosphorylated Tau induced neurofibrillary tangles and impaired cholinergic transmission in the brain. Prevalence rates of TDP-43 pathology in AD roughly vary between 20-50% of cases; however, in advanced stage AD cases it could be as high as 72%. It is generally accepted that TDP-43 pathology in AD is frequent and contributes to the neurodegenerative process both in amyloid-beta (Aβ) dependent as well as independent mechanisms. (Amador-Ortiz et al., 2007; Josephs et al., 2008; Arai et al., 2009; Davidson et al., 2011; Wilson et al., 2013; Josephs et al., 2014a; Josephs et al., 2014b; Josephs et al., 2015; Uchino et al., 2015). The presence of TDP-43 pathology in AD has been shown to modify the clinical and radiological phenotype: AD subjects with TDP-43 pathology tend to have more severe cognitive impairment, with specific deficits in episodic and working memory and language domains and greater hippocampal atrophy, as seen on MRI, compared to AD subjects lacking TDP-43 pathology. This could be related to the fact that in AD TDP-43 pathology is prominently visible in the limbic system of the brain and the amygdala seems to be the most vulnerable region (Uryu
et al., 2008).
In the Aβ-dependent pathology of TDP-43 in AD, the cleavage of amyloid precursor protein (APP) by β-secretase and γ-secretase generates β-amyloid and intracellular domain of APP (AICD), respectively. TDP-43 co-localizes with AICD in neuronal nucleus and upregulated P53 mRNA expression thereby promoting AICD-induced apoptosis (Wang et
al., 2014). Aβ also accelerates TDP-43 phosphorylation and aggregation in neuronal cytosol
and it has been reported that TDP-43 oligomers and Aβ are capable of cross seeding with each other to form amyloid oligomers (Herman et al., 2011; Xu et al., 2013). Apart from this,
TDP-43 also has Aβ-independent roles in AD. TDP-43 inclusions had been found to coexist with tau-positive neurofibrillary tangles in the same neuron in AD cases (Higashi et al., 2007). However, it may be possible that these two proteins mediate two distinctive pathology independent of each other where TDP-43 can also induce perturbation of the physical neuronal function, mitochondria, and Ca2+ homeostasis; dysregulation of the stress response;
and inflammation (Robinson et al., 2014).
1.3.3 Parkinson’s disease and Parkinson-dementia complex
Parkinson’s disease (PD) is a predominantly an adult onset neurodegenerative disorder primarily affecting the motor system. The hallmark of PD is death of dopaminergic neurons in the substantia niagra, particularly affecting the ventral component of the pars compacta. Prior to death, accumulation of Lewy bodies, which are ubiquitinated aggregates of alpha (α)-synuclein and other neurofilament proteins, are observed in these neurons (Davie, 2008). Mutations in the α-synuclein gene are responsible for some familial forms of PD in which Lewy bodies are also seen. However, mutations in the E3-ubiquitin ligase Parkin results in development of parkinsonian syndrome but without Lewy body formations, thereby indicating a role in formation of the inclusion bodies (Dawson and Dawson, 2010). Apart from motor disturbances, 60% patients suffering from PD develops dementia within 12 years of initiation of motor symptoms and more than 80% PD cases eventually lead to dementia (Wood et al., 2016).
In a mouse model adapted to overexpress human TDP-43, a concomitant expression of a mutant form of α-synuclein resulted in heightened loss of dopaminergic neurons thereby indicating that TDP-43 potentiated α-synuclein toxicity to dopaminergic neurons in living animals (Tian et al., 2011). In human cases, studies has indicated the involvement of TDP-43 pathology in Lewy body associated neurodegenerative disorders (Higashi et al., 2007; Nakashima-Yasuda et al., 2007). Amongst Sardinian population with a TARDBP mutation (c.G1144A; p.A382T), in approximately 2.5% cases, parkinsonism has been reported as a clinical manifestation (Quadri et al., 2011). The A315E mutant form of TARDBP has also been reported to manifest PD-like symptoms with loss of dopaminergic neurons in the substantia niagra (Fujita et al., 2011). A study conducted in the Mayo Clinic in Florida, USA
also resulted in the identification of the ALS-associated TDP-43 p.N267S mutation to be associated with PD (Rayaprolu et al., 2013). However, the presence of TDP-43 mutation in PD is extremely rare. In large population-based studies on French-Canadian PD patients, no TDP-43 could be detected whereas in a similar study on Dutch cohort, even though a silent mutation (p.S332S) was identified in TDP-43, no missense mutations could be detected, thereby concluding that TDP-43 mutations do not generally contribute to PD pathology (Kabashi et al., 2009; van Blitterswijk et al., 2013).
