Ecole doctorale Biologie - Santé - Biotechnologie
THESE
Pour l’obtention du grade de
DOCTEUR DE L’UNIVERSITE TOULOUSE III Discipline : Biologie Cellulaire et Moléculaire
Présentée et soutenue par
Charlotte Mailhat
Le 31 Octobre 2007Etudes des modifications post-traductionnelles de la
phosphatase Cdc25C lors de la régulation de la transition
G2/M du cycle cellulaire
Directeur de thèse : Bernard DUCOMMUN
JURY
M. Bernard DUCOMMUN, Professeur Hospitalo-Universitaire, Toulouse Directeur de thèse Mme May MORRIS, Chargée de Recherche CNRS, Montpellier Rapporteur M. Patrick JOUIN Directeur de Recherche CNRS, Montpellier Rapporteur M. John HICKMAN, Directeur Cancérologie SERVIER, Croissy sur Seine Examinateur Mme Anastassia HATZOGLOU, Professeur des Universités, Toulouse Président du jury
Laboratoire de Biologie Cellulaire et Moléculaire du Contrôle de la Prolifération Université Paul Sabatier- CNRS- UMR5088- IFR109
5 Je tiens tout d’abord à remercier l’ensemble des membres du jury pour avoir accepter de juger mon travail de thèse.
Ce travail de thèse a été financé par le laboratoire pharmaceutique SERVIER et je remercie le Docteur John Hickman pour son accueil chaleureux au sein du laboratoire de cancérologie de l’institut de recherche SERVIER. J’en profite également pour remercier le Docteur Roy Golsteyn et les membres de son équipe “Cycle cellulaire” : Aurélie et Céline qui ont accompagnés mon projet de recherche pendant un an au sein de l’IdRS et m’ont permis de découvrir le monde de la recherche en industrie.
Je remercie le Professeur Bernard Ducommun pour m’avoir accueillie dans son équipe au sein de LBCMCP et pour avoir endossé la casquette de directeur de thèse.
Bernard je te remercie tout particulièrement pour ta patience, tes encouragements et ta disponibilité, mais également pour m’avoir permis de terminer au mieux ce travail.
Je remercie également tous les membres de l’équipe BDuc, en particulier Jean-Pierre pour son aide précieuse pour la purification de Cdc25C et ses nombreux conseils aussi bien scientifiques que techniques qui m’ont permis d’avancer tout au long de ma thèse. Merci aussi à Odile, Martine et Valérie pour m’avoir aidée à avancer dans mes manipes de dernières minutes. Et je n’oublie pas l’ensemble des membres du LBCMCP pour m’avoir accompagnée et soutenue tout au long de ma thèse.
Je tiens aussi à remercier Bernard Monsarrat et son équipe pour m’avoir permis de découvrir les joies de la spectrométrie de masse et m’avoir permis d’accéder à la plateforme de spectrométrie de masse de la génopôle de Toulouse. Je remercie chaleureusement Carine pour ses formations, ses conseils et son aide précieuse dans les déchiffrages des spectres MS/MS.
Pour terminer, je souhaite également remercier mes amis et ma famille :
Tout d’abord, mes amies du labo : Corine T, Isa, Corine L, Christelle et Bernie qui m’ont permis de passer trois supers années, et m’ont permis d’oublier les moments de stress et de découragement. Je regretterais non seulement toutes nos pauses thé ou café, les moments de détentes que nous avons passées ensemble mais aussi tout simplement votre présence et votre compagnie qui m’ont apporté beaucoup tout au long de ma thèse.
Je pense également à Chrys avec qui nous nous sommes suivies tout au long de nos thèses respectives et qui a partagée avec moi chaque étape de nos thèses.
Et tous les Ginos qui me suivent maintenant depuis de nombreuses années et qui m’ont toujours soutenue. Un grand merci à : Tibo, Annef, Mathieu, Marie-Alix, Marie-Pierre, Fabien, Sophie, Xavier, Laurent, Florent, Charlotte, Valérie et Gilles pour vos encouragements et votre amitié sans faille.
mes frères : Guillaume, Cyril et Maxime pour leur patience et leur gentillesse et leur compréhension…
Et pour terminer, toutes mes pensées vont à mon mari Nicolas qui m’a toujours soutenue et qui a joué une grande part dans la réussite de ce projet !
7
Etudes des modifications post-traductionnelles de la phosphatase
Cdc25C lors de la régulation de la transition G2/M du cycle
cellulaire
Directeur de thèse : Bernard Ducommun
Résumé :
La phosphatase Cdc25C est un acteur critique de la progression du cycle cellulaire par son rôle jouée dans le contrôle de l’activation du complexe CDK1-Cycline B lors de l’entrée en mitose et de la mise en place des mécanime de surveillance. Son activité est régulée par de nombreuses kinases impliquées dans les cascades de signalisation cellulaire résultant dans la phosphorylation de nombreux résidus. Au cours de cette étude, nous avons purifié Cdc25C à partir de cellules humaines et réalisé une approche protéomique globale dans le but d’identifier de nouvelles modifications régulatrices. Nous présentons dans ce manuscrit la mise au point des conditions de purification de la phosphatase et les différentes modifications post-traductionnelles que nous avons identifiées par spectrométrie de masse, en particulier deux phosphorylations sur les résidus S168 et S263 et une méthylation sur le résidu R35. Pour terminer nous avons réalisé une analyse fonctionnelle des phosphorylations sur les S168 et S263. Nous avons montré par imagerie cellulaire que la mutation de la S263 en alanine conduisait à l’accumulation nucléaire de Cdc25C. Nous proposons ainsi que la phosphorylation de la S263 soit impliquée dans un mécanisme de régulation qui module l’import nucléaire de Cdc25C.
Mots clés :
Cycle cellulaire, points de contrôle, Cdc25C, phosphatase, modification post-traductionnelle, localisation.
