Étude de la différenciation des lymphocytes B mémoires
en milieu sans sérum
Influence du microenvironnement cellulaire
Mémoire
Catherine Gervais St-Amour
Maîtrise en biochimie
Maître ès sciences (M.Sc.)
Québec, Canada
Résumé
Les infections opportunistes sont l’une des principales causes de mortalité associées à la greffe de cellules souches. Afin d’aider à reconstituer le système immunitaire des patients greffés, nous proposons d’exploiter les plasmocytes autologues générés in vitro dans notre modèle de culture basé sur l’interaction CD40-CD154. Pour ce faire, les milieux de culture doivent être exempts de protéines animales. Deux milieux sans sérum ont été préalablement développés à Héma-Québec. Notre hypothèse est qu’il serait maintenant possible de promouvoir la différenciation des lymphocytes B et de maintenir la survie des plasmocytes en modifiant la composition de ces milieux sans sérum. Nos résultats montrent que notre milieu sans protéines animales permet de générer un grand nombre de plasmocytes et que ceux-ci ont un profil d’expression de CD38 qui est stabilisé par la vitamine A. En conclusion, la formulation de ce milieu représente un progrès quant à la possibilité d’utiliser ces plasmocytes en immunothérapie.
TABLE DES MATIÈRES
RÉSUMÉ ... III TABLE DES MATIÈRES... V LISTE DES TABLEAUX ... IX LISTE DES FIGURES ... XI LISTE DES ABRÉVIATIONS ... XIII REMERCIEMENTS ... XVII
1 INTRODUCTION ... 1
1.1 GREFFE DE CELLULES SOUCHES HÉMATOPOÏÉTIQUES ... 1
1.1.1 Applications thérapeutiques ... 1
1.1.2 Reprise de la greffe et immunosuppression ... 2
1.1.3 Reconstitution du système immunitaire ... 3
1.2 LE LYMPHOCYTE B ... 4
1.2.1 Ontogénie du lymphocyte B ... 4
1.2.2 Activation dans les ganglions lymphatiques ... 5
1.2.3 L’interaction CD40-CD154 ... 6
1.2.4 Maturation de l’affinité : le centre germinatif ... 6
1.2.5 Les immunoglobulines ... 8
1.2.6 Sous-populations de lymphocytes B dans le sang ... 9
1.3 LE PLASMOCYTE ... 10
1.3.1 Différenciation en plasmocytes ... 10
1.3.2 Niches de survie ... 11
1.3.3 Métabolisme sécrétoire ... 13
1.3.4 La cyclase ADP ribosyle CD38 ... 15
1.4 SIGNALISATION INTRACELLULAIRE ... 17
1.4.1 Homéostasie chez le lymphocyte B ... 17
1.4.2 Le potentiel redox comme élément de signalisation ... 18
1.5 CULTURE IN VITRO DES LYMPHOCYTES B ... 19
1.5.1 Modèle de culture en deux phases ... 19
1.6 PROTÉINES ANIMALES ET MILIEUX DE CULTURE ... 23
1.6.1 Problématique du sérum bovin ... 23
1.6.2 Des alternatives au sérum en culture cellulaire ... 24
1.6.3 Milieux sans sérum ... 24
1.7 HYPOTHÈSE ET OBJECTIFS ... 25
2 MATÉRIELS ET MÉTHODES ... 27
2.1 SOURCE DES CELLULES MONONUCLÉES DU SANG ... 27
2.1.1 Isolement des PBMC à partir de sang frais et congélation ... 27
2.1.2 Purification des lymphocytes B mémoires à partir des PBMC congelées ... 28
2.2 LIGNÉES HUMAINES ... 28
2.2.1 Lignée Ramos, RPMI-8226 et Jurkat ... 28
2.2.2 Cellules de support L4.5 ... 29
2.3 CULTURE DES LYMPHOCYTES B ... 29
2.3.1 Modèle de culture CD40-CD154 ... 29
2.3.2 Milieux de culture ... 30
2.3.3 Facteurs solubles ... 31
2.3.4 Banque de lymphocytes B mémoires ... 31
2.4 ANALYSES PHÉNOTYPIQUES ... 31
2.4.1 Prolifération et viabilité ... 31
2.4.2 Phénotype ... 32
2.5 ANALYSES FONCTIONNELLES ... 33
2.5.1 Évaluation de la sécrétion par dosage ELISA ... 33
2.5.2 Évaluation de la sécrétion par dosage Bioplex ... 34
2.5.3 Évaluation du nombre de cellules sécrétrices par dosage ELISPOT ... 34
2.6 ANALYSES MOLÉCULAIRES ... 35
2.6.1 Évaluation de la signalisation cellulaire ... 35
2.6.2 Quantification protéique... 36
2.6.3 Analyse des ARNm pour le CD38 en PCR semi-quantitatif ... 38
2.7 ANALYSE DU POTENTIEL REDOX ... 39
2.7.1 Concentration des ROS intracellulaires ... 39
2.8 ANALYSES STATISTIQUES ... 40
3 RÉSULTATS ... 41
3.1 EXPRESSION DE LA PROTÉINE CD38 EN MILIEU SANS SÉRUM ... 41
3.1.1 Pourcentage en CO2 pour la culture des lymphocytes B CD19+ ... 41
3.1.2 Expression de CD38 à la surface des lymphocytes B cultivées ... 43
3.1.3 Corrélation entre les résultats de cytométrie en flux et d’immunobuvardage ... 46
3.1.4 Expansion, viabilité et sécrétion d’immunoglobulines ... 47
3.1.5 Phénotype ... 47
3.1.6 Influence de l’albumine dans la régulation de CD38... 49
3.1.7 Transcription de CD38 ... 51
3.1.8 Environnement redox ... 52
3.2 LE MILIEU BPFM SUPPORTE LA DIFFÉRENCIATION EN PLASMOCYTE ... 54
3.2.1 Dose-réponse pour l’IGF ... 54
3.2.2 Dose-réponse pour le Trolox ... 57
3.2.3 Expansion sur 21 jours ... 57
3.2.4 Caractérisation des plasmocytes générés en présence et en absence de sérum ... 58
3.2.5 Sécrétion d’immunoglobulines ... 61
3.2.6 Hétérogénéité dans la capacité de sécrétion ... 63
3.2.7 Viabilité ... 65
3.2.8 Réponse aux cytokines de culture ... 66
3.3 AMÉLIORATION DU MILIEU BPFM ... 67
3.3.1 Vitamine C ... 67
3.3.2 Vitamine A ... 68
3.3.3 Sécrétion d’immunoglobulines ... 71
3.3.4 Propriétés antioxydantes des vitamines ... 73
3.3.5 Signalisation des vitamines ... 75
3.3.6 Réponse des lymphocytes B au NAC ... 77
4 DISCUSSION ... 79
4.1 CD38, UN SENSEUR DU MICROENVIRONNEMENT CELLULAIRE ... 79
4.2 UN MILIEU SANS PROTÉINES ANIMALES POUR LA DIFFÉRENCIATION EN PLASMOCYTE .... 82
5 CONCLUSION... 85
LISTE DES TABLEAUX
LISTE DES FIGURES
Figure 1.1 Rencontre de l’antigène dans les follicules lymphatiques. ... 5
Figure 1.2 Structure dynamique du centre germinatif ... 7
Figure 1.3 Populations de lymphocytes B dans le sang ... 10
Figure 1.4 Niches de survie des plasmocytes. ... 12
Figure 1.5 Le stress cellulaire chez le plasmocyte. ... 15
Figure 1.6 Rôles multiples de CD38 ... 16
Figure 2.1 Modèle de culture in vitro des lymphocytes B ... 30
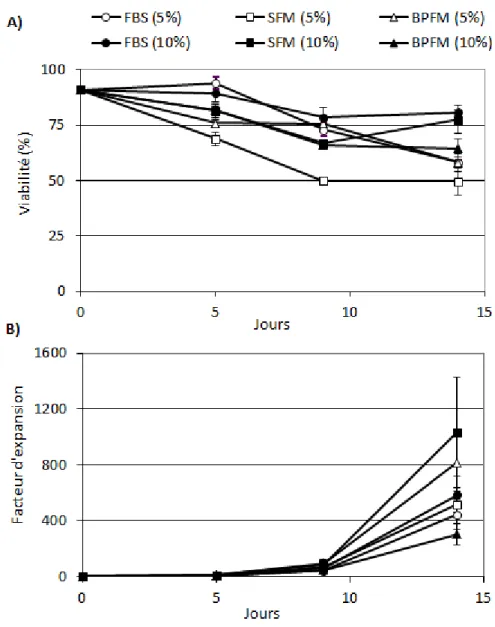
Figure 3.1 Expansion et viabilité des lymphocytes B en présence de 5% et de 10% CO2. ... 42
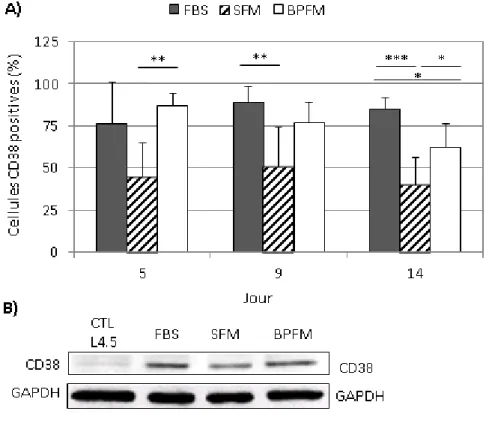
Figure 3.2 Évolution de l’expression de CD38 chez les lymphocytes B cultivés en absence ou en présence de sérum. ... 44
Figure 3.3 Modulation de CD38 à la surface des lymphocytes B selon le milieu de culture. ... 45
Figure 3.4 Expression relative de CD38. ... 46
Figure 3.5 Expansion et sécrétion des lymphocytes B dans les trois milieux de culture. ... 48
Figure 3.6 Phénotype des lymphocytes B. ... 49
Figure 3.7 Fréquence des cellules CD38+ en présence d’albumine bovine ou humaine. ... 50
Figure 3.8 Transcription de CD38. ... 51
Figure 3.9 Potentiel redox des cultures de lymphocytes B. ... 52
Figure 3.10 Niveaux relatifs intracellulaires des ROS. ... 53
Figure 3.11 Détermination de la concentration optimale d’IGF. ... 55
Figure 3.12 Évaluation du trolox comme antioxydant de remplacement de l’α-tocophérol. ... 56