1.3.4 Huntington’s disease
Huntington’s disease is another progressive adult-onset neurodegenerative disease characterized by progressive psychiatric disruption, cognitive deficits, and loss of motor coordination. The root cause of the disease results from mutation in the HTT gene leading to the trinucleotide CAG repeats. HTT codes for the huntingtin protein (Htt) whose normal function is not clearly elucidated but appears to be crucial in normal functioning of brain neurons. Htt reportedly interacts with multiple effector proteins to mediate a host of physiological processes including axonal trafficking, regulation of gene transcription and cell survival(Schulte and Littleton, 2011). The CAG repeats in the gene leads to the formation of an aberrant elongated huntingtin protein with a polyglutamine tail that affects the normal functioning of the protein. A post-mortem analysis of 10 HD cases reveled that TDP-43 colocalized with huntingtin in dystrophic neurites and various intracellular inclusions, but not in intranuclear inclusions(Schwab et al., 2008). This is a unique observation as in cases of other neurodegenerative disorders, TDP-43 does not always co-localize with immunoreactive inclusions in neurons. A possible connection interlinking Htt function with TDP-43 proteinopathy could be based on maintenance of cytoskeletal stability and axonal transport in which both proteins are known to be involved. However, based on current data, it is difficult to conjecture whether the Htt-TDP-43 interlink is due to common functional pathways or merely incidental to their accumulation after pathological transformation.
Thus, it is pretty clear from these studies that TDP-43 proteinopathy is intricately associated with neurodegeneration. Mainly this is based on the fact that cytoplasmic accumulation of TDP-43 results in loss of normal nuclear function of this protein in neurons
which makes them vulnerable for degeneration. Thus, it is quite plausible that this could be the basis for neurodegeneration in more direct neurodegenerative changes resulting from injury or ischemia.
1.4 Cerebral ischemia
Stroke remains one of the leading causes of death and disability in the world. No medical treatment has been approved apart from intravenous injection of tissue plasminogen activator, which needs to be administered within 3h after stroke. An estimated 28% of annual stroke victims are under age 65 and it is presumed that by 2050 in USA, rate of incidence of stroke cases would increase to over 1 million per year (Fisher and Bogousslavsky, 1998; Stephenson, 1998). Generally, risk factors for stroke are primarily associated with cardio-vascular disease and correlates with hypertension, diabetes, atherosclerosis, hypercholesterolemia as well as age. Stroke can be very dangerous and immediate medical care is necessary if warning signs appear. The major symptom of stroke is hemiparesis, specifically with drooping or numbness of the face and/or arm and speech deficits (Pancioli
et al., 1998).
Stroke is mainly an outcome of obstruction of blood flow in cerebral blood vessels (usually middle cerebral artery) which, if not resolved in short span of time, leads to a core damage in the brain tissue. If the blood supply in the zone of tissue around the core (penumbra) is falls below 25% of normal level, unavoidable expansion of the ischemic core occurs. Thus, blood flow plays a significant role in determining the size of the core infarct by providing the conditions essential to maintain cellular homeostasis. Eventually ischemic stroke involves a cascade of pathophysiological events (explained below) ultimately leading to death and dysfunction of brain cells and depending on the location of the compromised brain area, neurological deficits will be manifested (Villringer and Dirnagl, 1999).
1.4.1 Pathophysiology of stroke
Loss of blood supply during stroke results in pathophysiological response resulting in neuronal damage. Various mechanisms, including excitotoxicity, mitochondrial
alterations, protein misfolding, and inflammatory changes have been reported to contribute to cell death (Hossmann, 2006).
1.4.1.a) Excitotoxicity
Deficiency of glucose and oxygen makes the neurons vulnerable to maintain normal ionic gradients. Subsequently, an increased influx of calcium and sodium ions causes depolarization of the neurons leading to release of excitatory neurotransmitters like glutamate which acts on N-methyl-D-aspartate receptors causing more influx of calcium ions into the cell. Intracellular increase in calcium triggers mitochondrial dysfunction, free radical generation which generates peroxy nitrate radicals and causing peroxidation of lipids on the cell membrane, and activation of enzymes like phospholipases and proteases that degrade the membranes and proteins that are essential for cellular integrity and finally causing cell death (Dirnagl et al., 1999; Szydlowska and Tymianski, 2010).