Discipline administrative : Biologie Cellulaire et Moléculaire
Laboratoire de Biologie Cellulaire et Moléculaire du Contrôle de la Prolifération Université Paul Sabatier- CNRS- UMR5088- IFR109
118 route de Narbonne - Bâtiment 4R3 entrée B1 31062 TOULOUSE cedex 9
9 ADN : Acide DéoxyriboNucléique
UV : Ultra- Violet
CDK : Cycline Dependent Kinase MPF: M-phase promoting factor MCM : Mini-Chromosome Maintenance CAK : CDKs Activating Kinase
CKIs : CDK inhibitors
CRS : Cytoplasmique Retention Signal CAK : CDK Activating Kinase INK4 : Inhibitors of CDK4 ATP : Adenosyl tri phosphate MAT1 : Ménage à Trois Cks : CDC kinase subunit
RINGO : Rapid Inducer of G2/M progression in Oocytes Cdc25 : Cycle de Division Cellulaire
DSPases : Dual Specificity Phosphatase NES : Nuclear Export Signal
NLS : Nuclear Localisation Signal APC : Anaphase Promoting Complex SCF : Skp1/Cullin/F-box)
CK2 : Caséine Kinase 2
C-TAK1 : Cdc Twenty-five C Associated Kinase 1
CaMKII : Calcium calmoduline-dependent protein Kinase II Plk : Polo kinase
Chk : Checkpoint Kinase IR : Radiation ionisante RPA : Replication Protein A MRN : Mre11/Rad50/NBS1
NHEJ : Non Homologous End Joining ATM : Ataxia Telangiectasia Mutated ATR : ATM and Rad3 related
PIKK : Phospho-Inositide 3-Kinase related Kinases p38SAPK
: p38 Stress Ativated Protein Kinase MAPK : Mitogen Activated Protein Kinase ERK : Extracellular-signal Related Kinase SAPKJNK
: Stress Activated Protein Kinase/ c-Jun N-terminal Kinase MK2 : MAPKAP-K2 - MAP Kinase Activated Protein Kinase 2 Tap-tag : Tandem Affinity Purification tag
BIA-MS : Biomolecular Interaction Analysis – Mass Spectrometry MPT : Modification Post-Traductionnelle
MS : Mass Spectrometry ESI : Electrospray
LC : Liquid chromtography
SDS-PAGE : Sodium Dodécyl Sulfate- PolyAcrylamide Electrophoresis PI : point isoelectrique
MALDI :Matrix Assisted Laser Desorption/Ionisation TOF : Time Of Flight
IT: Ion Trap Q : Quadrupole
NCBI :National Center for Biotechnology Information IMAC : colonne d’ion metalliques immobilisé
NTA : acide nitrilo-acétique
HA : hemagglutinin du virus Influenza A IDA : Information-Dependant Acquisition PCC : Condensation prématurée de la chromatine
11
INTRODUCTION GENERALE ... 15
Préambule ... 17
I- Biologie du cycle cellulaire ... 19
I-2-A : Les complexes CDK-Cyclines ... 21
I-2-A-a : Les complexes CDK-Cycline dans le cycle cellulaire ... 22
I-2-A-b : Les autres rôles des complexes CDK-Cycline ... 23
I-2-B : La régulation des complexes CDK-Cyclines ... 24
I-2-B-a : La liaison avec les Cyclines ... 24
I-2-B-b : Les inhibiteurs CKIs ... 27
I-2-B-c : Les modifications post-traductionnelles ... 28
(i) Phosphorylations activatrices ... 28
(ii) Phosphorylations inhibitrices ... 29
(iii) Déphosphorylations activatrices ... 29
I-2-B-d : Les autres mécanismes de régulation ... 30
I-3-A : Présentations des phosphatases Cdc25 humaines ... 33
I-3-B : Caractérisation des phosphatases Cdc25 humaines ... 34
I-3-B-a : Le rôle des phosphatases Cdc25 dans le cycle cellulaire ... 34
I-3-B-b : Régulation des phosphatases Cdc25 ... 36
(i) Cdc25A ... 36
(ii) Cdc25B ... 39
(iii) Cdc25C ... 42
II- La réponse du cycle cellulaire aux dommages sur l’ADN ... 47
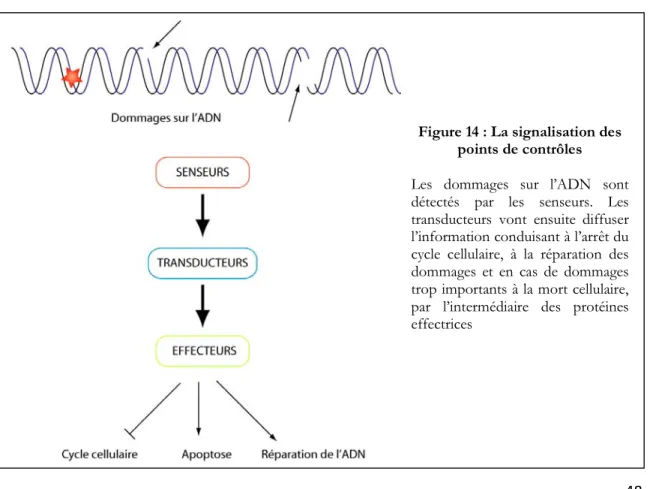
II-1-A : Dommages sur l’ADN et fonctionnement des points de contrôles ... 49
II-1-A-a : Les senseurs ... 49
II-1-A-b : Les transducteurs ... 49
(i) Les kinases apicales ... 50
(ii) Les kinases distales ... 51
II-1-A-c : les effecteurs ... 51
II-1-B : Arrêt à la transition G1/S ... 52
II-1-C : Arrêt à la transition G2/M ... 53
II-1-C-a : Les événements de régulation post-traductionnels ... 53
II-1-C-b : les événements de régulation transcriptionnelle ... 54
II-2-A : Modèle d’activation du point de contrôle G2/M en réponse aux stress génotoxiques ... 55
II-2-B : La sortie des points de contrôle ... 56
II-2-B-a : Retour dans le cycle cellulaire ... 57
II-2-B-b : Apoptose ... 57
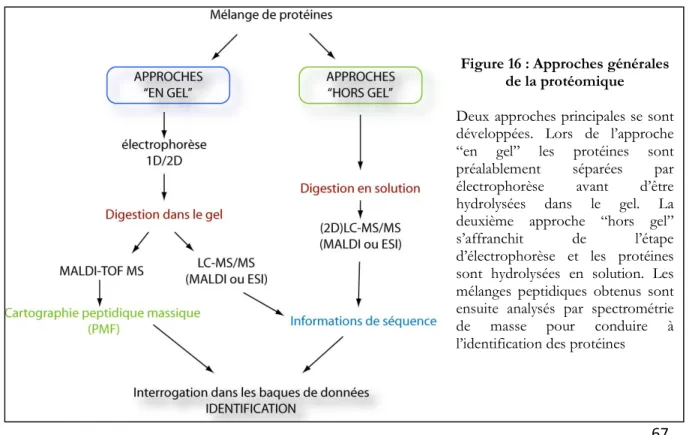
III- L’analyse protéomique ... 61
III-1-A : Les interactions entre les protéines ... 62
III-1-B : La recherche de modifications post-traductionnelles ... 63
III-2-B : L’identification et la caractérisation des protéines ... 70
III-2-B-a : Histoire et principe de la spectrométrie de masse ... 71
III-2-B-b : MALDI-TOF MS ... 72
III-2-B-c : LC-ESI-MS ... 73
III-2-B-d : La spectrométrie de masse en tandem ... 75
III-2-C : Les stratégies mises en œuvre pour l’identification des protéines ... 77
III-2-C-a : La protéolyse ... 78
III-2-C-b: Spectrométrie de masse en protéomique ... 78
III-2-C-c : L’interrogation dans les banques de données ... 79
III-3-A : Détection des phosphopeptides ... 81
III-3-B : Détermination des sites de phosphorylation par MS/MS. ... 82
III-3-C : Méthode d’enrichissement ... 82
RESULTATS ... 85
Projet de thèse ... 87
A- Situation du projet ... 87
B- Présentation des travaux de thèses... 88
I- Construction des outils ... 89
I-1 : Description de la lignée cellulaire ... 90
I-2 : Description de la stratégie de purification ... 91
I-3 : Construction et clonage de la phosphatase Cdc25C ... 91
I-3-A : Construction du plasmide pUHD10 ::HA-Cdc25C-His ... 91
I-3-B : Validation de la construction de HA-Cdc25C-His dans les U2OS ... 93
I-4 : Activation du point de contrôle en G2/M ... 96
I-5 : Les Voies de signalisation en réponse au traitement étoposide ... 100
I-6 : Validation de la stratégie de purification de HA-Cdc25C-His ... 101
I-6 : Conclusion ... 102
II- Analyse protéomique ... 105
II-1 : Spectrométrie de masse et recherche de MPT ... 105
II-1-A : préparation des échantillons ... 106
II-1-B : Recherche des modifications par spectrométrie de masse ... 107
II-2 : Séparation par gels bidimensionnels ... 114
II-2-A : Cartographie par électrophorèse en gel bidimensionnel ... 114
