UFR SCIENCES DE LA VIE ET DE LA TERRE
THESE
Présentée en vue de l’obtention du grade de
DOCTEUR DE L’UNIVERSITE TOULOUSE III
Spécialité : BIOLOGIE MOLECULAIRE ET BIOCHIMIE
Présentée et soutenue Par
Chrystelle LACROIX
THAP1, UN REGULATEUR CLE DE LA PROLIFERATION
DES CELLULES ENDOTHELIALES :
RELATIONS STRUCTURE/FONCTION ET GENES CIBLES
Soutenue le 22 novembre 2007
Directeurs de thèse
Jean-Philippe GIRARD et Vincent ECOCHARD
JURY
Pr. Hervé PRATS Professeur, Université Paul Sabatier, Toulouse Président
Dr. Danièle MATHIEU Directeur de recherche INSERM, I.G.M, Montpellier Rapporteur
Dr. Fabrice SONCIN Directeur de recherche INSERM, I.B.L, Lille Rapporteur
Dr. Gilles PAGES Maître de conférences, Université de Nice Rapporteur
Dr. Jean-Philippe GIRARD Directeur de recherche INSERM, I.P.B.S, Toulouse Co-Directeur de thèse
Au terme de ces quatre années de thèse, le moment est venu de remercier tous ceux qui m’ont, à
divers titres, aidée.
Avant toute chose, je tiens à exprimer toute ma gratitude et mes sincères remerciements au Dr Danièle MATHIEU, Dr Gilles PAGES et Dr Fabrice SONCIN pour avoir accepté d’être les rapporteurs de cette thèse et consacré une part précieuse de leur temps pour la lecture et l’analyse de ce manuscrit ainsi que pour l’intérêt qu’ils ont porté à mon travail. Je remercie également le Pr Hervé PRATS d’avoir présidé ce jury et d’avoir accepté de porter un œil critique sur ce travail. Merci au Ministère de la Recherche et à l’Association pour la Recherche contre le Cancer qui ont financé ce travail et m’ont permis de le réaliser dans de bonnes conditions.
Cette thèse a été réalisée au sein de l’Institut de Pharmacologie et de Biologie Structurale, CNRS UMR 5089 dirigé par le Pr. François AMALRIC. Je le remercie de m’avoir accueillie au sein de cet institut et de m’avoir ainsi donné la possibilité d’effectuer ma thèse dans un environnement scientifique et technique de qualité.
Ce travail a été dirigé par Messieurs les Dr Jean-Philippe GIRARD et Vincent ECOCHARD. Merci de m’avoir fait confiance jusqu’au terme de ce travail.
Merci à Jean-Philippe pour m’avoir donné l’opportunité d’effectuer cette thèse et d’avoir su réorienter efficacement mon travail dans les moments difficiles. La rigueur et l’enthousiasme scientifique dont il fait preuve ainsi que la richesse de ses connaissances, resteront pour moi une référence.
Merci à Vincent pour son soutien depuis le DEA jusqu’à la fin de cette thèse. Son optimisme épatant, sa bonne humeur et ses nombreux et pertinents conseils techniques et scientifiques ont largement participé au bon déroulement de ce travail.
Je remercie tous les membres du laboratoire de Biologie Vasculaire qui depuis le début des années 2000 contribuent à l’avancée des connaissances sur les protéines THAP.
Merci à Corinne CAYROL pour sa contribution essentielle à ce travail et à sa valorisation.
Merci à Myriam ROUSSIGNE et Thomas CLOUAIRE, doctorants pionniers de la famille THAP.
Enfin, merci à Vincent ECOCHARD, Raoul MAZARS, Nathalie ORTEGA, Pascale MERCIER, Arnaud DUJARDIN, Anne-Claire LAVIGNE, Catherine MATHE et différents stagiaires pour leur contribution passée et présente à la caractérisation fonctionnelle des protéines THAP. Je tiens également à remercier les personnes qui ont collaboré à ces travaux de thèse.
Tout d’abord, merci aux structuralistes de l’IPBS. Merci à l’équipe de « RMN et Interactions Protéines-Membranes » du Pr. Alain MILON et plus particulièrement à Damien BESSIERE, Virginie GERVAIS et Sébastien CAMPAGNE pour la détermination de la structure tridimensionnelle du domaine THAP de THAP1 et les études RMN de l’interaction entre THAP1 et sa séquence cible. Un merci tout particulier à Damien et Virginie pour avoir su rendre plus clair ce monde obscur qu’était pour moi la RMN.
Concernant la deuxième partie de mes travaux, merci à Pascale BOUILLE, Luc AGUILAR et Emilie LOREAU pour leur contribution aux transductions rétrovirales et aux expériences de PCR en temps réel.
Merci à Vladimir LAZAR et Philippe DESSEN de l’institut Gustave Roussy pour l’étude de génomique comparative.
Enfin merci à toute l’équipe du Pr Roberto MANTOVANI à Milan pour m’avoir accueillie chaleureusement dans une très bonne ambiance italienne et plus particulièrement à Michele CERIBELLI pour m’avoir formé à la technique d’immunoprécipitation de la chromatine.
Merci à Françoise VIALA pour son aide précieuse dans la réalisation de l’iconographie des articles et pour m’avoir véritablement sauvé la vie le jour de l’impression de mon manuscrit !!! Merci à Marie-Hélène Faye pour son travail, sa gentillesse et sa disponibilité lors de mes nombreuses recherches bibliographiques.
Enfin, je voudrais finir par remercier, de façon individuelle, toutes ces personnes qui m’ont
témoigné leur soutien et ont su rendre ces quatre années agréables.
J’adresse bien sûr mes sincères remerciements à tous les membres du laboratoire de Biologie Vasculaire.
Une mention toute particulière pour mes amies Virginie et Lucie, pour avoir été là dans les bons comme les mauvais moments et pour avoir su être patientes quand je n’étais pas dans de bons jours, et désolée parce qu’il y en a eu quelques-uns !!!! Merci Vir pour ton soutien sans failles depuis le début jusqu’à aujourd’hui même depuis Londres… pour nos discussions tardives où l’on a refait le monde et pour tes re-stimulations. Merci Lulu pour ton sourire en toutes circonstances, pour ta grande capacité à relativiser et à très peu stresser, qualités que j’ai certaines fois bien enviées… Ma thèse aurait été bien différente sans votre présence à toutes les deux et tous ces bons moments partagés avec vous deux… Merci pour tout !
Merci Vincent pour avoir supporté tous mes petits moments difficiles, merci pour ton écoute et tes encouragements. Merci également au duo Gégé/Steph pour leur bonne humeur communicative, Gégé pour ta culture (pas seulement) scientifique très enrichissante et Steph pour ta gentillesse (et tu verras, cette année c’est l’année des Auvergnats…Allez Clermont !!!!!!!). Merci Nath pour tes conseils scientifiques et autres, pour ta gentillesse, ton écoute et ton soutien et pour être toujours là quand on en a besoin. Merci Pascale pour ta gentillesse et ton soutien. Merci Corinne pour ta gentillesse, tes nombreux et très enrichissants conseils techniques et pour m’avoir transmis quelques-unes de tes connaissances. Merci Raoul, le grand sportif de l’équipe, pour ton enthousiasme et ta bonne humeur. Merci Christine et Arnaud, les spécialistes des souris de l’équipe (même si ce n’est pas tous les jours faciles de travailler avec ces petits rongeurs, hein Christine !!!!) pour votre gentillesse et votre bonne humeur.
Je n’oublierai pas les anciens avec qui j’ai passé de très belles années. Merci Anne-Claire pour ta gentillesse, ton naturel et pour tous tes conseils. Merci Cathy pour ta gentillesse et pour avoir su répondre à mes questions la veille de la soutenance. Merci DelphA, car si nous avons partagé que trop peu de temps, j’ai pu apprécier ta générosité et ton extrême gentillesse. Merci Thomas parce que tu as été un peu mon modèle de thésard. Merci Carine et Babeth, pour vos délires, vos bonbons et votre bonne humeur. Enfin merci à deux stagiaires fort sympathiques, notre petite Nancéenne Emilie et notre petite anglaise préférée Jackie.
Je remercie également l’équipe de Biophysique Structurale du Dr Lionel MOUREY, chez qui j’ai passé quelques jours voire semaines à essayer de purifier tant bien que mal certaines de ces protéines THAP et qui m’ont accueilli très chaleureusement, merci pour votre extrême
pendant ces 4 années, pour ta disponibilité et ta gentillesse (je suis heureuse d’avoir fait ta connaissance).
Ces structuralistes sont décidément fort sympathiques et je tiens à ajouter quelques remerciements particuliers à Damien et Virginie avec qui j’ai passé une dernière année de thèse très agréable…
Et pour ne pas les citer, je remercie la joyeuse bande à Lucie, qui ont passé quelques temps dans notre bureau, pour leur bonne humeur permanente… c’est à vous maintenant ! Merci également à Anne-Lise (pour avoir partagé des vacations très agréables avec toi…), Aurélie, Lolo, les deux Fanny, mes collègues de DEA qui sont presque tous partis…. les filles de l’accueil et toutes ces autres personnes rencontrées à l’Institut que j’aurai oubliées et qui ont fait en sorte que cette période de thèse soit une période très riche.