1.4.1.b) Mitochondrial alterations
The energy balance in the cell are critically maintained by mitochondria. In ischemic injury, because of excessive accumulation of calcium which activates calpains. These calpains act on Bid complex in the cytoplasm and truncated Bid interacts with Bax-Bcl2 complex on the mitochondrial membranes which causes mitochondrial permeability transition pore opening and cytochrome C release. These events cause mitochondrial swelling and membrane collapse and initiate apoptotic cell death. Mitochondrial integrity and minimizing the activation of apoptotic pathways in the cell are important aspects which should be under consideration to prevent widespread cell toxicity from an ischemic insult (Liu et al., 1996). Reactive oxygen species generated during ischemic injury by abnormal mitochondria cause damage to lipids, DNA and proteins and contributes to cell death (Moskowitz et al., 2010).
1.4.1.c) Protein misfolding
Endoplasmic reticulum plays an important role in the protein synthesis and is the largest repository of intracellular calcium and responds to protein misfolding (Zhang et al., 2015). In the endoplasmic reticulum, the misfolded proteins are either repaired or degraded
by molecular chaperones. This prevents the formation of insoluble aggregates in the cell. In ischemic injury, loss of energy supply disturbs this protein homeostatic mechanism leading to the formation of insoluble aggregates which can alter the physiological state of cell (Nakka
et al., 2010; Raghubir et al., 2011).
1.4.1.d) Inflammation
In ischemic injury, activation of secondary messenger systems by intracellular accumulation of calcium, increase oxygen free radicals as well as hypoxia itself trigger activation of transcription factors like NF-κB leading to the expression of pro-inflammatory genes (O'Neill and Kaltschmidt, 1997). Mediators of inflammation like tumor necrosis factor alpha, IL-1β and platelet activating factor are produced by injured brain cells (Rothwell and Hopkins, 1995). As a result of expression of adhesion molecules like intercellular adhesion molecule 1 (ICAM-1) and selectins on the cell surface is increased, making way for the entry of blood derived macrophages and neutrophils into the ischemic zone (Iadecola, 1997; Zhang
et al., 1998). Resident brains cells like microglia and astrocytes are activated after ischemic
injury. There is an increasing evidence that post ischemic inflammation contributes to ischemic brain injury and comprises of cellular and molecular components (Barone et al., 1998). Cytokines such as IL-1β and TNF-α released by microglia, astrocytes and neurons in the ischemic region influence this process (del Zoppo et al., 2000). Systemic inflammatory response also occurs along with the central inflammatory response after ischemic injury. Various cellular and molecular mediators of inflammation are explained in brief in the following sections.
1.4.2 Molecular mediators of neuroinflammation in cerebral ischemia
During ischemic injury, pro-inflammatory genes are upregulated, including transcription factors, reactive oxygen species (ROS), nitric oxide (NO), heat shock proteins, cytokines, chemokines and cell adhesion molecules. Most of them are regulated by NF-κB, including TNF-α, IL-1β, and intercellular adhesion molecule-1 (Baeuerle and Henkel, 1994).