II-2-B : Effet de l’étoposide sur la cartographie des isoformes de Cdc25C par gels bidimensionnels ... 116
II-3 : Conclusion ... 117
III- Analyse fonctionnelle des phosphorylations S168p et S263p ... 119
III-1 : Le développement des outils ... 119
III-1-A : Construction des mutants ... 119
III-1-B : Génération d’anticorps phospho-spécifiques ... 120
III-2 : Effet des mutants de Cdc25C ... 122
IV-1-A : Etude de la phosphorylation de la S263 ... 124
IV-1-B : Etude de la phosphorylation de la S168 ... 128
13
DISCUSSION ... 133
I- Validation du modèle d’étude ... 135
I-1 : La stratégie de purification ... 136
I-2 : Le contrôle du cycle cellulaire ... 137
II- Analyse protéomique ... 138
II-1 : La préparation de HA-Cdc25C-His pour la LC-MS/MS ... 139
II-2 : Le taux de couverture de séquence ... 140
III- Caractérisation fonctionnelle des phosphorylations identifiées... 144
CONCLUSION GENERALE ... 149
MATERIELS ET METHODES... 153
I- Analyse des protéines ... 155
I-1 : Western blot ... 155
I-2 : Immunofluorescence ... 155
I-2-A : Indirecte ... 155
I-2-B : Directe ... 156
I-3 : Cytométrie en flux ... 156
I-4 : Essais kinases in vitro ... 156
II- Culture cellulaire ... 157
II-1 : cellules U2OS ... 157
II-2 : Transfection transitoire ... 157
III- Extraction des protéines et purification ... 158
III-1 : Protocole classique ... 158
III-2 : Protocole adapté à la spectrométrie de masse et purifiactions ... 158
IV- Protéomique ... 159
IV-1 : spectrométrie de masse ... 159
IV-2 : Electrophorèse bidimensionnelle ... 160
ANNEXES ... 161
BIBLIOGRAPHIE ... 173
17
Préambule
Le cancer est défini comme une prolifération anormale de cellules saines ayant subi des altérations spécifiques sur leurs génomes, les conduisant à acquérir les capacités nécessaires pour échapper à la régulation homéostatique de la division cellulaire, envahir les tissus voisins et générer des métastases vers des sites plus éloignés dans le corps humain. En France, les cancers représentent la première cause de mortalité chez les hommes et la deuxième chez les femmes après les maladies cardiovasculaires. 280 000 nouveaux cas sont détectés chaque année et 150 000 personnes en meurent. Un homme sur trois et une femme sur quatre décèderont d’un cancer. Un thème central dans les efforts pour contrôler et éliminer les cancers est de promouvoir la recherche en science fondamentale afin de mieux comprendre la biologie du cancer avec l’espoir de générer de nouvelles interventions thérapeutiques qui pourraient être transformées en traitements cliniques contre le cancer. Les cancers humains présentent de nombreuses altérations génétiques qui peuvent conduire à plus d’une centaine de type de tumeurs différentes. Douglas Hanan et Robert Weinberg ont proposé que six mutations essentielles du processus cellulaire normal sont responsables de l’apparition de la majorité des cancers. Ces mutations sont : « l’indépendance vis-à-vis des signaux prolifératifs, l’insensibilité aux signaux inhibiteurs de la prolifération, l’abolition de la mort cellulaire par apoptose, une capacité proliférative illimitée, une capacité à susciter l’angiogénèse et la capacité d’invasion de tissu et de prolifération dans des sites étrangers (métastases) » (Hanahan and Weinberg, 2000). Afin de bien comprendre comment l’instabilité du génome humain peut contribuer au phénotype tumoral, il est important de connaître le fonctionnement du processus biologique fondamental avant qu’il ne dérive. Le travail que nous présentons ici fait parti de l’effort compréhension de la biologie du cycle cellulaire et la régulation de la machinerie de division cellulaire. Cette étude a également été entreprise en collaboration avec un partenaire industriel dans le but de mettre en avant les mécanismes conduisant aux dérégulations de la machinerie de division cellulaire qui pourraient définir de nouvelles cibles de traitements contre les cancers.
19
I- Biologie du cycle cellulaire
I-1 : Le cycle cellulaire
C’est au cours du cycle cellulaire qu’une cellule se divise pour donner naissance à deux cellules filles identiques. La division cellulaire est à l’origine de la croissance et du développement de tous les êtres vivants et est au centre de leurs hérédités et de l’évolution. Depuis que Rudolph Virchow (1821–1902) a proclamé son célèbre “Omnis cellula e cellula”1
, la compréhension des mécanismes contrôlant la division cellulaire et la transmission fidèle de l’information génétique entre chaque génération est considérée comme un problème majeur en biologie.
Les principaux évènements du cycle cellulaire sont définis par la réplication du matériel génétique et la ségrégation du génome répliqué conduisant à la division de la cellule (Mitchison and Creanor, 1971). Chez les eucaryotes ces évènements moléculaires et cellulaires sont séparés dans le temps et l’espace et sont régulés suivant un ordre chronologique précis.
C’est au cours de la phase S (Synthesis) qu’à lieu la réplication semi-conservative des chromosomes et la ségrégation des chromosomes répliqués est réalisée durant la phase M ou mitose (le terme vient du grec mito qui signifie fil) où les chromosomes, visibles et condensés, ségrègent pour donner deux cellules filles après la cytokinèse (Mitchison and Salmon, 2001). Les phases G1 (Gap 1) et G2 (Gap 2) précédant respectivement la phase S et la mitose ont pour but de préparer la cellule aux processus fondamentaux du cycle cellulaire. Au cours de la phase G1 la cellule prépare la duplication de l’ADN et intègre les signaux environnementaux (mitogènes ou facteurs de croissance) qui conduiront soit à sa division, soit à son entrée en phase G0 de pause ou quiescence, soit à sa sortie du cycle cellulaire pour se différencier. La phase G2 est une étape de transition qui permet à la cellule de se préparer à la division mitotique (figure 1).