Figure 3.13 Temps de génération des lymphocytes B mémoires cultivés ... 58
Figure 3.14 Phénotype des plasmocytes générés. ... 59
Figure 3.15 Sous-populations de plasmocytes dans le milieu BPFM. ... 60
Figure 3.16 Sécrétion des sous-classes d’immunoglobulines dans les milieux FBS et BPFM ... 62
Figure 3.17 Patron de sécrétion des plasmocytes générés ... 63
Figure 3.18 Détermination du pourcentage de cellules sécrétrices en culture. ... 64
Figure 3.19 Survie des lymphocytes B en culture. ... 65
Figure 3.20 Réponse intracellulaire des lymphocytes B aux cytokines. ... 66
Figure 3.21 Viabilité des plasmocytes en présence de vitamine C ... 68
Figure 3.22 Viabilité des plasmocytes en présence de vitamine A ... 69
Figure 3.23 Phénotype des lymphocytes B en présence de vitamine A dans le milieu BPFM. ... 70
Figure 3.24 Quantité de protéine CD38 totale chez les lymphocytes B cultivés avec la vitamine A. 71 Figure 3.25 Capacité de sécrétion des plasmocytes cultivés en présence de vitamine A ... 72
Figure 3.26 Niveaux relatifs de ROS intracellulaires chez les lymphocytes B en réponse aux
vitamines A et C. ... 74
Figure 3.27 Signalisation cellulaire des lymphocytes B en milieu FBS et en milieu BPFM ... 76
Figure 3.28 Effet du NAC sur STAT3 ... 78
LISTE DES ABRÉVIATIONS
ADPRc : adénosine diphosphate ribose cyclique
AID : activation –induced cytidine deaminase Akt : AK transforming
APRIL : a proliferation inducing ligand BAFF : B-cell activating factor
BCR : B-cell receptor
BLIMP1 : B lymphocyte-induced maturation
protein-1
BMK1 : big map kinase-1
BPFM : milieu sans protéines bovines (bovine-free protein medium)
BTK : Bruton’s tyrosine kinase
CAR : CXCL12-abundant reticular cell CCR : chimiokine (motif C-C) récepteur CD : cluster of differentiation
CDF : cellule dendritique folliculaire CMH-II : complexe majeur
d’histocompatibilité II CMV : cytomégalovirus
CSH : cellules souches hématopoïétiques CXCL : chimiokine (motif C-X-C) ligand CXCR : chimiokine (motif C-X-C) récepteur
DAG : diacylglycérol Ero1 : ER-oxidoreductin 1
ERK : extracellular signal-regulated kinase G-CSF : granulocyte-colony stimulating factor FBS : foetal bovine serum
ICOS : inducible costimulator Ig : immunoglobuline
IGF : insulin-like growth factor IL : interleukine
IKK : IκB kinase
IP3 : inositol triphosphate JAK : Janus kinase
JNK : c-jun N-terminal kinase
MAPK : mitogen-activated protein kinase MITF: microphthalmia-associated
transcription factor
NAC : N-acétyl cystéine
NAD/NADH : nicotinamide adénine dinucléotide
NFκB : nuclear factor κB NK : cellules natural killer PAX5 : paired box protein 5
PBMC : peripheral blood mononuclear cells PD1 : programmed death-1
PDI : protein disulphide isomerase PI3K : phosphatidyl-inositol-3’ kinase PK : protein kinase
RE : réticulum endoplasmique ROS : reactive oxygen species
SOCS : suppressor of cytokine signalling SFM : milieu sans sérum (serum-free
medium)
STAT : signal transducers and activators of
transcription
TBHP : tert-butyl hydroxyperoxide TCR : T-cell receptor
TNF : tumor necrosis factor UNG : uracil DNA glycosylase UPR : unfolded protein response XBP1 : x-box binding protein
« Le voyage de la découverte ne consiste pas à chercher de nouveaux horizons, mais à voir avec
des yeux nouveaux. » - Marcel Proust, 1871-1922
REMERCIEMENTS
Mon parcours de maîtrise vient à peine de commencer qu’il se termine déjà! Cette belle expérience n’aura pas été aussi enrichissante sans l’appui de nombreuses personnes. Je tiens à remercier particulièrement le Dre Sonia Néron pour m’avoir permis de réaliser ma maîtrise dans son laboratoire et au sein d’une équipe stimulante. Elle m’a guidée tout au long de mon cheminement et m’a donné de précieux conseils sur l’interprétation de mes résultats. J’ai grandement apprécié sa disponibilité. Elle réussissait toujours à se libérer même lorsqu’elle semblait débordée! Je la remercie également pour son intérêt envers les autres sphères de ma vie et pour son aide dans ma vie professionnelle. J’aimerais aussi remercier les membres de mon comité d’encadrement, André Darveau Ph.D., Louis Thibault Ph.D. et Éric Petitclerc Ph.D., qui m’ont encouragée et conseillée. J’ai apprécié discuter avec eux durant nos rencontres et voir leur intérêt pour mon projet de maîtrise. Un merci chaleureux à toute l’équipe des B pour nos enrichissantes discussions de science et sur n’importe quoi! Je remercie notamment Guillaume Bonnaure avec qui j’ai adoré argumenter tout au long de ma maîtrise, Rayelle Itoua Maïga et Jennifer Lemieux pour nos conversations étonnantes. Un grand merci à ma stagiaire, Christine Jobin, pour son apport dans mon projet de maitrise sur le N-acétyl cystéine en milieu sans sérum et la sécrétion d’immunoglobulines en présence de vitamine A, et à David Gaumont et Jennifer pour avoir dosé la sécrétion d’immunoglobulines dans les surnageants de culture avec la vitamine C. Je voudrais également remercier Annie Roy et Philippe Nadeau qui ont tous les deux contribué à ma formation en culture cellulaire. Merci également à Marc Cloutier et Carl Simard pour leurs nombreux conseils. C’est avec fierté que je poursuis mon cheminement avec mon casque et mon guide de survie vers la grande ville!
Je voudrais également profiter de l’occasion pour remercier ma famille qui m’a supportée tout au long de mes études, mes parents Benoit Gervais et Céline St-Amour et mes frères et sœurs Roxane, Mathieu, Philippe et Marie. Merci aussi à mon copain, Nicolas Martin, qui m’a patiemment encouragée malgré la distance.
Finalement, je tiens à remercier l’aide financière que j’ai reçue par la bourse Héma-Québec et la bourse BMP Innovation du FQRNT et du CRSNG.
1 Introduction
1.1 Greffe de cellules souches hématopoïétiques 1.1.1 Applications thérapeutiques
Au 18e siècle, le médecin anglais Edward Jenner (1749-1823) a été le premier à poser les
bases scientifiques de la vaccination et à promouvoir son efficacité. Ses observations ont introduit le concept de l’immunité humaine, mais ont également ouvert la porte à une panoplie d’applications possibles [1]. Louis Pasteur (1822-1895) poursuit dans la même voie que Jenner et développe d’autres vaccins à partir de souches moins virulentes. C’est le début de la manipulation du système immunitaire. Quelques décennies plus tard, les travaux de Georges Snell (1903-1996), de Baruj Benacerraf (1920-2011) et de Jean Dausset (1916-2009) sur l’histocompatibilité immunitaire permettent d’entrevoir la réussite des transplantations d’organes et de mieux comprendre les rejets de greffe [2].