Je voudrais remercier également l’équipe du Dr Bernard MONSARRAT, dans laquelle j’ai effectué l’un de mes stages de DEA, pour leur très bon accueil et leur gentillesse.
Merci à mes amis qui sont présents depuis de nombreuses années ou depuis mes années toulousaines, Cécile et Damien, Nathalie et Mikaël pour les plus anciens mais aussi Charlotte, Sonia, Eric, Fred, Gilles et Marion pour les plus récents et tous ceux que j’aurai oubliés, merci pour votre amitié !
Cette thèse est aussi l’occasion de remercier mes parents qui m’ont permis de faire de si longues études, qui ont cru en moi et m’ont soutenu tout au long de ces années. Merci également à ma sœur et à ma famille pour leur soutien et encouragement.
THAP1, un régulateur clé de la prolifération des cellules endothéliales : relations structure/fonction et gènes cibles
Un nouveau facteur nucléaire humain, la protéine THAP1 (Thanatos Associated Protein 1) a récemment été isolé et cloné par le laboratoire à partir de cellules endothéliales cuboïdales. Il a été montré au sein de l’équipe, que THAP1 contient un nouveau domaine de liaison à l’ADN dépendant du zinc séquence-spécifique, de signature C2CH (CX2-4C-X35-53-CX2H), qui a été nommé domaine THAP. Le domaine THAP est conservé au cours de l’évolution et est retrouvé dans une centaine de protéines chez l’homme et les organismes animaux modèles (souris, poulet, xénope, poisson zèbre, nématode et drosophile). De manière intéressante, le domaine THAP a été identifié chez l’orthologue du régulateur du cycle cellulaire E2F6 chez le poisson zèbre et chez plusieurs protéines du nématode Caenorhabditis elegans interagissant génétiquement avec l’orthologue de la protéine du rétinoblastome pRb, telles que les régulateurs du cycle cellulaire LIN-36 et LIN-15B.
En collaboration avec des équipes de structuralistes, nous avons montré que le domaine THAP de THAP1 est un doigt de zinc atypique d’environ 80 résidus, qui se distingue par la présence entre les deux paires de ligands de coordination du zinc du motif C2CH, d’un court feuillet β anti-parallèle dans lequel s’intercale une longue insertion de type boucle-hélice-boucle. Mes travaux de thèse, nous ont conduits, par des expériences de mutagenèse dirigée ciblant une trentaine de résidus au sein du domaine THAP de THAP1, notamment au sein de ce motif boucle-hélice-boucle, à l’identification de résidus critiques pour la reconnaissance de l’ADN. Ces données de mutagénèse combinées à des analyses de perturbation de déplacements chimiques issues d’expériences RMN, nous ont permis de localiser l’interface de liaison à l’ADN du domaine THAP au niveau d’une zône fortement chargée positivement à la surface de la protéine. Ces travaux rapportent ainsi la première étude des relations structure/fonction d’un domaine THAP pour lequel une activité de liaison à l’ADN séquence-spécifique a été démontrée. Ils fournissent de plus, des données importantes pour une meilleure compréhension du mode de reconnaissance de l’ADN de ce nouveau domaine de liaison à l’ADN dépendant du zinc.
Dans l’optique de déterminer le rôle fonctionnel de la protéine humaine THAP1, nous avons également démontré par interférence d’ARN, que THAP1 est un régulateur endogène majeur de la prolifération des cellules endothéliales et de la progression dans le cycle cellulaire à la transition G1/S. Afin de préciser son mécanisme d’action, nous avons recherché les gènes régulés par THAP1 par une approche de génomique comparative effectuée à grande échelle. Nous avons ainsi montré que THAP1 contrôle la prolifération des cellules endothéliales via la régulation coordonnée de plusieurs gènes impliqués dans la prolifération cellulaire et le cycle cellulaire, cibles des complexes pRB/E2F, dont le gène RRM1, un gène requis pour la synthèse d’ADN en phase S. Nous avons pu identifier, au niveau de la région promotrice du gène RRM1 humain, deux sites consensus de liaison à THAP1 qui sont conservés chez la souris. In vitro, des expériences de protection à la DNAseI et de gel retard, nous ont permis de mettre en évidence la liaison de THAP1 sur l’un de ces sites. De plus, nous avons pu démontrer par immunoprécipitation de la chromatine (ChIP) dans des cellules endothéliales prolifératives et dans des fibroblastes murins, l’association in vivo de THAP1 endogène au niveau des promoteurs des gènes RRM1 humain et murin. Ces travaux fournissent ainsi le premier lien chez l’homme entre les protéines THAP, la prolifération cellulaire et la voie pRB/E2F et ont permis d’identifier le gène RRM1 comme la première cible transcriptionnelle directe de THAP1.
ADN Acide Désoxyribonucléique
AMPc Adénosine Monophosphate Cyclique APC Anaphase Promoting Complex
ARN Acide Ribonucléique ARNm ARN messager ARNr ARN ribosomique
ATP Adenosine 5'-triphosphate BB Binding Buffer
bHLH basic Helix-Loop-Helix
BSA Bovine Serum Albumin
bZIP basic region leucine zipper
CAP Catabolite gene Activator Protein
CBP CREB-binding Protein
CDC-2 Cell Division Control-2, aussi appelé CDK-1 (Cyclin-Dependent Kinase-1)
CDK Cyclin-Dependent Kinase
CENP-B Centromere Protein B
ChIP Chromatin Immunoprecipitation (Immunoprécipitation de chromatine)
CKI Cyclin-dependent Kinase Inhibitor
CREB cAMP response element-binding protein
CREM cAMP responsive element modulator
CtBP C-terminalBinding Protein
CTE Carboxy-Terminal Extension (Extension carboxy-terminale)
DBD DNA Binding Domain (Domaine de liaison à l’ADN)
DMEM Dulbecco’s Modified Eagle’s Medium DNaseI Deoxyribonuclease I
dNDP 2′-deoxyribonucleoside diphosphate
dNTP Déoxynucléotide triphosphate DP E2F dimerization partner
DR Direct Repeat (Répétition directe)
EDTA Acide éthylène-diamine-tétraacétique EKLF Erythroid Krüppel Like Factor
ER Estrogen Receptor (Récepteur à l’oestradiol)
ER Everted Repeat (Répétition éversée ou palindromique)
ERE Estrogen Response element
FACS Fluorescence-Activated Cell Sorter
GR Glucocorticoid receptor (récepteur aux glucocorticoïdes)
GRE Glucocorticoid response element
GTP Guanine triphosphate HAT Histone Acétyltransférase HBM HCF-1 Binding Motif
HCF-1 HSV Host Cell Factor 1 HDAC Histone Désacétylase HLH Helix-Loop-Helix
HMGA High Mobility Group-A HMT Histone Méthyltransférase HRE Hormone Response Element
Inr Initiateur
IPTG Isopropyl-β-D-thiogalacopyranoside
IR Inverted Repeat (Répétition inversée)
kDa kilo Daltons KLF Krüppel Like Factor
KO Knock-Out
LB Luria Broth
LBD Ligand Binding Domain (Domaine de liaison au ligand)
LIN abnormal Lineage
MEF Murine Embryonic Fibroblasts
Mg Magnésium mL milli litre mM milli molaire NaCl Chlorure de sodium
NCoR Nuclear-receptor-corepresor
NDP Nucleoside diphosphate NF-Y Nuclear Factor Y
Ni Nickel
NTA NitriloTriAcetate
NURD Nucleosome Remodeling and Deacetylase
PBS Phosphate Buffer Saline
PCR Polymerase Chain Reaction
PDB Protein Data Bank
pRb protéine du rétinoblastome
PurR Purine Repressor (Répresseur purine) pb Paire(s) de Bases
qPCR quantitative Polymerase Chain Reaction
RAR Récepteur à l’Acide Rétinoïde Rb Rétinoblastome
RMN Résonance Magnétique Nucléaire RNase Ribonucléase
RNR Ribonucleotide Reductase
ROR RAR-related Orphan Receptor
RRL Rabbit Reticulocyte Lysate (Lysat de réticulocyte de lapin)
RRM1 Ribonucleotide Reductase M1 subunit
RXR Retinoid XReceptor (récepteur de l’acide rétinoïque-9-cis)
SDS-PAGE Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis
SELEX Systematic evolution of ligands by exponential enrichment
SVF Serum de veau Fœtal TBP TATA Binding Protein
THABS THAP1 Binding Sequence
THAP Thanatos Associated Protein