Afin de s’assurer que chaque cellule nouvellement formée reçoive un génome complet, le déroulement du cycle cellulaire est contrôlé de telle manière que chaque phase du cycle cellulaire n’ait lieu qu’une seule fois par cycle et qu’aucune phase ne puisse commencer sans que les précédentes ne se soient correctement déroulées. Ce sont les points de contrôle ou en anglais checkpoint qui sont mis en place aux étapes clés du cycle cellulaire et assurent la coordination des transitions entre les différentes étapes. Par exemple une cellule ne pourra pas se diviser si tous les
chromosomes ne sont pas correctement alignés sur la plaque métaphasique ou ne pourra pas répliquer son ADN avant d’avoir franchi la mitose (Hartwell and Weinert, 1989; Murray, 1993; Smith et al., 2002). D’autre part, ces points de contrôle peuvent également provoquer une pause dans la progression du cycle cellulaire pour permettre à la cellule de se défendre contre les agressions extérieures à la cellule (radiations ionisantes, UV, drogues, etc.) ou provenant de la cellule elle-même (radicaux oxygénés, erreurs de réplication, fourches de réplication arrêtées, etc.) qui peuvent endommager l’ADN et perturber l’intégrité du matériel génétique.
I-2 : les complexes CDK-Cycline
Les kinases dépendantes des Cyclines (Cyclin-Dependent Kinases ou CDK) en association avec les Cyclines régulatrices conduisent les événements du cycle cellulaire eucaryote et son horloge interne (figure 2) (Morgan, 1997). Bien que la liaison avec les cyclines soit le premier
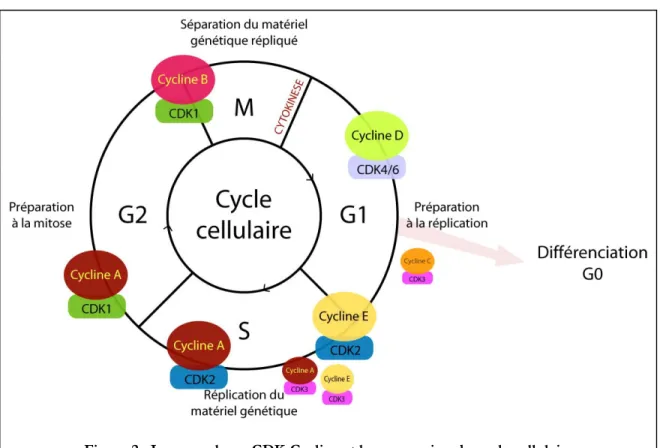
Figure 1 : Le cycle cellulaire des cellules eucaryotes
Représentation schématique du cycle cellulaire : la réplication fidèle du matériel génétique a lieu en phase S et la ségrégation du matériel génétique dupliqué a lieu en mitose ou phase M. Les phases G1 et G2 préparent respectivement la progression de la cellule vers la phase S et la phase M.
21 déterminant de l’activité des CDK, dont l’expression et la dégradation cyclique donnent le tempo pour chaque phase du cycle, d’autres facteurs régulent l’activité des complexes CDK-Cyclines tels que la phosphorylation des CDKs et des cyclines, la déphosphorylation des CDKs, la liaison à des protéines inhibitrices ou encore la localisation subcellulaire des complexes CDK-Cyclines.
I-2-A : Les complexes CDK-Cyclines
Les travaux réalisés dans les années 1970 sur les ovocytes d’amphibiens ont conduit à la découverte d’une activité enzymatique présente dans le cytoplasme, le MPF (M-phase promoting factor) qui est capable d’induire l’entrée en méiose ou en mitose lorsqu’il est micro-injecté dans un ovocyte ou une cellule (Ecker and Smith, 1971; Masui and Markert, 1971). Les études biochimiques et génétiques menées en parallèle sur de nombreux organismes ont à leur tour montré que le MPF est en fait constitué de la sous-unité enzymatique kinase dénommée maintenant CDK1 (Russell and Nurse, 1986) et d’une sous-unité régulatrice correspondant à la cycline de type B (Labbe et al., 1989). Le complexe CDK1-Cycline B qui est conservé de la levure jusqu’à l’homme a alors été défini comme le régulateur universel de l’entrée en mitose.
A la suite de la découverte du complexe CDK1-Cycline B, de nombreux homologues des CDKs et des cyclines ont été caractérisés chez divers organismes et sont impliqués dans la progression de l’ensemble du cycle cellulaire, une CDK pouvant s’associer à différentes cyclines. Il a également été montré que les complexes CDK-Cycline participaient aussi à la régulation d’autres événements que le cycle cellulaire.
Actuellement chez l’homme 11 CDKs (CDK1 à CDK11) ont été caractérisées et au moins 29 gènes codant pour des protéines apparentées à la famille des cyclines par la présence d’une séquence conservée de 150 acides-aminés : “la Cyclin box” ont été mis en évidence. Les cyclines peuvent être classées en trois groupes : (i) Le groupe des cyclines de phase G1, auquel appartiennent les Cyclines D et E et qui régulent la progression en phase S et la transition G1/S ; (ii) Le groupe des cyclines de phase G2 auquel appartiennent les Cyclines A et B, impliquées dans la transition G2/M (Murray and Marks, 2001) et (iii) le troisième groupe de cycline (notamment les Cyclines C, K, L et T) qui est impliqué dans la régulation de la transcription (Murray and Marks, 2001).
I-2-A-a : Les complexes CDK-Cycline dans le cycle cellulaire
C’est l’association de la Cycline D, activée par les signaux mitogènes, avec les kinases CDK4 et CDK6 qui assurent la progression de la phase G1 du cycle cellulaire. Ces complexes phosphorylent et inhibent la protéine de sensibilité au rétinoblastome (Rb) qui libère alors le facteur de transcription E2F et permet la transcription des gènes nécessaires à la transition G1/S. Parmi les cibles de transcription du facteur E2F se trouvent les gènes codant pour les Cycline E, A et B. La Cycline E est essentielle à l’activation de CDK2 et ainsi à la progression de la phase G1 vers la phase S (Cobrinik, 2005; Malumbres and Barbacid, 2001, 2005). Ce processus est supposé rendre les cellules indépendantes aux signaux mitogènes et correspond au fameux “point de restriction” ou point de non retour, qui est défini comme un passage après lequel la cellule n’a plus recours aux stimuli mitogènes pour engager sa division cellulaire (Dannenberg et al., 2000; Malumbres and Barbacid, 2001; Sage et al., 2000). De récentes études ont montré que CDK3 pouvait, elle aussi, phosphoryler et inhiber la protéine Rb, il semblerait ainsi que le complexe CDK3-Cycline C permette aux cellules de sortir de l’état de quiescence (G0) et assure le retour des cellules à l’état prolifératif lors de la transition G0/G1 (Ren and Rollins, 2004).