De nos jours, la greffe de cellules souches hématopoïétiques (CSH) constitue une approche thérapeutique prometteuse pour le traitement d’immunodéficiences, de certaines maladies auto-immunitaires et de tumeurs malignes. Par exemple, chez les enfants, la greffe de CSH est indiquée pour les leucémies aigües lymphoblastiques et myéloblastiques, ainsi que pour des aplasies médullaires sévères. Chez les adultes, elle est indiquée pour les leucémies, les myélomes multiples, la maladie de Hodgkin, les lymphomes non-Hodgkinien, certaines tumeurs solides et les leucémies myéloïdes chroniques [3-5]. La greffe peut être autologue, c’est-à-dire que ce sont les cellules souches du patient qui sont prélevées et réinjectées; ou allogénique, c’est-à-dire que les cellules proviennent d’un autre individu, apparenté ou non. La greffe de CSH vise principalement à contrebalancer les effets cytotoxiques des fortes doses de radiothérapie et de chimiothérapie qui détruisent les cellules malignes ou malades, mais détruisent également le système hématopoïétique. L’injection de cellules souches permet de contrer cette myéloablation en favorisant la régénération du système sanguin [4].
La greffe de CSH comporte plusieurs étapes clés. D’abord, les CSH peuvent être prélevées directement dans la moelle osseuse (greffon médullaire) ou récoltées par cytaphérèse dans le sang (greffon de CSH périphériques) [6]. Le sang de cordon constitue une source de CSH plus primitives [7]. Dans le cas de la cytaphérèse, les cellules souches doivent être mobilisées dans le sang par injection sous-cutanée de la chimiokine G-CSF (granulocyte colony-stimulating factor) quelques jours précédant le prélèvement [8, 9]. Les
cellules souches prélevées sont injectées chez le patient par voie intraveineuse, après les traitements de radiothérapie et de chimiothérapie requis. Ces traitements vont permettre d’éliminer les cellules malades, mais également de préparer le patient à recevoir des cellules souches allogéniques le cas échéant [10]. Après la greffe, les CSH doivent migrer dans la moelle osseuse avant de pouvoir régénérer le système hématopoïétique. Ce processus très rapide implique plusieurs mécanismes communs de migration et d’adhésion cellulaires, tels que l’interaction de CXCL12 (CXC ligand 12) à son récepteur CXCR4 (CXC récepteur 4) [11].
1.1.2 Reprise de la greffe et immunosuppression
Alors que la migration du greffon vers la moelle osseuse dure de quelques heures à 1-2 jours, la repopulation de la moelle osseuse peut prendre des mois, voire des années pour retrouver une homéostasie normale [12]. Les premiers 100 jours post-greffe sont caractérisés par des déficiences immunitaires dues au délai de la production des cellules immunitaires. Selon la source des cellules souches, les délais sont plus ou moins longs. Par exemple, les neutrophiles et les macrophages vont apparaitre dans la circulation sanguine durant les premières semaines post-greffe. Les lymphocytes B apparaissent en circulation environ 3 à 6 mois après une greffe allogénique de cellules souches, alors que les lymphocytes T n’apparaissent qu’après 9 à 12 mois. De plus, puisque l’activation des lymphocytes B naïfs requiert l’aide des lymphocytes T CD4+ [13], la réponse humorale
n’est donc pas optimale dans la première année et peut prendre deux ans avant d’être complètement effective. Des différences au niveau des sous-populations de lymphocytes, dont le ratio lymphocytes T CD4+/CD8+, persistent jusqu’à dix ans post-greffe. Finalement,
certains virus endogènes comme le cytomégalovirus (CMV), se réactivent et contribuent aux défis de la reprise de la greffe [14]. Ces longs délais augmentent la susceptibilité des patients greffés aux infections opportunistes, qui sont l’une des causes majeures de mortalité post-greffe [7, 14, 15].
Quelques études ont montré que la greffe allogénique de CSH pouvait avoir des effets « greffe contre tumeur ». Ces effets sont souhaitables, puisque les cellules immunitaires greffées avec les cellules souches peuvent réagir contre la tumeur de l’hôte et contribuer à l’éliminer. Cependant, cet effet est généralement associé avec la maladie du greffon contre l’hôte [4]. La prise d’immunosuppresseurs est indispensable pour réduire cette réaction, ce qui contribue à l’état immunosupprimé du patient.
1.1.3 Reconstitution du système immunitaire
Plusieurs groupes s’intéressent à développer des thérapies pour améliorer ou accélérer la reconstitution du système immunitaire des patients greffés en cellules souches (revue dans [14]). La majorité des études portent sur le renforcement du système immunitaire endogène et sur le transfert adoptif de lymphocytes T régulateurs, de lymphocytes T éduqués in vitro contre un antigène, de cellules Natural killer (NK) ou de cellules souches mésenchymateuses. La reconstitution de la réponse humorale est généralement étudiée au niveau des programmes de vaccination contre certains microorganismes communs, tels que Streptococcus pneumoniae et Haemophilus influenzae. Les lymphocytes B pourraient offrir un avantage non négligeable aux patients dans le combat des pathogènes opportunistes par leur capacité à se différencier en plasmocytes. Une étude récente a montré que les plasmocytes nichés dans la moelle osseuse étaient mobilisés dans le sang par le G-CSF et injectés avec les cellules souches lors de la greffe. Le phénotype de ces plasmocytes mobilisés ressemblaient au phénotype de plasmocytes provenant du sang ou générés in vitro. Il a été proposé que ces plasmocytes puissent aider à maintenir une certaine mémoire immunologique du donneur chez le patient greffé [16], puisque l’équivalent de 8% du nombre total de ses plasmocytes lui serait redonné lors de la greffe des CSH.
L’équipe du Dre Sonia Néron possède une expertise dans la culture in vitro des lymphocytes B mémoires humains [17, 18]. Notre modèle de co-culture basé sur l’interaction CD40-CD154 permet d’activer les lymphocytes B mémoires en absence de leur antigène, de stimuler leur prolifération et d’initier leur différenciation en plasmocytes. Avec cette expertise, nous suggérons d’exploiter les plasmocytes générés ex vivo comme thérapie cellulaire dans le cadre de greffe autologues de cellules souches pour aider à la reprise de la greffe et combattre les infections opportunistes. Les lymphocytes B mémoires du patient pourraient être prélevés, expansionnés et différenciés en plasmocytes ex vivo, puis réinjectés au patient en même temps que les cellules souches. Le patient pourrait alors récupérer sa propre mémoire immunologique.
1.2 Le lymphocyte B
1.2.1 Ontogénie du lymphocyte B
Le lymphocyte B est la cellule responsable de l’immunité humorale. Sa différenciation en cellule productrice d’immunoglobuline, le plasmocyte, contribue à la reconnaissance des pathogènes et des éléments étrangers dans les fluides biologiques et donc à la défense du corps humain [19]. Le lymphocyte B, le lymphocyte T et la cellule NK, se développent à partir d’un progéniteur commun dans la moelle osseuse. L’engagement de la cellule souche vers le lymphocyte B dépend de plusieurs facteurs de transcription, dont PU.1, E2A et PAX5 (paired box protein 5) [20]. De plus, la molécule CD38 (CD; cluster of
differentiation) est exprimée durant le développement [21]. La production d’un récepteur
membranaire BCR (B-cell receptor) fonctionnel correspond au développement précoce du lymphocyte B. Ce processus est responsable de la très grande diversité du répertoire antigénique reconnu (plus de 1011 immunoglobulines (Ig) uniques chez un individu), mais il
est fortement contrôlé pour empêcher la reconnaissance d’antigènes du soi [19, 22]. Les premières étapes du développement se caractérisent par des réarrangements géniques des chaînes lourdes et des chaînes légères des immunoglobulines [23, 24]. Durant tout ce processus qui a lieu dans la moelle osseuse, les lymphocytes B immatures sont soumis à une sélection négative où les cellules portant des récepteurs de lymphocytes B (BCR,
B-cell receptor) autoréactifs deviennent apoptotiques, anergiques ou sont remodelés. Ces
lymphocytes B expriment également des molécules d’IgD à leur surface par épissage alternatif [25]. Environ 75% des lymphocytes B en développement meurent par apoptose. Le lymphocyte B transitionnel quitte la moelle osseuse et rejoint les organes lymphoïdes secondaires par la circulation sanguine où il poursuivra sa maturation en lymphocyte B naïf, folliculaire ou marginal [20, 24]. Le lymphocyte B naïf est latent et n’exprime plus le marqueur CD38 à sa surface.
1.2.2 Activation dans les ganglions lymphatiques
Les lymphocytes B naïfs entrent dans les ganglions lymphatiques où ils vont migrer vers les follicules et patrouiller pendant environ 24 heures à la reconnaissance d’un antigène. S’ils ne rencontrent pas un antigène, ils retournent dans la circulation sanguine par les canaux lymphatiques, puis entrent dans un autre ganglion lymphatique [26]. La demi-vie du lymphocyte B naïf est d’environ 3 jours. Les antigènes peuvent diffuser librement dans les follicules ou y sont transportés par les macrophages et les cellules dendritiques (figure 1.1). La reconnaissance spécifique d’un antigène par le lymphocyte B augmente l’expression du récepteur de chimiokine CCR7 (CC récepteur 7) et favorise sa migration vers la zone des lymphocytes T [26]. De plus, au contact d’un antigène, il y a activation du lymphocyte B et réexpression de CD38 [21].
Figure 1.1 Rencontre de l’antigène dans les follicules lymphatiques
L’antigène de petite taille diffuse librement à travers les pores du sinus sous-capsulaire et dans les conduits folliculaires, alors que l’antigène de grande taille doit être transloqué et présenté par les macrophages et les cellules dendritiques aux lymphocytes B. Adapté de [27].