TR Thyroid hormone receptor (récepteur aux hormones thyroïdiennes) UAS Upstream Activation Sequence
VPC Vulval Precursor Cell (Cellule précurseur de la vulve)
XGal 5-bromo-4-chloro-3-indolyl-b-D-galactopyranoside
YY Ying and Yang
Zn Zinc µL Micro-litre
Code à
3 lettres Code à 1 lettre Nom
Ala A Alanine Arg R Arginine Asn N Asparagine Asp D Aspartate Cys C Cystéine Glu E Glutamate Gln Q Glutamine Gly G Glycine His H Histidine Ile I Isoleucine Code à
3 lettres Code à 1 lettre Nom
Leu L Leucine Lys K Lysine Met M Méthionine Phe F Phénylalanine Pro P Proline Ser S Sérine Thr T Thréonine Trp W Tryptophane Tyr Y Tyrosine Val V Valine
Liste des bases
Nom
A Adénine
T Thymine
S
OMMAIREPREAMBULE... 1
INTRODUCTION...2
I. SPECIFICITE DE RECONNAISSANCE DES DOMAINES DE LIAISON A L’ADN... 3
I.1. La reconnaissance spécifique de l’ADN ... 3
I.1.A. Comment la protéine repère-t’elle sa cible ? Importance de l’interaction non spécifique ...3
I.1.B. Complémentarités structurale et chimique...5
I.1.B.1. Complémentarité structurale ...5
I.1.B.1.a. L’ADN... 5
I.1.B.1.b. La protéine et son domaine de liaison à l’ADN... 6
I.1.B.1.b.1 Reconnaissance par une hélice alpha...6
I.1.B.1.b.2 Interactions avec des feuillets β ...7
I.1.B.1.b.3 Les boucles et les queues terminales...8
I.1.B.2. Nature des liaisons mises en jeu dans un complexe protéine/ADN ...9
I.1.B.2.a. Les liaisons hydrogènes dictent la spécificité de reconnaissance ...10
I.1.B.2.b. Importance des autres types d’interactions protéine/ADN...11
I.1.C. Existe-t-il un code de reconnaissance ? ...12
I.1.D. Importance des contacts avec le squelette sucre/phosphate...13
I.1.E. La courbure de l’ADN pour s’accommoder à la structure de la protéine ...14
I.1.F. Conclusion ...14
I.2. Reconnaissance spécifique par les principaux domaines de liaison à l’ADN... 16
I.2.A. Le domaine de liaison à l’ADN : un motif protéique pour lier l’ADN de façon séquence spécifique ...16
I.2.B. Les principaux domaines de liaison à l’ADN...18
I.2.B.1. Le motif Helice-Tour-Hélice (HTH) ...18
I.2.B.2. L’homéodomaine ...20
I.2.B.3. Les protéines à motif basique bZIP et bHLH...23
I.2.B.3.a. Le motif bZIP ...24
I.2.B.3.b. Le motif bHLH...26
I.2.B.4. Les domaines de liaison à l’ADN dépendant du zinc...28
I.2.B.4.a. Le domaine C2H2 ou doigt de zinc classique...29
I.2.B.4.a.1 Structure du doigt de zinc classique...29
I.2.B.4.a.2 Liaison à l’ADN du doigt de zinc classique...31
I.2.B.4.b. Les motifs multi-cystéine ...33
I.2.B.4.b.1 Les récepteurs nucléaires...33
I.2.B.4.b.2 Les modules à zinc des facteurs GATA...40
I.2.B.4.c. Les domaines de liaison à l’ADN dépendant du zinc reconnaissant l’ADN de manière originale...42
I.2.B.4.c.1 Le domaine de liaison à l’ADN de Gal4...42
I.2.B.4.c.2 Les domaines DM et GCM ...44
I.2.B.4.c.3 Le domaine de liaison à l’ADN de p53...48
I.2.C. Les modules à zinc comme motifs d’interaction protéine-protéine ...51
II. LE DOMAINE THAP : UN NOUVEAU DOMAINE DE LIAISON A L’ADN DEPENDANT DU ZINC QUI DEFINIT UNE NOUVELLE FAMILLE DE PROTEINES CELLULAIRES... 54
II.1. Un nouveau motif de liaison à l’ADN évolutivement conservé... 54
ii
II.1.B. Le domaine THAP, un domaine de liaison à l’ADN partagé entre des protéines cellulaires et la transposase d’un
élément génétique mobile ...56
II.1.B.1. La transposase de l’élément P ...56
II.1.B.2. Relation évolutive entre les protéines THAP et la transposase de l’élément P...58
II.1.B.2.a. L’élément P à l’origine de la formation de gènes nouveaux ...58
II.1.B.2.b. Un néogène P dans le génome humain : le gène codant pour la protéine THAP9 ...60
II.2. Le domaine THAP constitue un motif de liaison à l’ADN dépendant du zinc atypique et séquence spécifique... 61
III.LES PROTEINES THAP DE FONCTION CONNUES SONT IMPLIQUEES DANS LA REGULATION DE LA PROLIFERATION CELLULAIRE ET DU CYCLE CELLULAIRE ET DANS LA MODIFICATION DE LA CHROMATINE... 63
III.1.Les protéines THAP d’organismes animaux modèles ... 63
III.1.A.Chez le poisson zèbre, l’orthologue de la protéine E2F6 présente un domaine THAP ...64
III.1.B.Les protéines THAP de C. elegans ...65
III.1.B.1. L’orthologue du coréprésseur transcriptionnel CtBP présente un domaine THAP...65
III.1.B.2. Certains gènes SynMuv de classe B chez C. elegans sont impliqués dans la régulation de la transition G1/S ...65
III.1.B.2.a. Conservation de la voie Rb/E2F chez C. elegans pour le contrôle de la transition G1/S ...66
III.1.B.2.a.1La voie Rb/E2F chez les vertébrés ...66
III.1.B.2.a.2La voie Rb/E2F chez C. elegans...68
III.1.B.2.b. Les gènes SynMuv de classe B codant pour les protéines THAP LIN-36 et LIN-15B, des régulateurs négatifs de la transition G1/S ...70
III.1.B.3. La phosphatase CDC-14 inhibe la progression en G1...71
III.1.B.4. La protéine GON-14, un régulateur positif de la prolifération cellulaire ...72
III.1.B.5. La protéine HIM-17...73
III.1.C.Modification de la chromatine et régulation du cycle cellulaire par certaines protéines THAP de C. elegans...74
III.2.Les protéines THAP humaines... 76
III.2.A.Les protéines humaines THAP0 et THAP1 ...76
III.2.B.La protéine humaine THAP7...77
PROJET DE THESE... 79
I. SITUATION DU PROJET... 80
II. PRESENTATION DES TRAVAUX DE THESE... 80
PARTIE 1 RELATIONS STRUCTURE/FONCTIONS DU DOMAINE THAP DE LA PROTEINE HUMAINE THAP1... 82
I. AVANT PROPOS... 83
II. ARTICLE 1 ... 86
III.RESULTATS COMPLEMENTAIRES :RECHERCHE PAR LA PROCEDURE SELEX DE SEQUENCES ADN RECONNUES PAR LES PROTEINES THAP HTHAP2, HTHAP3,CE-CTBP ET GON-14 ... 87
III.1.Analyse des séquences issues du SELEX à partir du domaine THAP de hTHAP2... 87
III.2.Analyse des séquences issues du SELEX à partir du domaine THAP de hTHAP3... 88
III.3.Analyse des séquences issues du SELEX à partir des domaines THAP de Ce-CtBP et de GON-14 ... 89
IV.DISCUSSION... 90
IV.1.Déterminants structuraux du module à zinc du domaine THAP... 90
IV.1.A.La structure du module C2CH est atypique...90
IV.1.B.Résidus impliqués dans la structure du domaine THAP...91
IV.2.Le domaine THAP n’utilise probalement pas une hélice α pour la reconnaissance de l’ADN ... 92
IV.3.Différents domaines THAP ne reconnaissent pas la même séquence ADN cible... 93
PARTIE 2 IDENTIFICATION DE GENES REGULES PAR LA PROTEINE HUMAINE THAP1 .... 97
I. AVANT PROPOS... 98
II. ARTICLE 2 ... 102
III.RESULTATS COMPLEMENTAIRES... 103
III.1.Conservation homme/souris de la liaison in vivo de THAP1 sur le promoteur RRM1... 103
III.1.A.La protéine THAP1 murine reconnaît le site THABS ...103
III.1.B.La protéine THAP1 murine se lie sur le promoteur RRM1 de souris ...104
III.2.THAP1 est asociée constitutivement sur le promoteur RRM1... 105
III.3.Autres gènes cibles potentiels de THAP1... 106
III.3.A.Présence de sites THABS dans les régions promotrices de certains des gènes régulés par THAP1...106
III.3.B.Recherche d’une association de THAP1 in vitro et in vivo sur les promoteurs humains de certains gènes du cycle cellulaire...106
III.3.C.Recherche d’une association in vivo sur le promoteur cdc-2 murin ...107
III.4.Activité transcriptionnelle de THAP1 pour la régulation de ces gènes... 109
IV.DISCUSSION... 111
IV.1.La protéine THAP1 humaine est un régulateur clé de la prolifération des cellules endothéliales ... 111
IV.2.Gènes cibles de THAP1 ... 112
IV.3.Mécanisme de régulation de la prolifération et du cycle cellulaire par THAP1... 113
CONCLUSION GENERALE ET PERSPECTIVES...118
MATERIELS ET METHODES COMPLEMENTAIRES ... 123
I. PARTIE 1... 124
I.1. Expression des domaines THAP des protéines humaines THAP2 et THAP3 et des protéines CtBP et GON-14 de C. elegans ... 124
I.1.A. Clonage des domaines THAP...125
I.1.B. Transformation des bactéries BL21trxB(DE3) ...125