La transition G1/S est alors stimulée par le complexe CDK2-Cycline E en permettant, entre autre, aux protéines MCM (Mini-Chromosome Maintenance) de se fixer aux origines de réplication, ce qui déclenche l’initiation de la réplication de l’ADN (Hwang and Clurman, 2005; Pagano et al., 1993). Par ailleurs, CDK3 associée aux Cyclines A et E est peut-être aussi impliquée dans la transition G1/S (Hengstschlager et al., 1999). Puis, une fois les cellules installées en phase S, le complexe CDK2-Cycline E est réprimé afin de prévenir les phénomènes de “re-réplication” et le complexe CDK2-Cycline A achève correctement la phase S et prépare la transition vers la phase G2 (Hwang and Clurman, 2005; Malumbres and Barbacid, 2005). A la fin de la phase S la Cycline A s’associe avec CDK1 et assure la progression tout au long de la phase G2, puis la Cycline A est dégradée alors que la Cycline B est activement synthétisée. CDK1 s’associe alors avec la Cycline B et conduit à la progression des cellules en mitose. Ce complexe, essentiel à la transition G2/M, est responsable de la phosphorylation de plus de 70 protéines impliquées dans la progression du cycle des cellules de mammifères. Sa localisation subcellulaire est finement régulée et permet de contrôler son rôle dans la fragmentation du réseau golgien, la séparation des centrosomes, la nucléation des microtubules, la condensation des chromosomes et la déstructuration de l’enveloppe nucléaire (Nigg, 2001). Finalement la sortie de mitose est assurée par une chute
23 brutale de l’activité CDK1-Cycline B via la dégradation de la Cycline B (Malumbres and Barbacid, 2005).
I-2-A-b : Les autres rôles des complexes CDK-Cycline
Les autres CDK ne sont pas impliquées dans la progression proprement dite du cycle cellulaire. Les CDK7, 8, 9, 10 et 11 sont impliquées dans le contrôle de la transcription avec néanmoins pour certaines d’entre-elles un rôle direct dans le contrôle du cycle cellulaire (Loyer et al., 2005). Quant à CDK5, elle est activée par les protéines du cerveau p35 et p39 où elle aurait un rôle dans le développement et la maintenance du système nerveux central. La dérégulation de CDK5 est impliquée dans plusieurs maladies neurodégénératives et en particulier la maladie d’Alzheimer (Cruz and Tsai, 2004; Kesavapany et al., 2004).
Figure 2 : Les complexes CDK-Cycline et la progression du cycle cellulaire
La progression dans le cycle cellulaire est contrôlée par l’activation et l’inactivation successive des complexes CDK-Cycline.
I-2-B : La régulation des complexes CDK-Cyclines
Faisant suite aux études sur le cycle cellulaire qui ont menées à la découverte des CDKs, quatre mécanismes majeurs de régulation se détachent et gouvernent l’activité des CDKs. Le premier qui est aussi le principal est la liaison activatrice avec la sous-unité cycline régulatrice. Il semblerait cependant que l’activation complète de la plupart des CDKs requiert aussi la phosphorylation d’un résidu thréonine par la CAK (CDKs Activating Kinase). Enfin le complexe pleinement actif peut aussi être inhibé : par la liaison avec les sous-unités inhibitrices des CDKs (CKIs, CDK inhibitors) et par la présence de phosphorylations inhibitrices sur des résidus conservés dans la région amino-terminale des CDKs.
La détermination de la structure de CDK2 (De Bondt et al., 1993), puis du complexe CDK2-Cycline A (Jeffrey et al., 1995) associées à des études biochimiques, a permis de mieux comprendre les mécanismes impliqués dans la modulation de l’activité catalytique des complexes CDK-Cycline.
I-2-B-a : La liaison avec les cyclines
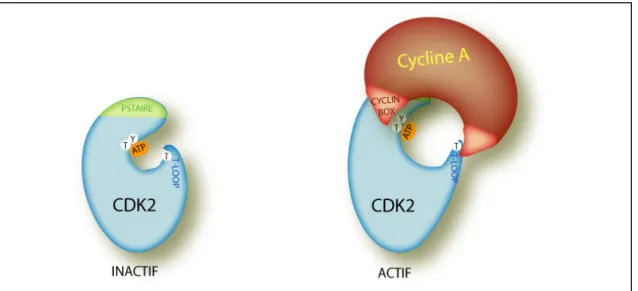
La structure de CDK2 humaine est, comme pour les autres protéines kinases, constitué d’un petit lobe amino-terminal, contenant le domaine PSTAIRE, associé à un lobe carboxy-terminal plus grand. L’étroite cavité située entre ces deux lobes constitue le site actif. La forme non modifiée et donc non active de CDK2 est alors caractérisée d’une part par la présence d’une boucle flexible ou “T-Loop” sortant du lobe carboxy-terminal et qui ferme l’entrée du site catalytique empêchant la liaison avec le substrat et d’autre part par le positionnement du domaine PSTAIRE, qui gène la réaction de phosphorylation par un désalignement des résidus clés du site catalytique (De Bondt et al., 1993 ; Morgan, 1995). Ainsi la protéine kinase doit subir un profond changement pour pouvoir être activée. La liaison avec une cycline est une étape essentielle au cours de l’activation des CDKs.
L’interaction entre les domaines “Cyclin box” de la Cycline A et le domaine PSTAIRE de CDK2, a pour principale conséquence d’abaisser la boucle flexible T-Loop, qui obstruait le site de liaison avec le substrat, et permet un réalignement des résidus importants pour la phosphorylation du substrat qui sont ainsi dans une position plus favorable (figure 3). Ce changement de conformation rend disponible aux phosphorylations un résidu thréonine
25 hautement conservé chez les CDKs (T161 sur CDK1 et T160 chez CDK2) (Jeffrey et al., 1995). D’autre part, en plus du changement conformationnel favorable à l’activité de la kinase, le choix de la cycline interagissant avec la kinase CDK va permettre de moduler la spécificité du substrat de la kinase (Cross et al., 1999; Pines, 1995).
Figure 4 : Régulation temporelle de l’activité des complexes CDK-Cycline La régulation fine de l’activité des complexes CDK-Cycline permet un contrôle de la progression dans chaque phase du cycle cellulaire (d’après Pines, Nature Cell biology 1999).
Figure 3 : Interaction entre CDK2 et la Cycline A
Représentation schématique de l’impact de la liaison de la Cycline A sur l’activation de CDK2. L’interaction se fait entre le domaine PSTAIRE de CDK2 et la séquence conservée des cyclines, la Cyclin box. L’impact majeur de cette interaction est le déplacement de la séquence T-loop qui obstruait le site catalytique de CDK2.