Dans un même temps, l’antigène de nature protéique reconnu par le BCR est internalisé, dégradé en courts peptides et présenté sur le complexe d’histocompatibilité II (CHM-II) à
un lymphocyte T. L’activation T-indépendante résulte de la reconnaissance d’antigènes non-protéiques ou multimériques, tels que les polysaccharides ou les glycolipides, qui ne peuvent être présentés sur le CMH-II [28]. Le lymphocyte B extrafolliculaire ainsi activé se différencie directement en plasmocyte à courte-vie et sécrète des IgM de faible affinité. De plus, les lymphocytes B naïfs ayant un BCR de grande affinité pour l’antigène sont généralement activés sans l’aide des lymphocytes T [28, 29]. L’activation T-dépendante requiert la reconnaissance du peptide apprêté dans le CHM-II par le récepteur du lymphocyte T (TCR pour T-cell receptor) et de la présence de plusieurs molécules co-stimulatrices. La signalisation subséquente mènera à l’activation de la transcription des gènes responsables de la prolifération cellulaire et à la formation d’un centre germinatif [25].
1.2.3 L’interaction CD40-CD154
Le contact initial entre le lymphocyte B et le lymphocyte T permet la formation d’une synapse immunologique, c’est-à-dire une polarisation des molécules de co-stimulation et d’adhésion vers le site de contact. La synapse immunologique permet de diriger la sécrétion de cytokines vers la cellule contactée. Les lymphocytes T expriment les molécules CD154, OX40, ICOS (inducible costimulator), CXCR5 et PD1 (programmed
death-1). Ils sécrètent localement plusieurs cytokines, dont l’interleukine-2 (IL-2), l’IL-4,
l’IL-21 et l’interféron-γ [30, 31]. Les lymphocytes B expriment notamment CD40, CXCR5 et les ligands des récepteurs des lymphocytes T, dont CD80 et CD86. L’interaction CD40-CD154 est indispensable pour induire la maturation de l’affinité du BCR. La présence de mutation dans les gènes CD40 ou CD154 peut causer le syndrome d’hyper-IgM chez les humains [25]. Cette interaction constitue le deuxième signal d’activation, suivant la liaison du CMH-II au TCR, et permet la maturation du lymphocyte B dans le centre germinatif.
1.2.4 Maturation de l’affinité : le centre germinatif
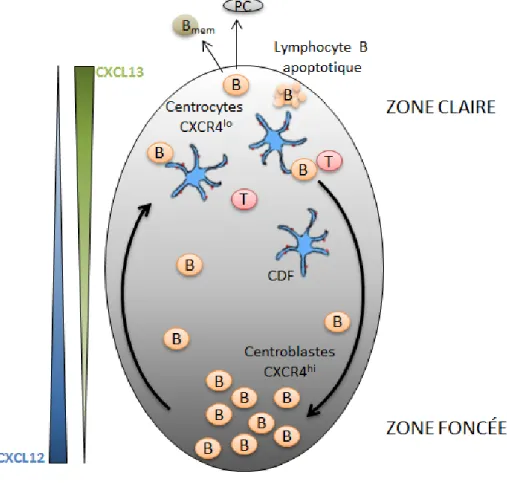
Le centre germinatif est une zone dans le follicule du ganglion où il y a une grande activité prolifératrice [32]. Cette structure est organisée en une zone claire et une zone foncée. La zone claire est caractérisée par une faible densité de lymphocytes B étant donné la présence d’un réseau de cellules dendritiques folliculaires. Les lymphocytes B sont continuellement en mouvement entre la zone foncée où ils prolifèrent (centroblastes) et la
zone claire où ils testent l’affinité de leur BCR pour l’antigène (centrocytes). Des gradients de chimiokines, soit CXCL12 et CXCL13, dirigent la migration des lymphocytes B dans le centre germinatif. De plus, l’expression différentielle de CXCR4 chez les centroblastes dirige ceux-ci vers la zone foncée. L’accumulation des centrocytes dans la zone claire est dirigée par CXCR5 et le niveau élevé de CXCL13 (revue dans [32]). Les centroblastes sont des lymphocytes B en prolifération qui ont une faible expression d’immunoglobulines membranaires. Les centrocytes, quant à eux, sont des lymphocytes B non-prolifératifs et expriment des immunoglobulines membranaires.
Figure 1.2 Structure dynamique du centre germinatif
Les lymphocytes B entament des cycles de prolifération (centroblastes dans la zone foncée), de maturation d’affinité et de sélection positive (centrocytes dans la zone claire). Les cellules dendritiques folliculaires (CDF) présentent les antigènes aux centrocytes. Les centrocytes compétitionnent ensuite pour le contact avec les lymphocytes T auxiliaires folliculaires. Les centrocytes qui réussissent à augmenter l’affinité de leur BCR sont sélectionnés et différenciés en lymphocytes B mémoires ou en plasmocytes. Les gradients des chimiokines CXCL12 et CXCL13 dirigent la migration des cellules. Figure adaptée de [32].
Dans la zone claire, les cellules dendritiques folliculaires (CDF) et les lymphocytes T auxiliaires folliculaires contribuent à la maturation du lymphocyte B et leur fournissent des signaux de survie [33]. Seulement un à six clones de lymphocytes B sont capables de mener à la formation d’un centre germinatif, ce qui semble favoriser une réponse humorale oligoclonale [32]. Deux évènements permettent la maturation de l’affinité et la production d’immunoglobulines de haute affinité. La commutation isotypique est un processus de recombinaison qui permet le réarrangement des segments géniques des chaînes lourdes et de produire des immunoglobulines de classe G, A et E. L’hypermutation somatique se déroule durant la prolifération des lymphocytes B activés et permet d’insérer aléatoirement des nucléotides dans les régions variables des immunoglobulines, et ainsi d’en modifier l’affinité. Ces deux processus sont sous le contrôle des enzymes AID (activation-induced cytidine deaminase) et UNG (uracil DNA
glycosylase) [25, 28, 32]. Les lymphocytes B qui réussissent à augmenter l’affinité de leur
BCR ont un avantage prolifératif et compétitionnent pour les signaux de survie tels que certaines interactions cellulaires. Ces lymphocytes B vont poursuivre leur différenciation en cellules mémoires ou en plasmocytes. Certains signaux spécifiques, tels que les lysophospholipides régulent l’émigration des cellules des ganglions vers la circulation sanguine [24, 25].
1.2.5 Les immunoglobulines
Les lymphocytes B peuvent produire plusieurs classes d’immunoglobulines qui seront exprimés à leur surface ou sécrétés dans l’environnement [19]. Les immunoglobulines sont catégorisées en cinq classes basées sur la chaîne lourde, soit IgM et IgD qui sont à faible affinité et IgG, IgA et IgE qui sont à haute affinité. De plus, chez l’humain, les IgG sont subdivisées en quatre sous-classes (IgG1 à IgG4) et les IgA, en deux sous-classes
(IgA1 et IgA2) [35]. Chaque classe a un rôle distinct dans la réponse humorale et
enclenche des processus immunitaires différents. Par exemple, l’IgM est généralement associée à la réponse primaire alors que l’IgA est associée à l’immunité des muqueuses [36]. Les IgG contribuent à la réponse immunitaire secondaire principalement par ses fonctions d’opsonisation et de neutralisation des pathogènes, qui mènent à l’activation du complément. Les IgE sont impliquées dans la réponse antiparasitaire, bien qu’elles soient majoritairement connues pour leur rôle dans les allergies [35]. Comme pour la spécificité qui est unique, chaque lymphocyte B ayant subi la commutation de classe ne peut
exprimer qu’une seule classe à la fois. Le lymphocyte B naïf et les lymphocytes mémoires des zones marginales peuvent cependant exprimer à la fois des IgM et des IgD [37]. Les IgG sont la classe la plus abondante dans le sang, dont les concentrations plasmatiques relatives entre les sous-classes sont en ordre décroissant (IgG1 étant la sous-classe la
plus abondante, et IgG4, la moins abondante) [38]. Des modifications post-traductionnelles
comme la glycosylation jouent des rôles importants pour la reconnaissance des immunoglobulines par les récepteurs Fc à la surface des cellules immunitaires effectrices et l’induction de la cascade du complément. Des défauts dans ces patrons de glycosylation des immunoglobulines peuvent être associés à des maladies auto-immunitaires [35].