1.4.2.a) Cytokines and Chemokines
Cytokines are the polypeptides that are associated with inflammation and cell differentiation or death. Activated microglia in CNS and macrophages in the periphery are the prime site of production of cytokines. Pro-inflammatory cytokines like IL-1 and TNF-α are known to exacerbate the cerebral ischemic injury (Allan and Rothwell, 2001). Studies on different stroke models have advanced our understanding of IL-1 in neurodegeneration (Simi
et al., 2007). Following brain ischemia, IL-1β mRNA expression has been shown to be
elevated (Minami et al., 1992). Another study has reported that exogenous IL-1β administration in the cortex region exacerbates the brain injury (Yamasaki et al., 1995). Several studies have shown the role for TNF-α in ischemic injury. Elevated levels of TNF-α mRNA and protein following middle cerebral artery occlusion have been reported (Liu et al., 1994). Cerebral ischemic injury can be minimized by administration of TNF-α antibody, and exacerbated by administration of recombinant TNF-α (Barone et al., 1997). A study conducted on post-mortem brain tissue of patients with acute cerebral infarction showed the upregulated levels of TNF-α and similar study in patients with acute ischemic stroke also detected elevated levels of TNF-α in the cerebrospinal fluid (Tomimoto et al., 1996; Zaremba
et al., 2001). Interestingly IL-10, an anti-inflammatory cytokine, decreased the infarct size in
brains of rats subjected to middle cerebral artery occlusion and thus could be assumed to play a role in neuroprotection following injury (Spera et al., 1998). IL-6 is an inflammatory cytokine known to enhance astrogliosis and exerts its effects by signaling through JAK-STAT pathway. It has been reported that it inhibits TNF-alpha, induce apoptosis in neutrophils, and recruit monocytes and T-cells which makes way for the transition between innate and adaptive immunity (Marz et al., 1996). IL-6 can be detrimental by enhancing the levels of prostaglandinE2 which is implicated in rising the body temperature which has been shown to increase the brain damage after stroke (Azzimondi et al., 1995). IL-4 a potent regulator of immunity secreted by T-cells and is a critical participant in higher brain functions such as learning and memory. A study conducted on mice reports that lack of IL-4 demonstrates cognitive impairment in spatial learning tasks which can be reversed by transplantation of IL-4-competent bone marrow (Derecki et al., 2010). IL-4 also plays an important role in leukocyte survival under both pathological and physiological conditions (Minshall et al., 1997), and macrophage activation (Gordon, 2003). IL-17 a
pro-inflammatory cytokine released by T-cells and acts in delayed phase of the stroke. it causes the blood brain barrier break by producing ROS. IL-17 promotes cell adhesion of leukocytes and promotes the production of CCL2 and CXCL1 from brain endothelial cells (Wojkowska
et al., 2017). Chemokines are regulatory peptides that plays roles in cellular communication
and inflammatory cell recruitment. IL-8 and monocyte chemoattractant protein-1(MCP-1) have been implicated to have a deleterious role in cerebral ischemia (Mennicken et al., 1999). In patients with acute stroke, plasma levels of IL-8 were increased and is believed to play a role in recruiting neutrophils to sites of cerebral ischemia (Kostulas et al., 1998). MCP-1 is a potent chemo-attractant, and it’s levels are increased in cerebrospinal fluid of the patients with acute ischemic stroke, indicating recruitment of immune cells to the site of injury (Losy and Zaremba, 2001). Granulocyte-macrophage colony stimulating factor has neuroprotective properties in experimental stroke and it was reported that it helps in reversing immunodepression in patients. It also helps in macrophage and leukocyte recruitment to the site of injury (Dames et al., 2018). In an acute mice model of stroke, administration of granulocyte colony stimulating actor intravenously after 30 minutes of induction of stroke has shown 27% reduction in infarct size and increase in the survival rate (Six et al., 2003).
1.4.2.b) Cell adhesion molecules
Presence of leucocytes at the site of injury is mainly dependent on expression of chemotactic factors and adhesion molecules (such as ICAM-1, selectin and integrins) on inflammatory cells and endothelium of capillaries (DeGraba, 1998). The levels of ICAM-1 mRNA are increased after middle cerebral artery occlusion in rats (Wang et al., 1994). Various reports have been suggested the presence of elevated levels of ICAM-1 in 24hrs of acute ischemic stroke (Shyu et al., 1997; Bitsch et al., 1998). The expression of ICAM-1 by brain micro vessels was also reported to be significantly increased in patients who succumbed to ischemic stroke (Lindsberg et al., 1996). During ischemic injury, reperfusion causes translocation of P-selectin on the surface of the platelets and endothelial cells blood leading to platelet aggregation and blood clotting. Glyco proteins like P-selectin glycoprotein ligand-1 expressed on leukocytes helps in firm binding of low affinity leukocytes on endothelial. Integrins like macrophage antigen complex-1 and leukocyte beta 2 integrins also play role in
firm adhesion of leukocytes on endothelial cells by binding with ICAM-1 (Iadecola and Anrather, 2011).
1.4.3 Cellular mediators of inflammation
Cerebral ischemia triggers a complex of cellular response which includes activation of both glial cells and recruitment of inflammatory cells from periphery (Dirnagl et al., 1999). Following cerebral ischemia, these activated resident microglial cells and infiltrating cells initiate a complex inflammatory cascade involving a variety of cyto/chemokines as well as free radicals that potentiate damage (Raivich et al., 1999).