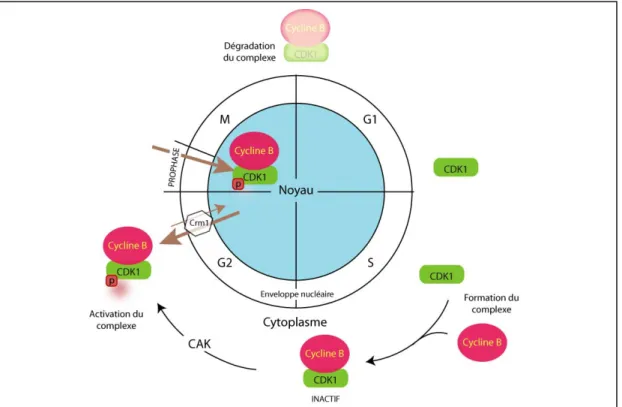
L’oscillation de la concentration des cyclines dans la cellule est en partie responsable des changements dans l’activité des CDKs au cours du cycle cellulaire (figure 4) (Pines, 1999). Ainsi la synthèse d’une cycline est responsable du passage à travers une phase particulière du cycle et sa dégradation conduit à la transition vers la phase suivante. Le niveau des cyclines est donc strictement contrôlé par un ensemble complexe de mécanismes actifs, aussi bien au niveau de la transcription des gènes (Breeden, 2000) que de la dégradation des protéines, dépendante du protéasome (Deshaies, 1995) et spécifique pour chaque étape du cycle cellulaire. En outre, certaines cyclines possèdent des séquences de localisation cellulaire qui permettent de cibler la kinase CDK associée dans un compartiment subcellulaire particulier et régulent son activité dans l’espace en la maintenant éloignée de son substrat (Yang and Kornbluth, 1999). Une séquence CRS (Cytoplasmique Retention Signal) permet à la Cycline B1 de se lier au facteur d’export nucléaire Crm1 et conduit ainsi à une localisation principalement cytoplasmique au cours de la phase G2 (Hagting et al., 1998; Porter et al., 2003). Lors de l’entrée en mitose, la Cycline B1 est phosphorylée par CDK1 et Plk1, elle ne peut plus être exportée ce qui conduit à la relocalisation nucléaire du complexe CDK1-Cycline B1 (figure 5) (Toyoshima-Morimoto et al., 2001).
Figure 5 : Localisation de CDK1-Cycline B au cours du cycle cellulaire
Le complexe CDK1-Cycline B, même après son activation par la CAK, est maintenu dans le cytoplasme par un export nucléaire supérieur à son import. En fin de phase G2 le complexe devient nucléaire pour assurer la mitose et est dégradé en fin de phase M, conduisant à la sortie de mitose.
27
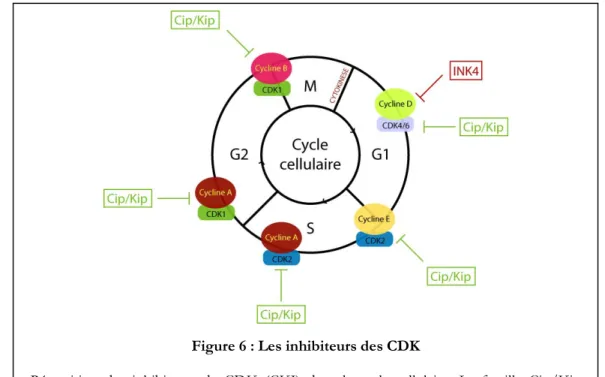
I-2-B-b : Les inhibiteurs CKIs
Les inhibiteurs des CDKs ou CKIs sont des petites protéines qui ont la capacité de se lier aux complexes CDK-Cycline afin d’inhiber leurs activités. Des analyses biochimiques ainsi que des études cristallographiques ont permis de révéler les mécanismes d’action par lesquels ces inhibiteurs abolissent l’activité des CDKs. Ils sont au nombre de trois : l’inhibition directe de l’activité kinase du complexe CDK, l’interférence contre l’activation des CDKs par la CAK (CDK Activating Kinase) et enfin la compétition avec les Cyclines pour la liaison aux CDKs (figure 6) (Arellano and Moreno, 1997). Chez les mammifères, il existe deux familles de CKIs : la famille Cip/Kip et la famille INK4 (Inhibitors of CDK4) (Elledge et al., 1996). La famille Cip/Kip comprend trois membres p21 WAF1/CIP1
, p27KIP1
et p57KIP2
, qui ont la capacité d’interagir avec tous les complexes CDK-Cyclines. La détermination de la structure du complexe CDK2-Cycline A-p27KIP1 a permis de mieux comprendre leurs mécanismes d’action (figure 7) (Russo et al., 1996). p27KIP1
se lie avec la Cycline A par son domaine amino-terminal (sans affecter sa structure) alors que sa région carboxy-terminale interagit fortement avec le lobe supérieur de CDK2, ce qui conduit à changement conformationnel et empêche la fixation de CDK2 à l’ATP. La famille INK4 comprend quatre membres : p16INK4a
, p15INK4b
, p18INK4c
, et p19INK4d
qui inhibent spécifiquement CDK4 et CDK6 en empêchant leur association avec la Cycline D (figure 6) (Russo et al., 1998).
Figure 6 : Les inhibiteurs des CDK
Répartition des inhibiteurs de CDK (CKI) dans le cycle cellulaire. La famille Cip/Kip inhibe tous les complexes CDK-Cycline et la famille INK4 inhibe uniquement les complexes CDK4/6-Cycline D.
I-2-B-c : Les modifications post-traductionnelles
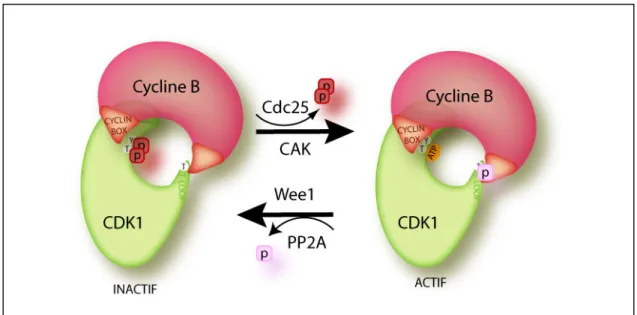
Les modifications post-traductionnelles sont impliquées dans la régulation de nombreux mécanismes du cycle cellulaire (voir plus loin). Dans le cas particulier de la régulation des complexes CDK-Cycline, un jeu de phosphorylations/déphosphorylations de la sous-unité catalytique CDK intervient, soit pour renforcer l’activité de la kinase, soit pour l’inhiber (figure 8).
(i) Phosphorylations activatrices
L’association avec la Cycline B n’est pas suffisante pour activer pleinement le complexe CDK1-Cycline B. L’activation nécessite la phosphorylation du résidu thréonine 161 (T161) présent sur la séquence T-Loop. La kinase CAK est responsable de cette phosphorylation durant la phase G2 (Solomon et al., 1992; Watanabe et al., 2005). Chez les vertébrés la CAK est un complexe trimérique constitué de la kinase CDK7 et des protéines Cycline H et MAT1 (Ménage à
Figure 7 : Inhibition des complexes CDK-Cycline par les CKI
Les CKI inhibent les CDK soit par une liaison directe avec le complexe CDK-Cycline, soit par une compétition avec la Cycline.
29 Trois) (Harper and Elledge, 1998). La déphosphorylation semble ensuite être assurée par les phosphatases de la famille PP2C et PP2A (Cheng et al., 2000; Lee et al., 1994; Lorca et al., 1992).