I.1.C. Culture et lyse des bactéries...126
I.2. Purification des domaines THAP utilisés pour l’approche SELEX... 126
I.2.A. Purification des domaines THAP des protéines humaines THAP2 et THAP3...126
I.2.B. Purification des domaines THAP des protéines THAP de C. elegans CtBP et GON-14 ...127
I.3. SELEX (Systematic Evolution of Ligands by Exponential Enrichment)... 129
II. PARTIE 2... 131
II.1. Immunoprécipitation de chromatine (ChIP) ... 131
II.1.A. Culture cellulaire...131
II.1.A.1. Cellules NIH3T3 ...131
II.1.A.2. Cellules HUVEC-THAP1...131
II.1.A.3. Cellules MEF/3T3 Tet-Off THAP1-FLAG-HA...132
II.1.B. Immunoprécipitation de chromatine (ChIP) ...132
II.2. Essais luciférase ... 133
L
ISTE DESF
IGURESFigure 1 Mécanismes proposés pour expliquer comment les protéines trouvent leurs cibles par « diffusion facilitée »
Figure 2 La formation d’un complexe non spécifique entre l’ADN et le répresseur au lactose, étape initiale à la reconnaissance spécifique de son opérateur (D’après Kalodimos et al., Science, 2004)
Figure 3 Représentation de la forme B de l’ADN Figure 4 Reconnaissance de l’ADN par une hélice α Figure 5 Reconnaissance de l’ADN par des feuillets β
Figure 6 Reconnaissance de l’ADN par des boucles et des extensions de DBDs
Figure 7 Liaisons hydrogènes possibles au niveau des sillons des paires de bases de l’ADN
Figure 8 Liaisons hydrogènes médiées par l’eau à l’interface protéine-ADN du complexe répresseur Trp/opérateur Figure 9 Contacts acides aminés/bases
Figure 10 Structure de la TBP (TATA binding protein) complexée à l’ADN Figure 11 Le motif HTH (Hélice-Tour-Hélice)
Figure 12 L’homéodomaine
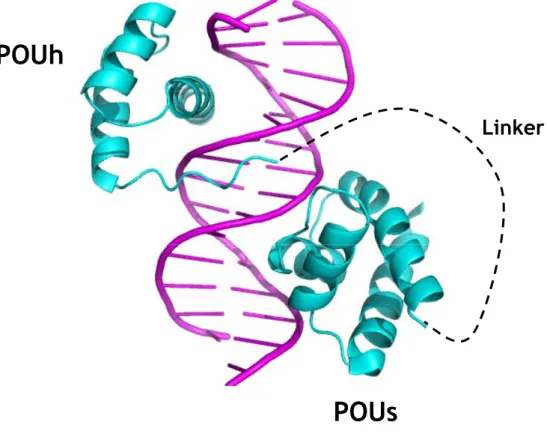
Figure 13 Structure des deux sous-domaines POU de la protéine Oct-1 en complexe avec l’oligonucléotide TGTATGCAAATAAGG
Figure 14 Le motif basique à leucine zipper bZIP
Figure 15 Reconnaissance de l’ADN par les protéines à motif bZIP Figure 16 Les domaines bHLH et bHLH-Z
Figure 17 Le C2H2 classique
Figure 18 Reconnaissance de la séquence GCGTGGGCG par la protéine à doigt de zinc Zif268
Figure 19 Contacts entre les chaînes latérales de résidus clés de l’hélice α et les bases du sous-site reconnu par différents doigts de zinc
Figure 20 Le domaine de liaison à l’ADN des récepteurs nucléaires
Figure 21 Reconnaissance des séquences héxamériques ACGTCA et AGAACA illustrées par les DBD du récepteur à l’oestradiol (ER) et du récepteur aux glucorcoticoïdes (GR) et leur élément de réponse correspondant (ERE et GRE)
Figure 22 La dimérisation des récepteurs nucléaires permet la reconnaissance de différentes séquences cibles Figure 23 Adaptabilité du DBD des récepteurs nucléaires et de l’ADN pour la dimérisation
Figure 24 Le domaine C4 des facteurs GATA Figure 25 Le domaine de liaison à l’ADN de GAL4 Figure 26 Le domaine DM
Figure 27 Le domaine GCM
Figure 28 Le domaine de liaison à l’ADN de p53
Figure 29 Le domaine N-terminal de GATA-1 interagit avec la protéine FOG sur une surface différente de la surface de liaison à l’ADN
Figure 30 Conservation évolutive des domaines THAP
v
Figure 32 Le domaine THAP de THAP1 est un motif de liaison à l’ADN séquence-spécifique, dépendant du zinc Figure 33 La protéine E2F6 présente un domaine THAP chez certains poissons
Figure 34 Les protéines caractérisées de la famille THAP chez le nématode Caenorhabditis elegans Figure 35 La transition G1/S chez les métazaoires
Figure 36 Implications des protéines THAP dans la régulation de la transition G1/S chez C. elegans Figure 37 Alignement des domaines THAP des protéines humaines THAP1, THAP2 et THAP3 Figure 38 Les protéines humaines THAP2 et THAP3 pleine taille ne lient pas la séquence THABS
Figure 39 Séquences des différents clones obtenus après 8 et 10 cycles de sélection par SELEX pour le domaine THAP de hTHAP2
Figure 40 Test de l’interaction in vitro entre le domaine THAP de hTHAP2 et différents clones issus du SELEX Figure 41 Séquences des différents clones obtenus après 6 et 8 cycles de sélection par SELEX pour le domaine
THAP de hTHAP3
Figure 42 Séquences des différents clones obtenus après 4, 5 et 6 cycles de sélection par SELEX pour le domaine THAP de Ce-CtBP
Figure 43 Séquences des différents clones obtenus après 4, 5 et 6 cycles de sélection par SELEX pour le domaine THAP de GON-14
Figure 44 Régions régulatrices des promoteurs des gènes RRM1 humain et murin Figure 45 Alignement de séquences entre les protéines THAP1
Figure 46 La protéine THAP1 murine interagit avec la séquence THABS in vitro
Figure 47 La protéine murine THAP1 s’associe in vivo sur le promoteur du gène RRM1 de souris Figure 48 L’association de THAP1 sur le promoteur RRM1 est constitutive
Figure 49 Présence de sites THABS dans les régions promotrices de certains des gènes régulés par THAP1
Figure 50 La protéine THAP1 humaine lie le promoteur Survivine in vitro et après expression ectopique s’associe in vivo sur les promoteurs des gènes RRM1, survivine, Cycline B1 et cdc-2
Figure 51 Identification d’un site THABS consensus dans le promoteur du gène murin cdc-2 Figure 52 La protéine THAP1 murine ectopique est associée in vivo sur le promoteur murin cdc-2
Figure 53 Étude de l’association in vivo de THAP1 murine endogène sur le promoteur murin cdc-2 dans des cellules synchronisées
Figure 54 Dans des essais transcriptionnels, THAP1 a un léger effet répresseur sur les promoteurs des gènes RRM1 et Survivine
Figure 55 L’effet répresseur de THAP1 observé dans les essais transcriptionnels n’est pas lié à sa liaison sur les sites THABS
Figure 56 L’effet répresseur de THAP1 observé pour les promoteurs RRM1 et Survivine n’est pas dépendant de son domaine de liaison à l’ADN
Figure 57 Modèle proposé pour l’activation par THAP1 de l’expression du gène RRM1
Figure 58 Purification des domaines THAP recombinants des protéines humaines THAP2 et THAP3 Figure 59 Purification des domaines THAP recombinants des protéines de C. elegans Ce-CtBP et GON-14 Figure 60 Principe de la technologie SELEX (systematic evolution of ligands by exponential enrichment)
L
ISTE DEST
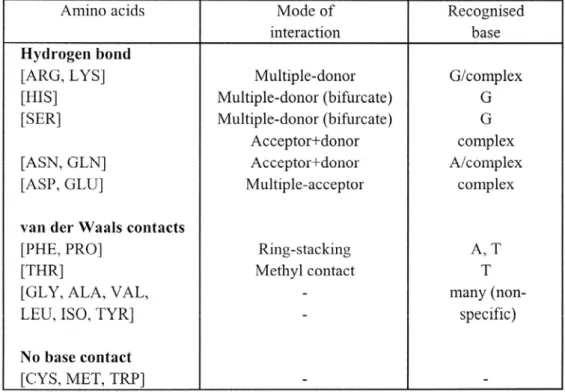
ABLEAUXTableau 1 Préférences d’interactions entre chaînes latérales d’acides aminés et bases de l’ADN Tableau 2 La famille THAP dans les organismes animaux modèles
Tableau 3 Séquences des oligonucléotides utilisés pour le clonage des domaines THAP de hTHAP2, hTHAP3, Ce-CtBP et GON-14 dans le vecteur d’expression pET21c
Tableau 4 Tampons de lyse utilisés pour la purification des domaines THAP recombinants utilisés en vue de la mise en œuvre de la technologie SELEX
Tableau 5 Séquences des oligonucléotides utilisés pour les PCR semi-quantitatives ou quantitatives après ChIP Tableau 6 Séquences des oligonucléotides utilisés pour les différentes constructions utilisées en vue des essais
Préambule
Les interactions ADN/protéine sont impliquées dans de nombreux processus cellulaires fondamentaux. Par exemple, la capacité d’une protéine à lier de façon spécifique un site ADN particulier dans le gènome constitue la base même de la régulation de la transcription des gènes. La compréhension des mécanismes généraux de la reconnaissance spécifique de l’ADN par les protéines est donc capitale. L’appréhension de ces mécanismes de reconnaissance passe par l’étude structurale de la protéine isolée ainsi que des complexes ADN/protéine. Depuis une vingtaine d’années, nous avons pu noter une grande expansion dans la détermination de telles structures qui ont pu souligner la diversité dans les stratégies mises en jeu par ces protéines pour se lier de façon spécifique à des séquences ADN particulières.