1.2.6 Sous-populations de lymphocytes B dans le sang
Les lymphocytes B représentent entre 5 et 10% des lymphocytes circulant dans le sang et se divisent en quatre catégories : lymphocytes B immatures/transitionnels, naïfs, mémoires et les plasmocytes (figure 1.3) [19]. Ces populations sont hétérogènes et peuvent être distinguées par des marqueurs de surface caractéristiques. Les lymphocytes B naïfs et immatures sont caractérisés par l’expression d’IgM et d’IgD et par l’absence d’expression de CD27. Les lymphocytes B immatures expriment fortement CD38, contrairement aux lymphocytes B naïfs [39]. Après maturation dans le centre germinatif, les lymphocytes B mémoires et les plasmocytes retournent dans le sang pour rejoindre leur niche respective. Les lymphocytes B mémoires résident dans les zones de drainage des antigènes, soit la rate et les amygdales, et vont régulièrement circuler dans le sang [39]. Deux grandes populations de lymphocytes B mémoires peuvent être distinguées, soit ceux à faible affinité (IgM+) et ceux qui ont commuté (IgG+, IgA+, IgE+) [40]. De plus, les
lymphocytes B mémoires expriment majoritairement CD27, bien qu’une proportion de moins de 25% ne l’exprime pas [41, 42]. Les lymphocytes B mémoires commutés (soit CD19+ CD27+/- IgG+/IgA+/IgE+) produisent des immunoglobulines de haute affinité et
modifient leur patron de récepteurs membranaires, ce qui leur permet de répondre plus rapidement lors d’un second contact avec l’antigène [39]. Les plasmocytes, quant à eux, transitent par le sang pour rejoindre la moelle osseuse. Les plasmocytes expriment fortement CD38. Selon le stade de développement, ils peuvent ou non exprimer CD138 [43]. Les travaux de maîtrise de Rayelle Itoua Maïga avec l’équipe du Dre Néron ont permis de caractériser les plasmocytes du sang basé sur l’expression de CD31 et CD39
[43, 44]. Ces plasmocytes semblent tous exprimer CD31, alors que l’expression de CD39 est partagée.
Figure 1.3 Populations de lymphocytes B dans le sang
Les lymphocytes B circulent dans le sang pour rejoindre les organes lymphoïdes primaires ou secondaires selon leur stade de développement. Figure adaptée de [39, 42, 44, 45].
1.3 Le plasmocyte
1.3.1 Différenciation en plasmocytes
À la sortie du centre germinatif, les lymphocytes B peuvent se différencier en plasmablastes, puis subséquemment en plasmocytes. Cette différenciation amène de nombreux changements dans le transcriptome cellulaire, notamment au niveau du facteur de transcription PAX5. L’expression de PAX5 est requise pour maintenir l’identité des lymphocytes B (section 1.2.1), mais doit être réprimée pour permettre la différenciation en plasmocytes. D’autres facteurs de transcription, tels que MITF (microphthalmia-associated
transcription factor), XBP1 (x-box binding protein 1) et BLIMP1 (B lymphocyte-induced maturation protein-1), jouent un rôle dans l’initiation de la différenciation [20]. Les
plasmablastes possèdent une forte capacité de prolifération et sécrètent des immunoglobulines. Ils se forment dans les organes lymphoïdes secondaires et transitent par le sang vers la moelle osseuse. Ils expriment fortement CD38, mais n’expriment peu ou pas CD138. Une troisième molécule, CD39, semble être un marqueur adéquat pour les plasmablastes [43, 44], bien qu’elle soit également exprimée chez d’autres types cellulaires, dont les macrophages, certaines sous-populations de lymphocytes T et les cellules endothéliales vasculaires [46]. L’arrêt du cycle cellulaire est caractéristique du passage de plasmablaste vers le plasmocyte. Les plasmocytes expriment fortement CD38, tout comme les plasmablastes, et sont couramment identifiés par l’expression de CD138 [43]. CD138 (ou syndecan-1) est une protéoglycane membranaire qui joue un rôle dans la migration des lymphocytes [47]. De plus, CD31 est une intégrine également retrouvée chez les plasmocytes [43] et pourrait servir à l’identification des plasmocytes [44]. D’autres molécules de surface, telles que CD45, CD126 et CD54, sont différentiellement exprimées chez les plasmablastes, les plasmocytes précoces et les plasmocytes matures. Notamment, les plasmocytes matures dans la moelle osseuse n’exprimeraient que faiblement CD45.
1.3.2 Niches de survie
L’immunité à long terme est obtenue à la fois par les lymphocytes B mémoires qui vont se différencier massivement en plasmocytes au contact de l’antigène et par les plasmocytes qui maintiennent un certain niveau d’anticorps dans le sang. Bien que ces deux populations aient des niches différentes (section 1.2.6), leur survie dépend de nombreux facteurs solubles et interactions cellulaires fournis par leur microenvironnement respectif [48, 49]. Plus de 80% des immunoglobulines du sérum proviennent des plasmocytes établis dans la moelle osseuse [50]. Les plasmocytes peuvent y survivre pendant des mois et des années où ils ne représentent que 0,5% de la population cellulaire [43]. Les lymphocytes B mémoires contribuent à régénérer la réserve de plasmocytes lorsque celle-ci s’épuise, mais ils ne sont pas requis pour leur maintien dans la moelle osseuse [22]. Le nombre de niches de survie est restreint, ce qui oblige les plasmocytes à compétitionner pour celles-ci [43]. Les plasmocytes peuvent être classifiés à courte-vie ou à longue-vie. Les plasmocytes à courte-vie sont majoritairement générés par la voie indépendante des
lymphocytes T et sécrètent des IgM ou des IgG [51]. Les plasmocytes de haute affinité générés à la fin de la réaction du centre germinatif semblent être davantage favorisés pour coloniser la moelle osseuse et deviennent à longue-vie. Cependant, seulement une faible proportion des plasmocytes générés réussissent à migrer vers la moelle osseuse et à recevoir les signaux de survie. La majorité des plasmocytes produisent intensément des immunoglobulines durant trois à six jours [52, 53]. Ces plasmocytes à courte-vie permettent de combattre le pathogène immédiatement, puis meurent par apoptose afin de freiner la réponse humorale à la fin de l’infection [54].
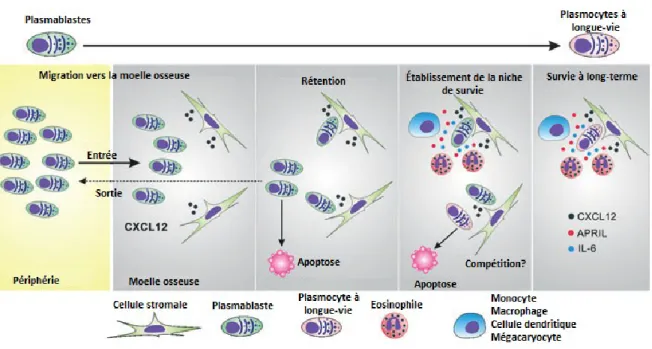
Figure 1.4 Niches de survie des plasmocytes
Les plasmocytes à longue-vie survivent dans la moelle osseuse pendant des années dans des microenvironnements spécifiques. Ces microenvironnements, ou niches, sont formés de cellules immunitaires et de cellules structurelles qui fournissent des facteurs solubles et des ligands membranaires pour diverses interactions cellulaires. Figure traduite de [51].
La migration des plasmocytes dépend de la perte d’expression des récepteurs CXCR5 et CCR7, qui les maintenaient dans les ganglions, et de l’acquisition de l’expression du récepteur CXCR4 [48, 55]. Les cellules stromales de la moelle expriment CXCL12, le ligand de CXCR4, et permettent la rétention des plasmocytes. CXCL12 est aussi exprimée par les cellules épithéliales des muqueuses et contribue à la localisation des plasmocytes
IgA+ dans ces sites [56]. D’autres récepteurs tels que CXCR6 sont important dans la
migration des plasmocytes vers la moelle osseuse. Les niches de survie des plasmocytes se composent de plusieurs types cellulaires, dont les basophiles, les cellules dendritiques, les monocytes et les neutrophiles, qui sécrètent de nombreux facteurs solubles [49] (figure 1.4). Les plasmocytes sembleraient également établir une relation avec les ostéoclastes et les ostéoblastes [43]. Plus de 95% des plasmocytes se localisent près d’une sous-population de cellules mésenchymateuses, les cellules CAR (CXCL12-abundant reticular
cells) [48, 49]. Ces cellules agiraient comme cellules supports [48]. Des études in vitro
suggèrent que le TNFα (tumor necrosis factor), BAFF (B cell activating factor) et APRIL (a
proliferation inducing ligand) sont d’importants facteurs de survie. De plus, 4, 5,
l’IL-6 et l’IL-10 semblent prolonger la survie des plasmocytes [57, 58]. Les mégacaryocytes et les éosinophiles contribuent aussi à l’établissement de la niche, puisqu’ils sécrètent APRIL et l’IL-6. La plupart de ces facteurs solubles promeuvent l’expression de molécules anti-apoptotiques [59]. La capacité de la moelle osseuse à offrir un refuge aux plasmocytes a été estimée à 109 plasmocytes [60]. Lors d’une réponse immunitaire, les nouveaux
plasmocytes formés peuvent forcer les plasmocytes établis dans la moelle osseuse à recirculer dans le sang [24].