1.4.3.a) Leukocytes
Leukocytes, especially neutrophils, rapidly migrate to site of injury and are known to exert detrimental effects in human stroke patients (Herz et al., 2015). Interestingly, leukocyte infiltration was found to be lower in experimental models utilizing older animals, even though CCL5 protein concentrations were found to be higher, suggesting an age-related defect in migratory ability (Desai et al., 2010). Neutrophils also play important role in the production of reactive oxygen species and matrix metalloproteinases, two potent drivers of neuronal injury and blood brain barrier damage (Rosell et al., 2006; Allen and Bayraktutan, 2009). In the ischemic region, recruited monocytes have the capacity to transform into phagocytic macrophages and all these changes occur within 1-7 days after initial ischemic insult (Schilling et al., 2003). In humans the prognosis of stroke depends on the ratio between pro- and anti-inflammatory macrophages in the peripheral blood (Urra et al., 2009).
1.4.3.b) Microglia
Microglia, principle resident immune cells of the brain, provide constant surveillance and coordinate various critical roles and maintain brain homeostasis (Nimmerjahn et al., 2005; Paolicelli and Gross, 2011). Microglia are the key players in the brain injury and disease states (Wolf et al., 2017) and activation of these cells is the first step of the inflammation and takes place within minutes after injury (Denes et al., 2007). The term activation includes changes in morphology, proliferation, and migration of the resident cell population (Hansson and Ronnback, 2003; Raivich, 2005; Hanisch and Kettenmann, 2007)
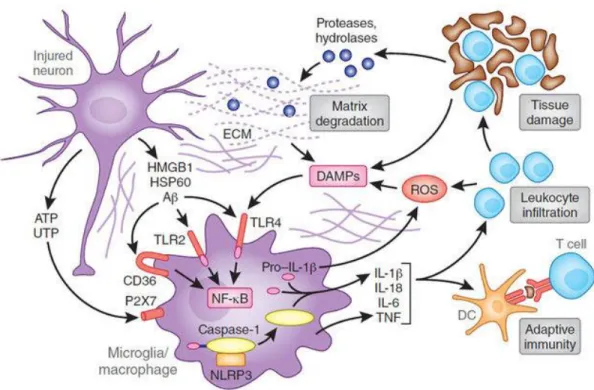
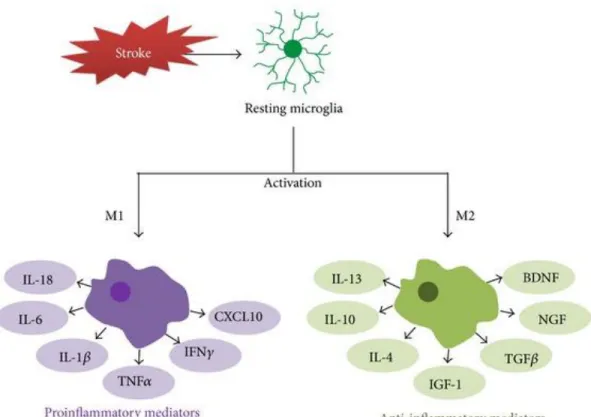
and the expression of surface markers like iba-1, CD11b, Mac1 and TLR2 were also upregulated (Lalancette-Hebert et al., 2009; Greter et al., 2015). Microglia play an important role in clearing the cellular debris and clear the potentially dangerous protein aggregates in the brain (Sierra et al., 2010). Microglia are capable of secreting immune modulators such as cytokines and chemokines, and there has been a strong belief that hyperactivation of these cells is responsible for pathogenesis of neuroinflammatory and neurodegenerative disorders. Similarly, in cerebral ischemia, it has been shown that activation of microglia by free radicals in the later stages of ischemic damage further results in secondary cell death in penumbra (Danton and Dietrich, 2003). Defective microglial activation or proliferation significantly increased the size of infarction and the neuronal apoptosis after stroke, suggesting the deleterious role of microglia after ischemic stroke (Lalancette-Hebert et al., 2012). Danger signals like ATP, heat shock proteins, amyloid beta, released from dying neurons activate various pattern recognition receptors like toll like receptor 2 in concert with CD36 on microglia activate various transcription factors like NF-κB pathway and causes release of inflammatory cytokines (Figure 1.3) (Iadecola and Anrather, 2011), and it has been shown that TLR2 signals were upregulated on microglia following ischemic injury (Lalancette-Hebert et al., 2009) and activation of these receptors by danger signals and substantial tissue damage leads to chronic inflammatory responses (Piccinini and Midwood, 2010). Aging drives the resident microglia to acquire more pro-inflammatory cellular phenotypes, and a study conducted on mice using live imaging technique has shown that TLR2 responses following MCAO were upregulated in aging (Rahimian et al., 2018). Based on the severity of the injury during ischemic stroke, activated microglia can act as a double edge sword (Figure 1.4) by providing either beneficial (M2 phenotype) or detrimental effects M1 phenotype).