Le domaine THAP récémment découvert dans l’équipe, est un nouveau domaine de liaison à l’ADN dépendant du zinc conservé au cours de l’évolution, qui définit une nouvelle famille de protéines nucléaires, la famille des protéines THAP. L’équipe a également pu montrer que le domaine THAP du membre prototype de cette famille, la protéine THAP1, lie l’ADN de manière séquence-spécifique. Suite à ces travaux, l’un des objectifs de l’équipe afin de caractériser de façon fonctionnelle cette nouvelle famille de protéines, était de mieux définir les mécanismes d’action de ces protéines en s’intéressant notamment aux interactions domaine THAP/ADN. C’est dans cette thématique que s’est inscrit mon projet de thèse.
Dans ce manuscrit, je présenterai donc dans une introduction générale divisée en trois parties, l’état des connaissances sur les mécanismes de reconnaissance spécifique de l’ADN par les protéines ainsi que sur le domaine THAP et les protéines présentant ce nouveau motif protéique de liaison à l’ADN dépendant du zinc. Dans une première partie, j’exposerai les contraintes architecturales et chimiques nécessaires à la reconnaissance spécifique entre une protéine et l’ADN en m’appuyant sur certains exemples précis. J’illustrerai notamment cette complexité en présentant plus en détail les variations dans la structure et le mode de reconnaissance spécifique de l’ADN, des principaux domaines de liaison à l’ADN des facteurs de transcription, en m’attardant plus particulièrment sur les domaines de liaison à l’ADN dépendant du zinc. Dans une seconde partie, je présenterai les travaux ayant conduit l’équipe à l’identification et à la caractérisation biochimique du domaine THAP en tant que nouveau domaine de liaison à l’ADN dépendant du zinc. Enfin, j’exposerai dans une troisième partie les différentes données disponibles à ce jour pour certaines protéines THAP notamment chez les organismes animaux modèles tels que le nématode Caenorhabditis elegans, qui nous ont conduits à suggérer un rôle des protéines THAP dans la régulation de la prolifération cellulaire et de la progression dans le cycle
I. Spécificité de reconnaissance des domaines
de liaison à l’ADN
I.1. La reconnaissance spécifique de l’ADN
Les protéines interagissant avec l’ADN de façon spécifique jouent un rôle central dans les différents aspects des activités génétiques d’un organisme tels que la réplication du génome, la transcription des gènes et la réparation de l’ADN endommagé. Nous nous intéresserons plus particulièrement à l’une des classes les plus importantes et variées de protéines de liaison à l’ADN, qui est celle des facteurs de transcription, responsables de la régulation de l’expression des gènes. La régulation de l’expression génique par les facteurs de transcription correspond au mécanisme principal de contrôle des processus de développement, de différenciation et de croissance cellulaire et requiert la fixation de ces protéines sur des séquences ADN spécifiques.
I.1.A. Comment la protéine repère-t’elle sa cible
?
Importance de l’interaction non spécifique
Les séquences ADN cibles reconnues spécifiquement par les facteurs de transcription ne représentent qu’une fraction minime de l’ADN cellulaire. Par conséquent les processus par lesquels les facteurs de transcription sont capables de localiser rapidement et de façon efficace leurs sites cibles parmi la quantité considérable d’ADN non spécifique sont de tout intérêt. Or, il a été montré que certains facteurs de transcription identifient leurs sites cibles 100 à 1000 fois plus rapidement que la vitesse maximale attendue pour la diffusion simple en trois dimensions, c'est-à-dire par simples collisions sur l’ADN par association/dissociation de la protéine (ou « hopping ») (Berg and von Hippel, 1985; Riggs et al., 1970). Dans ce cas, il est maintenant admis que ces protéines trouveraient leurs cibles par « diffusion facilitée » dont l’étape initiale implique une liaison aléatoire sur l’ADN par la formation d’interactions protéine/ADN non-spécifiques. Cette liaison non spécifique est alors suivie d’une translocation intramoléculaire sur le site de liaison spécifique (Halford and Marko, 2004; Shimamoto, 1999; von Hippel and Berg, 1989). La formation de tels complexes protéine/ADN non-spécifiques semble nécessaire à l’efficacité et à la rapidité avec lesquelles la protéine peut identifier sa (ses) séquence(s) ADN cible(s) spécifique(s) et est donc capitale dans le mécanisme de reconnaissance. Suite à cette association non spécifique, différents mécanismes de transfert sur la molécule d’ADN à la recherche des séquences cibles ont été proposés (Figure 1). La protéine peut se dissocier puis se réassocier sur un autre site proche ou distant (« hopping » ou diffusion en trois dimensions), ou
« Intersegmental transfer » « Hopping » « Hopping » à longue « Sliding» distance
Figure 1 : Mécanismes proposés pour expliquer comment les protéines trouvent leurs cibles par « diffusion facilitée »
A
B
DBD du répresseur
lac libre Complexe non-spécifique Complexe spécifique
Figure 2 : La formation d’un complexe non spécifique entre l’ADN et le répresseur au lactose, étape initiale à la reconnaissance spécifique de son opérateur (D’après Kalodimos et al., Science, 2004)
(A) Le domaine de liaison à l’ADN du répresseur au lactose présente la même surface d’interaction
dans le grand sillon de l’ADN, dans le complexe spécifique (en jaune) et dans le complexe non spécifique (en orange). Toutefois, la protéine est basculée de 25° entre les deux complexes
d i t à é t d t t téi /ADN
conduisant à un réarrangement des contacts protéine/ADN.
(B) La formation du complexe spécifique induit des changements conformationnels de la protéine
et de l’ADN. En effet, la région en rouge du DBD est désordonnée pour le répresseur sous sa forme libre ou en complexe avec de l’ADN non spécifique mais va se structurer en hélice α dans le complexe spécifique et former des contacts dans le petit sillon, conduisant à une courbure significative de l’ADN.
elle peut rester liée à l’ADN et glisser sur sa longueur (« sliding » ou diffusion en une dimension). Un autre mécanisme suggère le transfert direct de la protéine sur deux segments d’ADN non contigus qui sont liés transitoirement par la même protéine (« intersegmental transfert ») (Figure 1). Cependant, si les mécanismes cités précédemment peuvent être adoptés par tous les facteurs de transcription, ce n’est pas le cas du « transfert direct » qui n’est possible que si la protéine possède deux surfaces de liaison à l’ADN (Halford and Marko, 2004).