1.3.3 Métabolisme sécrétoire
La différenciation en plasmocyte entraine d’importants changements morphologiques et fonctionnels afin de produire de grandes quantités d’immunoglobulines. Un plasmocyte est en mesure de sécréter plus de 1 000 immunoglobulines par seconde [34]. L’un de ces changements est l’élargissement du réticulum endoplasmique (RE) et des organelles sécrétoires [53]. De plus, l’augmentation de la synthèse protéique associée à la production d’immunoglobulines impose des stress métaboliques et oxydatifs considérables [61]. Ces stress servent à la fois à la différenciation et au contrôle de la durée de vie des plasmocytes [62]. Le défi des plasmocytes est donc de produire de grandes quantités d’immunoglobulines riches en ponts disulfures tout en contrôlant les déséquilibres homéostatiques qui s’en suivent. [61]. La formation des ponts disulfures se déroule dans le RE durant le processus de repliement des protéines où deux cystéines sont oxydées par l’isomérase PDI (protein disulphide isomerase) [63]. La PDI est régénérée par les flavoprotéines Ero1 (ER-oxidoreductin 1), dont leur expression est augmentée en réponse à un stress du RE. Le débordement dans le traitement des immunoglobulines lors de
l’initiation de la différenciation en plasmocyte génère un signal de stress qui active la réponse des protéines non-repliées (UPR, unfolded protein response). Cette réponse adapte la capacité de chargement du RE et contribuerait à l’acquisition du phénotype sécrétoire des plasmocytes [34]. De plus, l’accumulation du facteur de transcription XBP-1 (section 1.3.1) dans les premières phases de différenciation plasmocytaire est un facteur clé de la réponse UPR [52, 61]. L’accepteur final d’électron lors de la formation des ponts disulfures est l’oxygène et mène à la génération de peroxyde (H2O2) en quantité
équivalente au nombre de ponts disulfures formés [54]. La quantité d’espèces réactives à l’oxygène (ROS, reactive oxygen species) intracellulaires augmente rapidement [64]. Le plasmocyte contrôle ce stress oxydatif par l’activation de ses défenses antioxydantes, notamment les peroxiredoxins et le facteur de transcription Nrf2 [61, 62]. Des études ont montré que la capacité du protéasome pour la dégradation des protéines mal-repliées diminuait au fur et à mesure que le plasmocyte mature. Les protéines polyubiquitinées destinées à la dégradation par le protéasome s’accumulent, ce qui stabilise les protéines pro-apoptotiques et conduit à l’apoptose (revu dans [61]). D’autres éléments sensibilisent les plasmocytes à l’apoptose, dont le déséquilibre de la balance redox, l’approvisionnement limité en acides aminés et une réponse UPR prolongée [61, 65] (figure 1.5).
Figure 1.5 Le stress cellulaire chez le plasmocyte
L’adaptation du plasmocyte au stress cellulaire lui permet de rencontrer les exigences de sécrétion d’immunoglobulines. Cependant, les différents types de stress auxquels le plasmocyte est soumis peuvent agir en synergie et déclencher la mort par apoptose. Figure traduite de [61].
1.3.4 La cyclase ADP ribosyle CD38
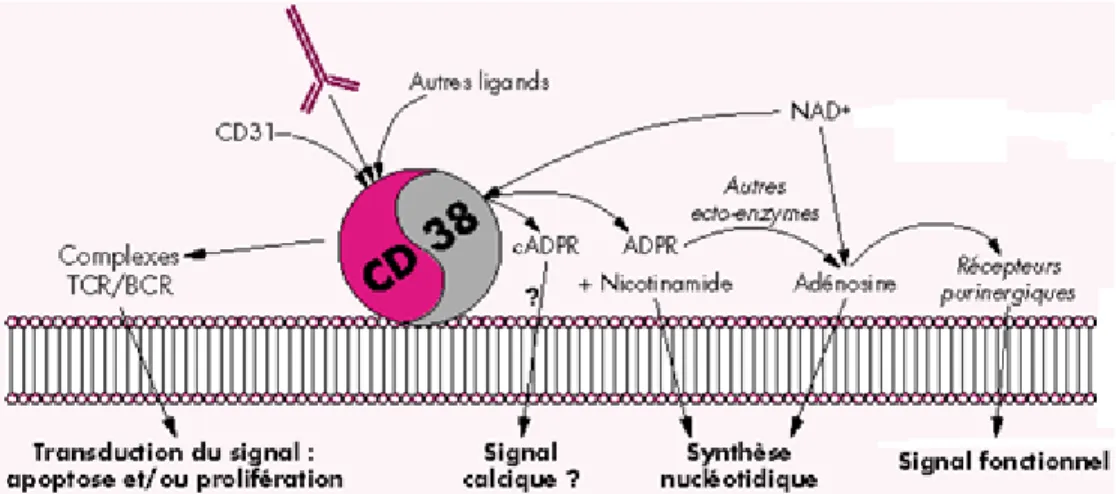
CD38 est une molécule exprimée à la surface des lymphocytes B en développement dans la moelle osseuse, des lymphocytes B activés et différenciés. Son expression n’est pas restreinte à ce seul type cellulaire, puisqu’elle est notamment exprimée chez les lymphocytes T activés et les cellules dendritiques [66]. CD38 est une ectoenzyme membranaire de type II et se localise dans la membrane plasmique et les membranes des organelles [66]. Cette enzyme catalyse la conversion du NAD+ (nicotinamide adénine
dinucléotide) en ADPRc (adénosine diphosphate ribose cyclique) à un pH neutre. L’ADPRc agirait comme second messager par la libération du Ca2+ intracellulaire [67, 68].
NAD+/NADH constitue un couple redox crucial. L’utilisation du NAD+ par CD38 affecte son
niveau intracellulaire et dépend de sa disponibilité [69]. Il a été proposé que CD38 puisse agir comme un senseur du potentiel redox extracellulaire [70]. Elle est aussi connue pour catalyser d’autres réactions enzymatiques à un pH acide [67]. CD38 agit également
comme récepteur et l’interaction avec son ligand CD31 mène à la transduction de signaux d’activation, de prolifération et d’adhésion [68]. Un groupe a d’ailleurs reporté l’implication de CD38 dans la migration des lymphocytes leucémiques chroniques par CXCL12 [71]. CD38 semblerait également favoriser l’apoptose chez certains lymphomes [72]. La transduction du signal par CD38 à la liaison de CD31 est dépendante de la dimérisation de CD38, de sa translocation dans les radeaux lipidiques et de son association avec le complexe transductionnel de CD19 et de CD81 [73]. De plus, sa localisation intracellulaire lui permet d’être impliqué dans d’autres processus fondamentaux et métaboliques, tels que la sécrétion d’insuline, l’activité des ostéoclastes et la contraction des muscles lisses pulmonaires [66].
Figure 1.6 Rôles multiples de CD38
La liaison de CD38 par son ligand CD31 ou des anticorps dirigés contre CD38 mène à son association avec les complexes du BCR (ou du TCR chez les lymphocytes T) et à la transduction du signal. Son activité enzymatique permet de convertir le NAD+ en plusieurs produits finaux qui agissent comme messagers intracellulaires. Figure tirée de [74].
1.4 Signalisation intracellulaire
1.4.1 Homéostasie chez le lymphocyte B
La capacité des lymphocytes B à percevoir et à répondre à leur microenvironnement passe par un réseau complexe d’interactions moléculaires et de modifications post-traductionnelles. Des dysfonctionnements dans la signalisation cellulaire mènent à un bris de l’homéostasie cellulaire, dont le développement de cancers et de maladies auto-immunitaires [75-77]. Ce réseau de signalisation comprend plusieurs familles de protéines signalétiques. La famille des MAPK (mitogen-activated protein kinase) est impliquée dans les réponses aux facteurs de croissance, aux cytokines et au stress. Les quatre principales voies des MAPK sont les voies de BMK1 (big map kinase-1), ERK (extracellular signal-regulated kinase), JNK (c-jun N-terminal kinase) et p38. Plusieurs isoformes existent pour ERK (soit ERK1 et ERK2), JNK (JNK1, JNK2, JNK3) et p38 (p38α, p38β, p38γ, p38δ). La phosphorylation des cibles des MAPK affectent leur localisation cellulaire, leur stabilité, leur capacité de lier l’ADN et d’interagir avec d’autres protéines. Généralement, ERK est associé aux signaux de facteurs de croissance, alors que p38 et JNK répondent aux signaux de stress [75].
Plusieurs autres intermédiaires ayant des activités de kinases sont aussi impliqués dans la régulation de la physiologie des lymphocytes B. La PKA (protéine kinase A) qui est régulée par l’adénylate cyclase, agit en association avec des récepteurs couplés aux protéines G [78]. La sérine/thréonine kinase Akt (AK transforming, ou PKB, protein kinase
B) devient active une fois recrutée à la membrane plasmique [79]. Akt mène à la survie
cellulaire en inactivant la protéine pro-apoptotique Bad et la suppression de l’expression de gènes pro-apoptotiques. Elle active également le facteur de transcription central qu’est NFκB (nuclear factor κB) [80].
La famille des kinases lipidiques, dont PI3K (phosphatidyl-inositol-3’ kinase), phosphoryle le groupement hydroxyle de certains phospholipides membranaires, soit les phosphoinositides (PIP, PIP2 et PIP3). La voie de PI3K est impliquée dans plusieurs processus de développement chez les lymphocytes B (revu dans [79]). De plus, PIP3 active BTK (Bruton’s tyrosine kinase) et mène à la phosphorylation de la protéine kinase C (PKC) et à la libération de calcium par IP3 (inositol triphosphate) et DAG (diacylgycérol) [81]. L’influx de calcium entraîne d’ailleurs l’activation de plusieurs voies signalétiques, dont NFκB et les MAPK [81]. NFκB joue des rôles cruciaux dans l’inflammation, l’immunité, l’apoptose et la prolifération cellulaire [82]. NFκB se situe dans le cytoplasme
où elle protéine est associée avec un complexe inhibiteur. La phosphorylation du complexe inhibiteur par IKK (IκB kinase) permet de libérer et d’activer NFκB. NFκB migre au noyau où il régule l’expression de multiples gènes anti-apoptotiques. Il est notamment impliqué dans la signalisation par CD40 et BAFF [83].