Post ischemic inflammation is a self-regulatory process that eventually subsides and prepares for the repair of the brain. Still it not clear regarding the factors involved in resolution of inflammation and restructuring the tissue homeostasis and it involves a large number of mediators that actively suppress the inflammatory response. The most important steps include removal of dead cells, development of anti-inflammatory milieu and generation of pro-survival factors responsible for tissue reconstruction and repair (Spite and Serhan, 2010). Microglia play an important role in phagocytosis of dead cells and tissue debris after
stroke (Schilling et al., 2005). Purines which acts as find me signals released from injured cells and chemokines, attract microglia to the site of injury. These phagocytic microglial cells are then presented with eat me signals associated with dying cells (Figure1.5) (Davalos et al., 2005). TGFβ and IL-10 are pleotropic cytokines whose production is upregulated during phagocytosis and occurs in concert with removal of dead cells (Nathan and Ding, 2010). TGFβ, which is upregulated after ischemia primarily in microglia has neuroprotective properties and plays an important role in suppressing the inflammation by inhibiting T-helper response and promoting T-regulatory cell development (Taylor et al., 2006). Production of growth factors Post-ischemic conditions creates an environment that supports for neuronal sprouting, neurogenesis, gliogenesis and matrix re-organization (Greenberg and Jin, 2006). Microglia are required for the full expression of insulin like growth factor-1which is a key factor in post-ischemic neuronal sprouting(Li et al., 2010). Vascular endothelial growth factor (VEGF) which is crucial for post-ischemic angiogenesis, is produced by glial cells and its action may require neutrophil matrix metalloproteinases suggesting a link between inflammatory cells and angiogenesis (Hao et al., 2007). VEGF administration early after ischemia or in excessive doses may enhance the damage (Manoonkitiwongsa et al., 2004). Evidences suggest that cells of the immune system have a significant role in all the phases of post-ischemic brain recovery. But only a limited data provides a glimpse into the complex events that helps in the structural and functional homeostasis of the brain after stroke.
Figure 1.3 Cell death and activation of pattern recognition receptors (Iadecola and
Anrather, 2011)
Release of nucleotides ATP, UTP from injured neurons, activates purinergic receptors on microglia leading to the production of pro-inflammatory cytokines. Although most of cytokines are transcriptionally induced, IL-1beta and IL-18 are processed from their pro-peptides by the activity of interleuikin-1 converting enzyme. This enzyme is embedded in a multiprotein complex (NLRP3, or inflammasome) and is activated by microglial P2X7 receptors. Ischemic cell death leads to the formation of DAMPs, which activate TLR2. TLR2 in concert with scavenger receptors such as CD36, upregulate pro-inflammatory gene expression through NF-κB pathway. DAMPs are also derived from matrix break down by lytic enzymes released from dead cells and by the action of ROS on lipids. Cytokine activation resulting from the events leads to increased leukocyte infiltration and enhance issue damage, which in turn produce more DAMPs. Antigens unveiled by tissue damage are presented to T-cells setting the stage for adaptive immunity.
Figure 1.4. Phenotypes and activation of microglia after ischemic stroke
Under ischemic conditions, microglia change their morphology and become activated. Activated microglia are characterized as either an M1 classically activated phenotype or an M2 alternatively activated phenotype. Microglial activation induces transcription associated with the inflammatory mediators. According to their phenotypes, microglia can promote proinflammatory (by M1) or anti-inflammatory (by M2) machinery. IGF-1: insulin-like growth factor 1; IL-1β: interleukin-1 beta; IL-6: interleukin-6; IL-10: interleukin-10; TGF-β: transforming growth factor-beta; TNF-α: tumor necrosis factor-alpha.