(ii) Phosphorylations inhibitrices
Les complexes CDK-Cycline impliqués dans le contrôle du cycle cellulaire sont inhibés par une phosphorylation de la kinase CDK au niveau du résidu tyrosine 15 (Y15) fortement conservé au cours de l’évolution (Gu et al., 1992). Chez les eucaryotes supérieurs, un deuxième site de phosphorylation sur la thréonine 14 (T14) est aussi impliqué dans l’inactivation du complexe et semble simplement renforcer l’inactivation de CDK1 (Krek and Nigg, 1991; Norbury et al., 1991). Les T14 et Y15 sont toutes les deux situées dans le site catalytique de la kinase à proximité du site de liaison de l’ATP sur CDK1 et leurs phosphorylations empêcheraient la fixation de l’ATP ou la reconnaissance du substrat (Berry and Gould, 1996; Morgan, 1997). Wee1 est la kinase responsable de la phosphorylation sur la Y15 de CDK1 dans les cellules en interphase afin d’empêcher l’activation du complexe CDK1-Cycline B avant l’entrée en mitose (Russell et al., 1989). Chez les vertébrés la kinase Myt1, une kinase transmembranaire localisée au niveau du réticulum endoplasmique apparentée à la famille de Wee1, phosphoryle aussi bien la T14 et la Y15 (Booher et al., 1997; Mueller et al., 1995). La modulation du taux de phosphorylation sur ces deux sites est particulièrement importante pour l’activation du complexe CDK1- Cycline B à l’entrée de la mitose, mais participe également au contrôle de l’activation des CDKs à la transition G1/S ainsi qu’en phase S.
(iii) Déphosphorylations activatrices
La Y15 et la T14 de CDK1 sont déphosphorylées par les phosphatases de la famille Cdc25, qui sont conservées au cours de l’évolution. Trois isoformes ont été identifiés chez les mammifères : Cdc25A, Cdc25B et Cdc25C, et feront l’objet de la partie suivante de l’introduction générale.
I-2-B-d : Les autres mécanismes de régulation
Suc1 identifiée chez Schizoaccharomyces pombe est une petite protéine adaptatrice qui appartient à la famille des Cks (CDC kinase subunit) et a la particularité de pouvoir s’associer aux CDKs. Les Cks sont conservées de la levure jusqu’à l’homme (Cksh1 et Cksh2), et malgré la structure cristallographique du complexe Cks1-CDK2 (Jeffrey et al., 1995), leurs rôles restent mal connus. Il semblerait cependant que la liaison de Cks1 créé un site de liaison aux phosphoprotéines pour le complexe CDK-Cycline et aurait un rôle activateur lors de l’entrée en mitose et inhibiteur en fin de mitose (Harper, 2001; Pines, 1996).
Enfin, plus récemment, il a été montré chez le xénope que les CDKs pouvaient être activées par leur association avec les protéines RINGO (Rapid Inducer of G2/M progression in Oocytes) sans recourir à une liaison avec une Cycline (Nebreda, 2006). Ces protéines sont nécessaires à la
Figure 8 : Régulation du complexe CDK1-Cycline B
L’activation complète du complexe CDK1-Cycline B nécessite en plus de la déphosphorylation des résidus Y15 et T14, préalablement phosphorylés par Wee1, la phosphorylation par la CAK du résidu T161 dans la séquence T-loop.
31 maturation de l’ovocyte et peuvent s’associer à CDK1 et CDK2 et l’interaction avec RINGO suffit à la pleine activation de CDK1-RINGO et CDK2-RINGO sans avoir recours à la phosphorylation par la CAK sur les thréonines 161 et 160 respectivement (Ferby et al., 1999; Lenormand et al., 1999).
I-3 : Les phosphatases Cdc25
La complémentation fonctionnelle du mutant Cdc25 chez la levure à fission Schizosaccharomyces pombe a permis de mettre en évidence une protéine jouant un rôle majeur dans le Cycle de Division Cellulaire (Cdc25) (Russell and Nurse, 1986). La délétion de Cdc25 dans la levure entraine une absence de division cellulaire accompagnée d’une croissance anormale de la cellule. Cela suggère que Cdc25, fonctionnant comme un antagoniste de Wee1, qui est un inhibiteur de la progression dans le cycle cellulaire (voir plus haut), est essentielle pour l’entrée en mitose (Gould and Nurse, 1989; Russell and Nurse, 1986). Plusieurs études ont permis de caractériser Cdc25 comme une phosphatase impliquée dans la déphosphorylation activatrice du résidu tyrosine 15 de la kinase CDK1 : (i) tout d’abord la preuve phénotypique que Cdc25 était un antagoniste de la protéine kinase Wee1 ; (ii) l’accumulation de complexes CDK1-Cycline B inactifs car phosphorylés sur la tyrosine 15 corrélée à l’augmentation du taux de phosphotyrosine dans les cellules délétées pour Cdc25 ; (iii) la mutation du résidu tyrosine 15 de CDK1 en phénylalanine qui rend les cellules de levure résistantes à la délétion de Cdc25 (Gould and Nurse, 1989) et (iv) l’ajout de Cdc25 purifiée à des extraits d’ovocyte de xénope qui induit leur entrée en mitose accompagnée d’une déphosphorylation du résidu Y15 de CDK1 et d’une augmentation de l’activité du complexe CDK1-Cycline B (Jessus and Beach, 1992; Kumagai and Dunphy, 1991). Mais il fut montré que le produit du gène cdc25 était en réalité une phosphatase atypique. Cdc25 est en effet capable de déphosphoryler à la fois un résidu tyrosine et un résidu thréonine sur la même molécule (Millar and Russell, 1992). Elle a donc été classée au sein d’une sous famille : les phosphatases à double spécificité ou DSPases (Dual Specificity Phosphatase) qui appartiennent à la famille des phosphatases à spécificité tyrosine (PTPases).
Il a été ensuite clairement établi dans les cellules humaines que Cdc25 catalyse la réaction de déphosphorylation des résidus tyrosine 15 et thréonine 14 de CDK1 préalablement phosphorylés par les kinases Wee1 et Myt1 (Honda et al., 1993; Sebastian et al., 1993). Trois homologues de Cdc25 : Cdc25A, Cdc25B et Cdc25C ont été identifiés chez les mammifères et plus
particulièrement chez l’homme soit par complémentation fonctionnelle dans la souche de levure Cdc25-22TS
, soit par PCR avec des amorces dégénérées dirigées contre les séquences conservées entre les Cdc25 (Galaktionov and Beach, 1991; Nagata et al., 1991; Sadhu et al., 1990).
Les trois homologues humains complémentent avec succès la souche mutante de levure Cdc25-22TS
. Il est maintenant connu que leur expression est dépendante du cycle cellulaire dans les cellules de mammifères : l’ARNm de Cdc25C est majoritairement exprimé au cours des phases G2 et M, l’expression de l’ARNm de Cdc25B oscille tout au long du cycle cellulaire et est prédominante en phase G2, enfin l’expression de l’ARNm de Cdc25A oscille aussi au long du cycle cellulaire avec un pic d’expression au cours des phases G1 et S (Jinno et al., 1994; Sadhu et al., 1990)(Nagata 1991).