Les kinases de la famille Src régulent plusieurs voies, dont les MAPK et la PI3K. La kinase Src peut phosphoryler des substrats dans le cytosol, à la membrane plasmique ou aux adhésions cellule-cellule et cellule-matrice. Elle régule la motilité et l’adhésion cellulaire, ainsi que la prolifération et la survie. Le gène src est perçu comme un proto-oncogène [84].
Une voie très importante dans la prolifération et la différenciation des lymphocytes B est la voie JAK/STAT (Janus kinase / signal transducers and activators of transcription). Elle représente l’une des voies les plus directes, dont la liaison de cytokines à leurs récepteurs entraine la phosphorylation des JAK qui phosphorylent les STAT. Les STAT activés entrent au noyau ou se dirigent dans la mitochondrie. Ils forment des dimères ou des oligomères afin de réguler l’expression des gènes cibles. Il y a quatre membres dans la famille des JAK et sept membres dans la famille des STAT [85]. Bien que la voie soit directe, elle communique avec plusieurs autres voies telles que celles des protéines ERK et Akt. Les différentes réponses qu’induit l’activation de la voie JAK/STAT dépendent du signal, de la localisation et du contexte cellulaire. En plus de la prolifération et de la différenciation, la voie JAK/STAT est impliquée dans la migration, l’apoptose et la survie cellulaire [85]. Cette voie est régulée négativement par l’internalisation du récepteur, la déphosphorylation par des tyrosines phosphatases et l’inhibition directe par les PIAS (protein inhibitor of activated STAT) et les SOCS (suppressor of cytokine signalling) [86].
1.4.2 Le potentiel redox comme élément de signalisation
Des molécules chimiques hautement réactives, les ROS, jouent d’importants rôles dans la régulation de la survie cellulaire et sont très importantes pour l’homéostasie chez le plasmocyte (section 1.3.3). Le potentiel redox est impliqué dans la régulation de plusieurs voies de signalisation. Certains facteurs de transcription, dont NFκB, sont sensibles aux fluctuations du potentiel redox. Leur oxydation semble inhiber leur capacité de se lier à l’ADN, ce qui les empêche d’activer l’expression de leurs gènes cibles, notamment l’expression de gènes anti-apoptotiques [87]. De plus, la peroxydation des lipides peut
mener à l’activation de JNK et de la caspase-3 et induire l’apoptose. Il est également connu que les lipides oxydés extracellulaires peuvent être reconnus [87, 88] et mener à une réponse immunitaire [89]. Le stress oxydatif peut induire des modifications des cystéines chez de nombreuses protéines telles que JNK, Akt, p38 et PKC. Ces modifications ont des rôles régulateurs dans la fonction de ces protéines [90].
1.5 Culture in vitro des lymphocytes B 1.5.1 Modèle de culture en deux phases
Plusieurs groupes de recherche s’intéressent depuis quelques années au développement d’un modèle basé sur l’interaction CD40-CD154 permettant la culture des lymphocytes B (revu dans [91]). Différentes manières d’activer la signalisation par CD40 ont été rapportées : anticorps monoclonaux anti-CD40, protéines CD154 recombinantes, membranes CD154+ solubles, ainsi que des lignées cellulaires transformées pour
exprimer CD154. L’utilisation des lignées cellulaires CD154+ se rapproche davantage de
l’interaction entre le lymphocyte B et le lymphocyte T in vivo et à la formation de synapse [91]. De plus, l’intensité de l’interaction CD40-CD154 peut être augmentée ou diminuée afin de moduler la prolifération et la différenciation des lymphocytes B humains [92]. Durant les dernières années, l’équipe du Dre Sonia Néron a travaillé sur la mise au point d’un modèle de culture utilisant une lignée de fibroblastes murins qui expriment de manière constitutive la molécule CD154 humaine à sa surface [93]. Ces cellules de la lignée L4.5 sont irradiées, ce qui bloque leur capacité de se diviser, mais n’affecte pas leur viabilité, ni leur métabolisme [94]. De plus, ce modèle permet de dévier la présentation antigénique, puisqu’aucun antigène n’est nécessaire pour l’activation des lymphocytes B. Dans ce modèle, l’activation des lymphocytes B est polyclonale et permet la production de toutes les sous-classes d’IgG dans les proportions comparables à celles du sérum humain [18]. À ce jour, le modèle de culture se divise en trois étapes, soit une phase d’expansion des lymphocytes B, qui est suivie de quelques jours de transition vers une phase de différenciation en plasmocytes [17, 18, 44, 95].
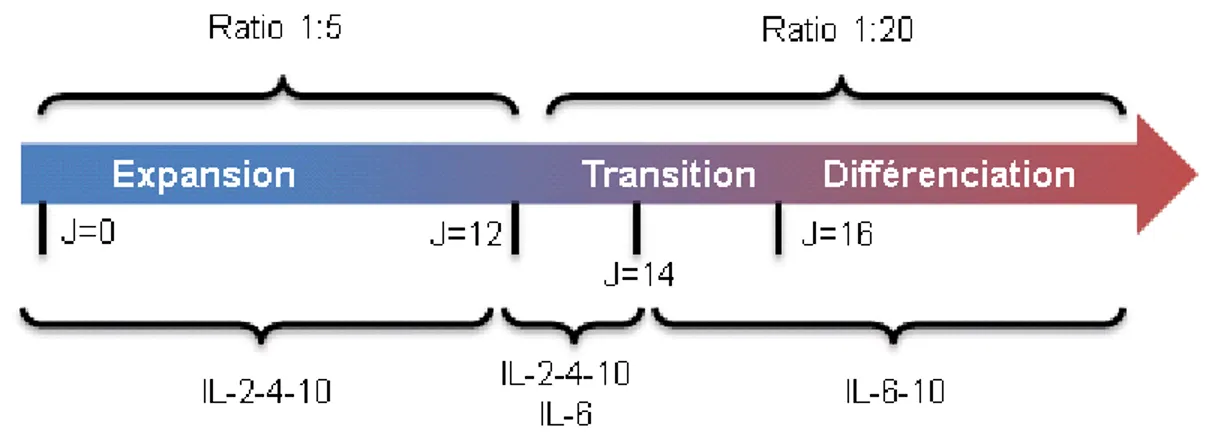
1.5.1.1 Phase d’expansion
La phase d’expansion permet d’augmenter le nombre de lymphocytes B mémoires provenant du sang d’individus en santé. Il a été montré que les lymphocytes B pouvaient être maintenus pendant une période de 50 à 65 jours et permettait de générer entre 107 et
109 fois plus de cellules qu’au départ [18]. Durant cette phase, les lymphocytes B sont
activés par un niveau élevé d’interaction CD40-CD154, ce qui correspond à la présence de 1600 à 2500 molécules CD154 par lymphocyte B [17]. La durée de la phase d’expansion dépend de la prolifération souhaitée des lymphocytes B. Elle peut être de courte-durée, soit entre 9 à 12 jours, ou de longue-durée, généralement entre 19 et 30 jours. Lors de cette étape, l’addition de trois cytokines, l’IL-2, l’IL-4 et l’IL-10 permet de soutenir la prolifération et la viabilité des lymphocytes B.
1.5.1.2 Phase de différenciation
L’un des buts de la culture de lymphocytes B est d’obtenir une différenciation terminale menant à la génération de plasmocytes. Pour permettre la différenciation, les lymphocytes B expansionnés sont soumis à un faible niveau d’interaction CD40-CD154, ce qui correspond à la présence de 300 à 500 molécules CD154 par lymphocyte B [92]. Les travaux de Josiane Tremblay-Rochette ont permis de conclure que la combinaison d’IL-6 et d’IL-10 favorisait la meilleure différenciation en plasmocytes, ainsi qu’une sécrétion accrue d’immunoglobulines confirmant la différenciation en cellules sécrétrices. Cependant, les lymphocytes B doivent être conditionnés quelques jours avant à l’IL-6 en présence d’IL-2 et d’IL-4 afin d’augmenter l’expression de son récepteur à l’IL-6 [95]. À ce jour, la survie des plasmocytes générés dans ce modèle n’est pas optimale et nécessite une investigation afin d’améliorer le microenvironnement.
1.5.2 Facteurs solubles
La relation du lymphocyte B avec son environnement est nécessaire et essentielle à son développement. Une panoplie de facteurs solubles influencent de près ou de loin le lymphocyte B tout au long de son cheminement dans le corps humain, de la moelle osseuse aux ganglions lymphatiques en passant par la circulation sanguine. Plusieurs ont été décrits aux sections précédentes. Cependant, en culture cellulaire, il est très difficile de reproduire avec exactitude la composition, la séquence ou la concentration en facteurs
solubles que le lymphocyte B rencontrerait in vivo. Les cytokines, chimiokines ou autres hormones sont largement étudiées et connues pour moduler le comportement des lymphocytes B [96, 97]. Cependant, d’autres facteurs solubles, dont les vitamines, peuvent avoir un potentiel pour améliorer la viabilité et la différenciation des plasmocytes, notamment pour leurs propriétés antioxydantes.