Comprendre comment les protéines se lient de façon non spécifique à l’ADN et comment elles diffusent ou se dissocient/réassocient sur l’ADN à la recherche de leur séquence cible spécifique est donc de grande importance pour une meilleure compréhension du mécanisme de reconnaissance spécifique. Toutefois, si de nombreux complexes protéine/ADN spécifiques sont disponibles à ce jour, seuls de rares travaux rapportent la structure de facteurs de transcription complexés à de l’ADN non spécifique. Deux études récentes ont néanmoins permis de mettre en évidence l’importance de la formation d’un complexe non spécifique en tant que prérequis au processus de reconnaissance séquence spécifique. La première a rapporté la comparaison de la structure du répresseur au lactose à l’état libre, complexée à son opérateur ou complexée à une séquence ADN non-spécifique (Figure 2) (Kalodimos et al., 2004). Ces structures ont révélé qu’un ensemble de résidus du répresseur avait un double rôle, réalisant des contacts essentiellement électrostatiques avec le squelette sucre/phosphate de l’ADN dans le complexe non spécifique afin de le stabiliser, et conférant la spécificité via des contacts directs avec les bases dans le complexe spécifique. Ainsi, le répresseur au lactose utilise la même surface d’interaction pour interagir à la fois avec le complexe spécifique et avec le complexe non-spécifique (Figure 2A). Néanmoins, la formation du complexe non-spécifique s’accompagne d’une rotation de 25° de la protéine par rapport à l’ADN, ce qui induit un réarrangement de la surface d’interaction protéine-ADN. En effet, les chaînes latérales des mêmes résidus qui sont mobiles dans le complexe non-spécifique vont interagir directement avec les bases résultant en une interface plus rigide dans le complexe spécifique. Le passage du mode non spécifique au mode spécifique s’accompagne également de la structuration en hélice alpha d’une région du répresseur qui est désordonnée en absence d’ADN et dans le complexe non-spécifique (Figure 2B). Cette hélice α va réaliser dans le complexe spécifique des contacts au niveau du petit sillon et induire une courbure de l’ADN au niveau du site de reconnaissance. Cette région est donc importante à la fois pour la spécificité et pour le passage structural du mode non-spécifique au mode spécifique (Kalodimos et al., 2004). Si ces travaux ont ainsi fournis des informations structurales sur cette reconnaissance non-spécifique, ils n’ont pas apporté de réponses quant à la dynamique de cette interaction puisque la protéine a été étudiée en interaction avec un seul site non spécifique sur l’ADN. Or, comme nous l’avons vu, l’interaction non-spécifique s’accompagne de différents états
d’échanges rapides via des mécanismes de transfert de la protéine sur la molécule d’ADN. Mais, plus récemment, de telles données cinétiques ont été rapportés pour des interactions entre l’ADN et une protéine à homéodomaine sur différents sites non spécifiques (Iwahara et al., 2006). Cette étude a ainsi suggéré que cette protéine utilise un mécanisme de diffusion rapide sur l’ADN non spécifique en utilisant la même interface de liaison que dans le complexe spécifique jusqu’à ce qu’elle trouve le site ADN cible. Certaines chaînes latérales très mobiles dans le mode non-spécifique vont alors réaliser des contacts directs avec les bases de cette séquence cible (Iwahara et al., 2006).
I.1.B. Complémentarités structurale et chimique
La reconnaissance d’une séquence nucléotidique spécifique par une protéine est déterminée par une complémentarité structurale entre l’ADN et la protéine, et par une reconnaissance chimique au niveau atomique. La reconnaissance structurale concerne l’architecture globale de la protéine alors que la reconnaissance chimique est déterminée par l’arrangement stéréochimique des acides aminés à la surface de la protéine (Rhodes et al., 1996).
I.1.B.1. Complémentarité structurale
Un des modes de reconnaissance structural les plus utilisés pour former le réseau complexe de contacts est l’insertion d’une hélice alpha de la protéine dans le grand sillon de l’ADN sous forme B. Toutefois, nous verrons que si la majorité des structures de complexes protéine/ADN présente un ADN sous forme B, les domaines de liaison à l’ADN peuvent utiliser différents éléments de structure secondaire voire même des boucles pour former les contacts spécifiques avec les bases.
I.1.B.1.a. L’ADN
La double hélice d’ADN peut adopter des conformations différentes en fonction de la composition en nucléotides ainsi que du degré d’hydratation et de salinité du milieu (Arnott and Selsing, 1974a; Arnott and Selsing, 1974b). Les formes A et B (qui différent dans leurs paramètres hélicoïdaux) sont les formes les plus connues. Une troisième forme, dite forme Z caractérisée par une hélice gauche a également été observée à forte concentration saline et en présence d’ions multivalents (Wang et al., 1979). La forme B de l’ADN correspond à la conformation de l’ADN retrouvée majoritairement in vivo. Cette forme est retrouvée dans une solution qui présente 90% d'humidité et une faible force ionique. Elle correspond à la forme décrite en 1953 par Crick et
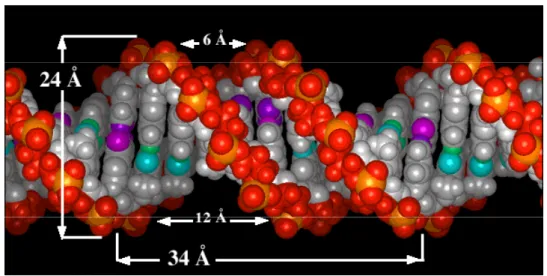
Figure 3 : Représentation de la forme B de l’ADN
La double hélice d’ADN sous forme B comporte 10 nucléotides par tour d’hélice, le pas de l’hélice est de 34 Å et son diamètre de 24 Å. La forme B de l’ADN présente un grand sillon large (12 Å) et un petit sillon étroit (6 Å).
Watson (Watson and Crick, 1953). La double hélice d’ADN sous forme B s’enroule à droite et présente environ 10 paires de bases par tour d’hélice, le pas de l’hélice est de 34 Å et le diamètre de l’hélice est de 24 Å. Dans les cellules, la forme de l’hélice est un peu plus compacte et comporte environ 10,4 paires de bases par tour d’hélice et les plans successifs de deux paires de bases sont éloignés de 3,4 Å. Dans cette forme B de l’ADN, toutes les bases puriques et pyrimidiques ont la même conformation par rapport à l’ose (ou anti). Les bases puriques et pyrimidiques sont à l’intérieur de l’hélice, les groupements phosphates et les désoxyriboses sont à l’extérieur. Les plans des bases sont perpendiculaires à l’axe de l’hélice. Les plans des oses sont presque perpendiculaires à ceux des bases. Une caractéristique importante de la forme B de l’ADN est la présence d’un grand sillon large (12 Å) et d’un petit sillon étroit (6 Å), les deux sillons étant peu profonds (Figure 3). Pour des interactions directes avec les bases de l’ADN, la protéine doit accéder aux groupements fonctionnels des bases au sein des sillons de l’ADN. Ainsi, pour la forme B de l’ADN, le grand sillon apparaît plus adapté pour une compatibilité avec les éléments de structure secondaire de la protéine.
I.1.B.1.b. La protéine et son domaine de liaison à l’ADN
Pour contacter les bases constituant le site de reconnaissance spécifique du domaine de liaison à l’ADN, celui-ci doit présenter des éléments structurels capables de s’insérer dans le grand sillon de l’ADN sous forme B.
I.1.B.1.b.1 Reconnaissance par une hélice alpha
L’hélice alpha est l’élément de structure secondaire protéique le plus utilisé pour la reconnaissance des bases car elle présente les proportions idéales pour une bonne complémentarité avec le grand sillon de l’ADN sous forme B. Son insertion dans le grand sillon avec son axe parallèle au squelette sucre/phosphate fournit le plus grand nombre de contacts (Suzuki and Gerstein, 1995). Ce type d’hélice est généralement nommé « hélice de reconnaissance » puisque les résidus exposés sur une des faces de l’hélice vont interagir directement avec les bases. Cependant, considérer par l’utilisation de ce terme que la reconnaissance n’implique que ces contacts est souvent incorrect et parler « d’hélice de reconnaissance » paraît alors inadéquat. En effet, il a été démontré que d’autres régions du DBD en dehors de cette hélice peuvent aussi former des contacts et contribuer à la reconnaissance spécifique en permettant de bien orienter l’hélice par rapport à l’axe de l’ADN (Pabo and Sauer, 1992). Nous verrons d’ailleurs que selon le type de domaine de liaison à l’ADN, l’orientation de
A
B
Motif HTH au contact du grand sillonHélice charnière dans le petit sillon
Figure 4 : Reconnaissance de l’ADN par une hélice α
(A) Différentes orientations des hélices α dans le grand sillon de l’ADN (D’après Garvie and
Wolberger, Mol Cell, 2001)
(B) Le petit sillon de l’ADN peut également s’accommoder à une hélice α
Par exemple, la structure du complexe du répresseur purine, PurR sous forme dimérique avec
é h è i d d i é é i
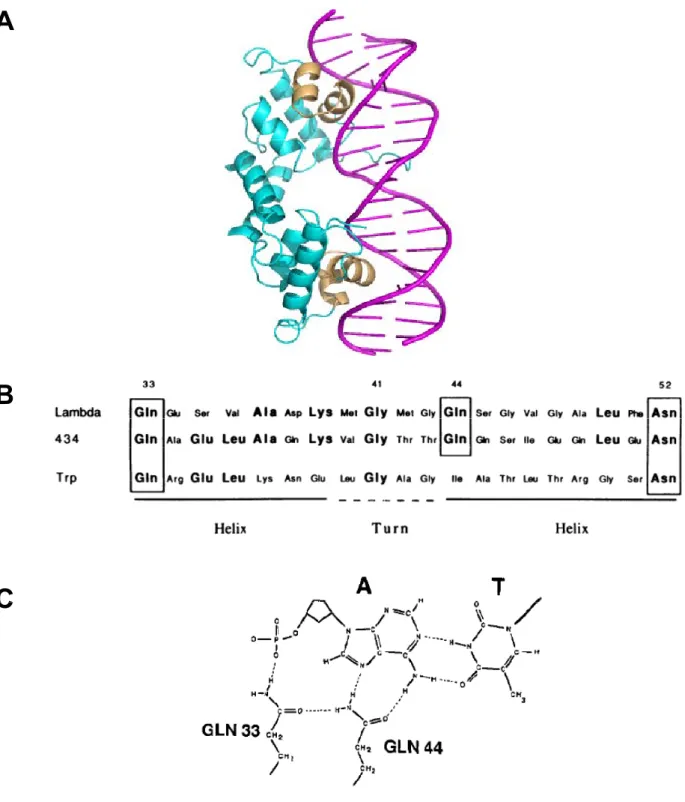
son opérateur, montre que chaque monomère contient deux domaines séparés qui contactent l’ADN : un motif hélice-tour-hélice (HTH) qui contacte les bases dans le grand sillon par son « hélice de reconnaissance » et une hélice charnière qui contacte les bases dans le petit sillon (Code PDB : 1PNR) (D’après Schumacher et al., Science, 1994).
l’axe de l’hélice alpha dans le grand sillon de l’ADN peut varier considérablement (Figure 4A) (Garvie and Wolberger, 2001).
Toutefois, il apparaît que la reconnaissance hélice alpha/grand sillon de l’ADN n’est pas universelle. En effet, le petit sillon est également capable de s’accommoder à une hélice α. Un membre de la famille LacI, le répresseur purine (PurR), fournit un bon exemple de ce mode de reconnaissance. PurR régule la synthèse de novo de purine et de pyrimidine en réprimant des gènes qui codent pour des enzymes qui participent à cette voie de biosynthèse. Ce répresseur se lie en présence de son co-répresseur (hypoxanthine) sous forme d’homodimère sur son site opérateur pseudo-palindromique de deux façons (Schumacher et al., 1994). Classiquement, la deuxième hélice d’un motif hélice-tour-hélice (HTH) contacte les bases dans le grand sillon. De plus, une hélice charnière (« hinge helix ») s’associe avec la même hélice de l’autre monomère pour interagir ensemble dans le petit sillon au centre du site de liaison (Figure 4B). Le petit sillon de l’ADN sous forme B étant trop petit pour s’accommoder à une et encore moins à deux hélices α, ces hélices charnières ouvrent le petit sillon et tordent l'ADN par l’intercalation de chaînes latérales de leucines. Le motif DM, qui constitue un domaine de liaison à l’ADN dépendant du zinc retrouvé dans des facteurs de transcription qui régulent la différenciation sexuelle, utilise également une hélice α pour interagir avec le petit sillon. En effet, ce motif se lie à l’ADN sous forme de dimère en réalisant des contacts avec le petit sillon par sa queue C-terminale qui se structure en hélice au contact de l’ADN (Zhu et al., 2000).
I.1.B.1.b.2 Interactions avec des feuillets β
Les hélices α ne sont pas les seuls éléments de structure secondaire responsables des contacts avec les bases. En effet, bien que les feuillets β soient moins utilisés que les hélices α pour la reconnaissance spécifique de l’ADN, plusieurs exemples existent.
Le premier concerne la famille des répresseurs MetJ qui régulent l’expression des enzymes de biosynthèse de la méthionine chez E. coli. Le répresseur MetJ lie l’ADN sous forme de dimère où chaque monomère donne un brin β pour former un feuillet β antiparallèle responsable de la liaison à l’ADN. En effet, ce feuillet s’insère dans le grand sillon de l’ADN et contacte les bases par les chaînes latérales d’une face du feuillet (Figure 5A) (Somers and Phillips, 1992).
Le mode de reconnaissance de l’ADN par le domaine de liaison à l’ADN dépendant du zinc GCM constitue un autre exemple. Les protéines GCM forment une petite famille de facteurs de transcription impliquée dans différentes étapes du développement. Le domaine de liaison à l’ADN de cette famille, le domaine GCM, consiste en deux modules à zinc enchâssés et utilise
A
B
Figure 5 : Reconnaissance de l’ADN par des feuillets β
(A) Interaction d’une face d’un feuillet β (en bleu foncé) formé par un dimère du répresseur MetJ
(en cyan) avec le grand sillon de l’ADN (en violet) (Code PDB : 1CMA) (D’après Somers and Philips, Nature, 1992).
(B) La protéine TBP (TATA binding protein) (en cyan) utilise une large surface en feuillet β (en bleu
foncé) pour interagir avec le petit sillon de l’ADN (en violet). L’insertion de ce feuillet β
f é d 10 b i ’ d’ i di i d l’ADN (C d PDB
concave formé de 10 brins s’accompagne d’une importante distorsion de l’ADN (Code PDB : 1YTB) (D’après Kim et al, Nature, 1993a et Kim et al, Nature, 1993b).
deux des cinq brins constituant l’un de ces feuillets β pour les contacts avec les bases dans le grand sillon de l’ADN (Voir partie I.2.B.4.c.2) (Cohen et al., 2003).
Outre l’interaction avec le grand sillon de l’ADN, il avait été prédit par la construction de modèles, que les feuillets β étaient également compatibles avec le petit sillon de l’ADN (Church et al., 1977). L’étude de l’interaction de la protéine TBP (TATA binding protein) avec l’ADN a permis d’illustrer cet arrangement. En effet, TBP utilise une large surface en feuillet β pour interagir avec le petit sillon de l’ADN (Kim et al., 1993b; Kim et al., 1993c). En revanche, contrairement à la reconnaissance de feuillets β dans le grand sillon qui s’accompagne d’une courbure modérée de l’ADN, l’insertion dans le petit sillon du feuillet β antiparallèle de TBP, composé de 10 brins, requiert une forte distorsion de l’ADN (Figure 5B). Les chaînes latérales de résidus phénylalanines s’intercalent dans l’ADN afin de favoriser cette courbure.
Toutefois, malgré ces différents exemples de liaison à l’ADN médiée par des feuillets β aussi bien dans le grand que dans le petit sillon, l’utilisation de feuillets β pour la reconnaissance de l’ADN par les domaines de liaison à l’ADN semble défavorisée par rapport aux hélices α. Ce phénomène pourrait s’expliquer par une évolution des feuillets pour l’acquisition de nouvelles spécificités de reconnaissance plus lente que celle des hélices. Les hélices α peuvent adopter différentes géométries dans le grand sillon de l’ADN et n’utilisent qu’un nombre restreint de chaines latérales pour interagir avec l’ADN de manière plus ou moins indépendante. Au contraire, dans le cas des feuillets β, en plus des résidus impliqués dans l’interaction avec l’ADN, de nombreuses interactions sont nécessaires pour maintenir la cohésion et le positionnement de la structure. Il apparaît donc que la génération de feuillets β avec de nouvelles spécificités requiert la coévolution de multiples positions, rendant l’adaptation plus lente que celle des hélices α (Connolly et al., 2000).
I.1.B.1.b.3 Les boucles et les queues terminales
Par ailleurs, alors que les éléments de structure secondaire tels que les hélices α et les feuillets β constituent des structures rigides pour les contacts avec les bases, certaines protéines de liaison à l’ADN interagissent avec l’ADN par l’intermédiaire de boucles. Les membres de la superfamille de facteurs de transcription apparentés à p53 (qui comprend les protéines à domaine d’homologie à Rel telles que NF-κB p50, les protéines à domaine runt, les protéines STAT et p53) partagent un élément commun : un domaine de structure similaire aux immunoglobulines dit en sandwich β qui sert de support à plusieurs boucles. Bien qu’il existe des variations dans l’orientation de
A
NGFI-B Antennapedia
B
Fi 6 R i d l’ADN d b l t d t i d DBD
Figure 6 : Reconnaissance de l’ADN par des boucles et des extensions de DBDs (A) Certaines protéines de liaison à l’ADN telles que NFκB (en cyan) reconnaissent l’ADN (en violet)
par des boucles (en bleu foncé) (Code PDB : 1NFK) (D’après Ghosh et al., Nature, 1995).
(B) Certains DBDs utilisent des extensions N- ou C-terminales pour réaliser des contacts spécifiques
avec l’ADN, complémentaires de ceux effectués par les éléments de structure secondaire du DBD.
Le domaine de liaison à l’ADN de la protéine à homéodomaine Antennapedia contacte le petit
ill d l’AD b i l (i di é flè h ) DBD d é lé i
sillon de l’ADN par un bras N-terminal (indiqué par une flèche). Le DBD du récepteur nucléaire orphelin NGFI-B présente une extension C-terminale (CTE) qui contacte le petit sillon de l’ADN. Cette interaction est réalisée par une séquence apparentée au motif AT hook (Arg-Arg-Gly-Arg en vert et indiquée par des flèches) qui réalise des contacts spécifiques avec une séquence A/T riche en 5’ du site de liaison (D’après Crane-Robinson et al, TIBS, 2006).
cette structure au niveau de l’ADN, toutes ces protéines reconnaissent l’ADN par des boucles (Figure 6A) (Garvie and Wolberger, 2001).