1.5.2.1 La vitamine E et son analogue hydrosoluble
La vitamine E est un antioxydant liposoluble et est considérée comme la première ligne de défense contre la peroxydation des lipides [98]. La vitamine E agit en association avec le glutathion et un facteur membranaire pour prévenir les dommages de la peroxydation des lipides [99]. Elle est retrouvée sous plusieurs formes, soit naturelle : α-, β-, γ- et δ-tocophérols; soit insaturée : α-, β-, γ- et δ-tocotriénols. L’α-tocophérol est le mieux assimilé par le foie et représente la forme la plus abondante dans les tissus [98, 100]. La concentration plasmatique normale de la vitamine E se situe entre 23 μM et 35 μM [99]. Son action antioxydante passe par la délocalisation de l’électron non-pairé au cycle aromatique de la vitamine E, rendant le radical beaucoup moins réactif. Cependant, la vitamine E peut également avoir des effets pro-oxydants, notamment en présence de métaux comme le cuivre ou lorsqu’il est oxydé après son action antioxydante [99]. La vitamine E joue aussi un rôle dans les membranes lipidiques et permet de stabiliser celles-ci [99]. La vitamine E semble avoir des effets immunomodulateurs, dont la formation de la synapse immunologique et son implication dans les processus d’activation et de prolifération. Des déficiences en vitamine E entrainent plusieurs problèmes immunitaires et ont été étudiées chez plusieurs espèces animales [98]. La vitamine E étant une molécule liposoluble, le choix du solvant organique peut être toxique pour les cellules en culture. Un analogue de la forme α-tocophérol, le Trolox, possède un groupement carboxyle au lieu de la queue lipophile, ce qui augmente sa solubilité dans l’eau [101]. Au point de vue physiologique, le Trolox comme la vitamine E peuvent être recyclés par l’albumine complexée à la bilirubine qui se retrouvent dans le plasma [102].
1.5.2.2 La vitamine C
La vitamine C (ou acide ascorbique) est une petite molécule, mais un puissant antioxydant biologique hydrophile [103]. La vitamine C peut interagir directement avec un large spectre de radicaux libres formés par les voies métaboliques [104, 105]. Sa forme oxydée est recyclée en acide ascorbique par le gluthation et certaines réductases. Indirectement, la vitamine C est aussi impliquée dans le recyclage de la forme oxydée de la vitamine E et permet ainsi de régénérer les réserves contre la peroxydation des lipides [105]. Tout comme la vitamine E, la vitamine C possède également des activités pro-oxydantes en présence de métaux. Son implication dans l’immunité se situe au niveau de la protection contre le stress oxydatif durant les infections [106]. Au point de vue des cellules cultivées, la majorité des milieux de culture ne contiennent presque jamais d’acide ascorbique, ce qui met les cellules à la merci des variations du potentiel redox [104].
1.5.2.3 Le N-acétyl cystéine
Le N-acétyl cystéine (ou NAC) est un analogue de la cystéine qui possède plusieurs applications thérapeutiques, dont le traitement de plusieurs désordres respiratoires [107]. Le NAC peut servir comme précurseur dans le cycle du glutathion, d’où provient son activité antioxydante. Cette molécule possède également une activité antioxydante intrinsèque [108]. Il a été démontré que l’addition de NAC au milieu de culture inhibe la différenciation des lymphocytes B naïfs activés par l’interaction CD40, diminue la sécrétion d’immunoglobulines et améliore la viabilité cellulaire dans la phase d’expansion [109].
1.5.2.4 La vitamine A
La vitamine A (ou acide rétinoïque) n’est pas considérée comme un antioxydant, bien qu’elle puisse inhiber la NADPH-oxydase de la membrane plasmique et ainsi influencer le potentiel redox [110]. La concentration plasmatique de la vitamine A se situe entre 5 et 20 nM [111]. Une trop grande concentration de vitamine A est toxique. Cette vitamine semble avoir un rôle crucial dans le système immunitaire, entre autres dans la régulation du développement du lymphocyte B et de la production d’immunoglobulines [112]. Sa propriété lipophile lui permet d’entrer rapidement dans les cellules par diffusion passive. La vitamine A peut inhiber la prolifération, probablement par une régulation négative de la voie de NFκB. Selon d’autres circonstances, elle favorise la prolifération et la
différenciation cellulaire par l’activation de la voie de p38 (section 1.4.1). Ses effets dépendent du stade de développement de la cellule et des stimuli [111]. De plus, la vitamine A induit l’expression de CD38 chez plusieurs types cellulaires [113, 114].
1.6 Protéines animales et milieux de culture 1.6.1 Problématique du sérum bovin
La culture de cellules humaines ou animales constitue un outil indispensable dans l’étude de la biologie cellulaire et moléculaire et pour les applications de la biotechnologie. La culture cellulaire vise à reproduire le microenvironnement in vivo en respectant les paramètres physicochimiques tels que la température, le pH et l’osmolarité. Les milieux de culture de base réussissent généralement à maintenir ces paramètres, mais requièrent d’être enrichis en facteurs solubles d’origine sérique pour permettre la croissance et la différenciation des cellules en culture. Le sérum bovin est le supplément le plus largement utilisé en culture. Il se compose d’un nombre important de molécules de faible et de haut poids moléculaire et possède des activités activatrices et inhibitrices de la prolifération cellulaire. Le sérum fournit notamment des hormones, des vitamines, des facteurs de croissance, des protéines de transport, des facteurs d’attachement et de migration, des facteurs de détoxication et de stabilisation (revu dans [115]).
Dans le cadre de thérapies destinées à l’humain, la qualité et de la sécurité des produits générés par la culture cellulaire sont des considérations très importantes [116]. Malheureusement, la composition du sérum bovin est chimiquement indéfinie et il peut même contenir des toxines [115]. Chez l’humain, l’utilisation du sérum bovin augmente les risques de contamination aux prions et aux virus, ainsi que le développement de zoonoses. De plus, les protéines animales pourraient être reconnues par le système immunitaire humain et pourraient causer des réactions secondaires indésirables [117]. Conséquemment, il se pourrait que la culture de cellules immunitaires en présence d’antigènes bovins puisse influencer leur croissance et leur différenciation. Des considérations éthiques entrent également en cause dans les désavantages du sérum bovin, entre autres les méthodes utilisées pour récolter le sérum du fœtus [115].
1.6.2 Des alternatives au sérum en culture cellulaire
Pour ces considérations biologiques et éthiques, il devient nécessaire de développer des milieux de culture sans sérum bovin. L’option de remplacer le sérum bovin par du sérum humain a été étudiée par plusieurs groupes. Bien que le sérum humain semble préserver la capacité de certaines cellules à proliférer et à se différencier, il demeure très variable entre les individus et sa disponibilité est limitée [118]. L’utilisation de sérum autologue a toutefois semblé efficace dans la culture de cellules souches du tissu adipeux chez la souris [119] et le lapin [120]. Il a d’ailleurs été proposé que le sérum autologue puisse être une alternative sécuritaire au sérum allogénique ou animal chez l’humain [120, 121]. Une autre option, beaucoup plus étudiée et faisant l’objet de nombreuse formulations commerciales, est de remplacer le sérum par un mélange de composantes prédéfinies. Ce type de milieux peuvent donc être classifié en catégorie soit, sans sérum, sans protéines animales, sans protéines ou chimiquement définis [122]. Des extraits de tissus dérivés d’animaux ou de plantes ou des lysats de plaquettes humaines ont été proposés comme substituts, mais ceux-ci ne règlent pas l’ensemble de la problématique du sérum [115]. Le remplacement des protéines animales par des protéines humaines ou recombinantes permettrait de réduire les risques d’activer le système immunitaire chez le patient et serait plus prometteur. L’utilisation d’un milieu sans sérum semble augmenter la stabilité des chromosomes in vitro comparativement à un milieu contenant du sérum [123]. Dans tous les cas, le développement d’un milieu complètement défini est un processus long et complexe [116].
1.6.3 Milieux sans sérum
Beaucoup de milieux sans sérum ont été développés depuis les dernières années et plusieurs sont disponibles commercialement. Aucun milieu de culture n’est actuellement commercialisé pour la culture des lymphocytes B. Généralement, ces milieux sont développés pour la culture de cellules souches, de cellules dendritiques ou de lymphocytes T. Plusieurs de ces milieux, dont la série des X-vivo et les milieux StemSpan développés par la compagnie Stem Cells Technologies ont été testés dans notre laboratoire pour la culture de lymphocyte B sans donner des résultats satisfaisants. Récemment, les travaux de l’équipe du Dre Néron ont mené au développement de deux formulations de milieux qui permettent la prolifération et la sécrétion d’immunoglobulines par les lymphocytes B humains. Ces deux milieux donnent en culture des résultats