Enfin, certains domaines de liaison à l’ADN présentent des extrémités N- ou C-terminales riches en lysine et/ou arginine et non structurées en solution, qui lient l’ADN de façon indépendante et souvent dans le sillon opposé au domaine structuré. On compte par exemple l’extension N-terminale du répresseur de phage λ qui s’étend au niveau du grand sillon sur la face de la double hélice opposée à celle liée par son domaine globulaire HTH. Les protéines à homéodomaine présentent quant à elles un bras N-terminal qui contacte le petit sillon de l’ADN et contribue à la spécificité (Figure 6B) (Voir partie I.2.B.2) (Crane-Robinson et al., 2006). Enfin, certains DBDs présentent comme extension le motif « AT hook » (d’environ 9 résidus). En fait, ce motif n’est pas une extension au sens strict, puisqu’il constitue un DBD autonome chez les protéines de la famille HMGA (High Mobility Group-A), mais est souvent associé à différents DBDs fonctionnels (Aravind and Landsman, 1998). Le motif AT-hook lie des séquences AT riches au niveau du petit sillon de l’ADN via un motif central Arg-Gly-Arg (Huth et al., 1997). Une séquence apparentée au motif AT-hook (la boîte A : Arg-Arg-Gly-Arg) est retrouvée dans l’extension C-terminale (CTE) du DBD du récepteur nucléaire orphelin NGFI-B et fournit des contacts supplémentaires au niveau du petit sillon avec 3 paires de bases A-T, en 5’ de son site de liaison (Figure 6B) (Voir partie I.2.B.4.b.1) (Meinke and Sigler, 1999). En fait ce motif « A/T hook » dégénéré participe à l’extension C-terminale (CTE) de plusieurs récepteurs nucléaires orphelins (Gearhart et al., 2003; Meinke and Sigler, 1999; Wilson et al., 1992; Zhao et al., 1998).
I.1.B.2. Nature des liaisons mises en jeu dans un complexe
protéine/ADN
Outre une reconnaissance structurale de la protéine avec l’ADN, les surfaces d’interaction, protéine et ADN, doivent également être compatibles chimiquement. Cette reconnaissance chimique concerne la nature des interactions mises en jeu entre les deux surfaces. Ainsi, la nature et les arrangements tridimensionnels des groupements fonctionnels de la protéine doivent être complémentaires de ceux de la cible ADN. C’est l’arrangement de ces groupements qui détermine la spécificité de reconnaissance au niveau atomique (Rhodes et al., 1996). La nature des contacts atomiques protéine/ADN est complexe et implique différents types d’interactions faibles dont les liaisons hydrogène, les forces de van der Waals, les interactions électrostatiques, les liaisons hydrophobes et les ponts hydrogènes faisant intervenir des molécules d’eau. Etant faibles, ces liaisons nécessitent un rapprochement des molécules. Les associations
Guanine/Cytosine
Adénine/Thymine
Grand sillon
A D AGrand sillon
A D A CH3 5 6 4 1 3 2 C C C 7 8 9 N N O H N H C N C H C N H H 4 5 3 6 2 1 C C N C C N H H C O 5 6 4 1 3 2 C C C 7 8 9 N N C N C H C N H 4 5 3 6 2 1 C C N C C N H C O N H H O H NPetit sillon
A N H D H O C APetit sillon
A H O C AGrand sillon
A H D AGrand sillon
A A D HCytosine/Guanine
Thymine/Adénine
5 4 6 3 1 2 CH3 H C C C O N N C O H H N H 6 5 1 4 2 3 C N C C C N H C N C N 7 8 9 H C 5 4 6 3 1 2 H C C C N N C O H 6 5 1 4 2 3 C N C C C N C N C N H C H N H O H N H 7 8 9Figure 7 : Liaisons hydrogènes possibles au niveau des sillons des paires de
Petit sillon
A APetit sillon
A H D A bases de l’ADNLes donneurs potentiels de liaisons hydrogènes sont indiqués en bleu et notés de la lettre D, les accepteurs potentiels de liaisons hydrogènes sont indiqués en rouge et notés de la lettre A. Les liaisons hydrogènes des paires de bases elles-mêmes sont indiquées sous forme de courtes lignes rouges parallèles. Les groupements méthyles qui forment des protubérances hydrophobes sont montrés en jaune.
liaisons covalentes, les liaisons faibles permettent une grande flexibilité et plasticité des macromolécules et structures cellulaires. Les interactions protéine/ADN peuvent être séparées en deux classes : celles impliquant le squelette sucre/phosphate de l’ADN et celles concernant les bases. Alors que les contacts avec le squelette participent à la stabilisation du complexe, les contacts avec les bases contribuent à la spécificité de reconnaissance (Luscombe et al., 2001). Dans cette partie, nous nous intéresserons plus particulièrement aux liaisons hydrogènes puisqu’il s’agit du type de liaison majoritairement impliqué dans la « lecture directe » des bases de l’ADN par les chaînes latérales de la protéine.
I.1.B.2.a. Les liaisons hydrogènes dictent la spécificité de reconnaissance
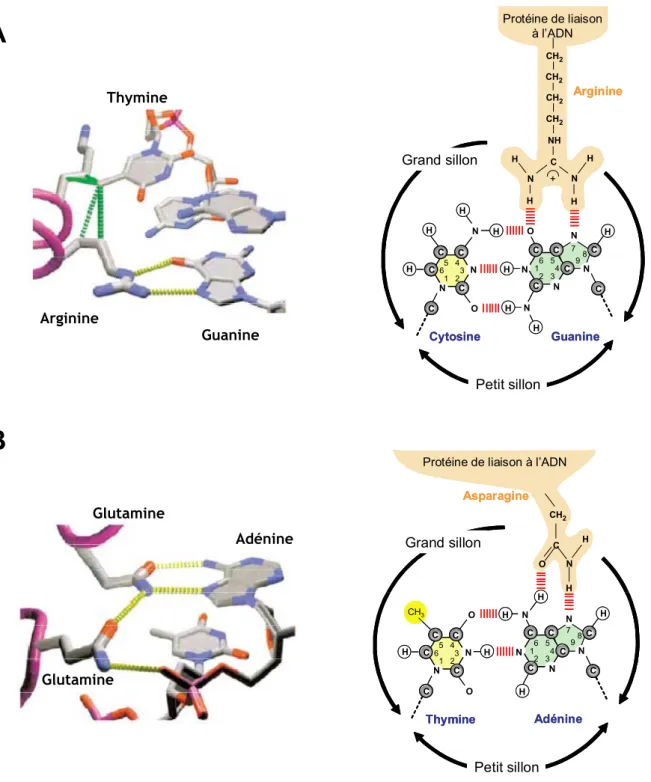
Une liaison hydrogène résulte de l’interaction électrostatique entre un atome d’hydrogène (H), lié par covalence à un atome électronégatif (O, N, S) (donneur) et un deuxième atome électronégatif possédant une paire d’électrons non partagée (accepteur). La liaison hydrogène est par exemple -O-H…O = C. Les liaisons hydrogènes peuvent s’établir entre les phosphates, sucres et bases de la double hélice d’ADN et les liaisons peptidiques ou les chaînes latérales des acides aminés. Celles formées entre les chaînes latérales des acides aminés et les groupements fonctionnels des bases dictent la « lecture directe » de la cible ADN par la protéine, c'est-à-dire la spécificité de la reconnaissance. En effet, les acides aminés et les bases possèdent à la fois des potentiels donneurs et des potentiels accepteurs d’hydrogène pour ce type d’interactions (Benos et al., 2002).
Les sillons de l’ADN sont riches en groupements fonctionnels de liaisons hydrogènes (Figure 7). La paire de bases A-T expose au niveau du grand sillon, deux accepteurs de liaisons hydrogènes au niveau des atomes N7 de l’adénine et O4 de la thymine ainsi qu’un donneur en 6-NH2 de l’adénine. Le petit sillon présente quant à lui deux accepteurs en N3 de l’adénine et O2 de la thymine. Pour la paire de base G-C, le grand sillon présente deux accepteurs de liaisons hydrogène au niveau de N7 et de O6 sur la guanine et un donneur en 4-NH2 de la cytosine. Le petit sillon de G-C expose un accepteur en N3 et un donneur en 2-NH2 de la guanine et un accepteur en O2 de la cytosine (Figure 7) (Seeman et al., 1976). La thymine peut quant à elle également accepter une liaison hydrophobe par son groupement méthyle dans le grand sillon. Ainsi, le petit sillon permet seulement de discriminer la guanine des autres bases par l’intermédiaire du groupement donneur en 2-NH2. En revanche, dans le grand sillon, le profil des sites accepteurs et donneurs de liaisons hydrogènes diffère pour chaque paire de bases. Par conséquent, dans la forme B de l’ADN, le grand sillon apparaît comme le meilleur candidat pour une reconnaissance spécifique (Garvie and Wolberger, 2001). Toutefois, la molécule d’ADN