Figure 9 : Les phosphatases Cdc25 au cours du cycle cellulaire
Les phosphatases Cdc25A, B et C sont impliquées dans la progression du cycle cellulaire par la déphosphorylation activatrice des CDK. Chaque phosphatase Cdc25 participe à plusieurs étapes clés cycle cellulaire.
33
I-3-A : Présentations des phosphatases Cdc25 humaines
La majorité des travaux qui ont été abordés ici se rapportera aux phosphatases Cdc25 humaines. Ceux qui concernent d’autres organismes, comme la levure ou le xénope seront présentés soit pour appuyer les résultats obtenus dans les cellules humaines, soit pour apporter des données complémentaires.
Les trois isoformes humains Cdc25 sont codés par trois gènes situés sur des chromosomes différents Cdc25A localisé en 3p21, Cdc25B en 20p13 et Cdc25C en 5q31. La famille des Cdc25 humaines est encore compliquée par un épissage alternatif qui génère au moins deux variants pour Cdc25A (Wegener et al., 2000) et cinq de chaque pour Cdc25B (Baldin et al., 1997b; Forrest et al., 1999) et Cdc25C (Bureik et al., 2000; Wegener et al., 2000). Bien que le rôle exact de ces différents variants d’épissage soit inconnu, il semblerait que dans les cas de Cdc25A et Cdc25C, les variants d’épissage jouent un rôle différent en fonction des lignées cellulaires ou diffèrent dans leur distribution au sein du cycle cellulaire. De plus l’épissage alternatif pourrait éliminer des sites consensus de phosphorylation spécifiques, soumettant ainsi les variants d’épissage à des mécanismes de régulation différents. La délétion de tels motifs chez Cdc25B conduit à une diminution de son activité phosphatase et l’activité de Cdc25C a été décrite comme étant augmentée à la suite de phosphorylation (Baldin et al., 1997b; Gabrielli et al., 1997; Theis-Febvre et al., 2003; Wegener et al., 2000).
Au niveau protéique, les DSPases Cdc25 sont structuralement divisées en deux domaines majeurs : un domaine carboxy-terminal hautement conservé représentant environ 30% de la protéine et délimité par le motif LIGD ; et un domaine amino-terminal dont la taille varie et qui est peu conservé entre les trois isoformes humains (Draetta and Eckstein, 1997). Le cœur catalytique des Cdc25 constitué du motif canonique des PTPases HCX5R (ou X représente
n’importe quel acide-aminé) est situé dans le domaine carboxy-terminal. Ainsi, alors que le domaine carboxy-terminal des Cdc25 renferme la machinerie catalytique de l’enzyme, le domaine amino-terminal est perçu comme la région régulatrice de la protéine contenant de nombreux sites de phosphorylation qui peuvent agir soit comme des activateurs, soit comme des inhibiteurs de l’enzyme (Bulavin et al., 2002; Draetta and Eckstein, 1997; Lyon et al., 2002; Nilsson and Hoffmann, 2000).
Les structures des cristaux des domaines catalytiques de Cdc25A et Cdc25B ont été résolus à 2,3Å et 1,9Å respectivement mais aucune structure de Cdc25 pleine taille n’a été décrite. Les deux phosphatases contiennent le motif consensus HCX5R caractéristique du site catalytique des
PSPases, niché au sein du motif structurel “P-loop” typique des phosphatases à spécificité tyrosine (Fauman et al., 1998; Reynolds et al., 1999; Zhang, 1998). Les données de structure semblent appuyer les études biochimiques qui suggèrent que dans le cas de Cdc25B sa spécificité avec le substrat est régulée par sa queue carboxy-terminale de 17 acides-aminés et que Cdc25A nécessite un degré de promiscuité plus élevé pour la sélection de son substrat (Wilborn et al., 2001; Xia et al., 1999).
I-3-B : Caractérisation des phosphatases Cdc25 humaines
I-3-B-a : Le rôle des phosphatases Cdc25 dans le cycle cellulaire
Les rôles de Cdc25A, Cdc25B et Cdc25C dans les différentes phases du cycle cellulaire ont été bien étudiés (figure 9).
La principale fonction attribuée à Cdc25A est de promouvoir la transition G1/S du cycle cellulaire et la progression en phase S par la déphosphorylation activatrice des complexes CDK2-Cycline E et CDK2-CDK2-Cycline A. En effet la surexpression de Cdc25A dans les cellules humaines accélère l’entrée en phase S avec une activation prématurée de la kinase CDK2, alors que la micro-injection d’anticorps dirigés contre Cdc25A dans des cellules synchronisées en phase G1 provoque un arrêt du cycle cellulaire (Blomberg and Hoffmann, 1999; Hoffmann et al., 1994; Jinno et al., 1994). Cependant le niveau protéique de Cdc25A ainsi que son activité restent élevés après la phase S et augmentent jusqu’à l’entrée des cellules en mitoses où Cdc25A est dégradée (Bernardi et al., 2000; Molinari et al., 2000), suggérant un rôle à la transition G2/M. En fait c’est le complexe CDK1-Cycline B1 qui, en phosphorylant Cdc25A, permet sa stabilisation jusqu’à l’entrée des cellules en mitose (Mailand et al., 2002). De plus l’inhibition de l’expression de Cdc25A par ARN interférence entraîne une diminution du nombre de cellules entrant en mitose alors que sa surexpression conduit à une entrée prématurée des cellules en mitose par déphosphorylation du complexe CDK1-Cycline B1 (Lindqvist et al., 2005; Mailand et al., 2002).
L’inhibition de Cdc25B par la micro-injection d’anticorps conduit à un arrêt des cellules en phase G2 (Honda et al., 1993; Lammer et al., 1998). Ainsi Cdc25B a été initialement décrite comme étant impliquée dans l’entrée des cellules en mitose en déphosphorylant le complexe CDK1-Cycline B1 (Gabrielli et al., 1997). De plus, Cdc25B est capable de déphosphoryler
35 d’autres complexes CDK-Cycline dont la spécificité est dépendante du cycle cellulaire : CDK2-Cycline A en phase S (Goldstone et al., 2001) (Lammer et al., 1998) et CDK1-CDK2-Cycline A en phase G2 (Sebastian et al., 1993) (Lammer et al., 1998). Enfin une autre étude a proposé un rôle de Cdc25B en phase S (Garner-Hamrick and Fisher, 1998).
Il a tout d’abord été admis que Cdc25C induisait l’entrée des cellules en mitose et catalysait la progression mitotique par la déphosphorylation du complexe CDK1-Cycline B1 (Gautier et al., 1991; Lee et al., 1992; Millar et al., 1991b; Strausfeld et al., 1991). En effet la micro-injection d’anticorps dirigés contre Cdc25C conduit à l’arrêt des cellules en phase G2 (Millar et al., 1991a) et son activité phosphatase est maximale en ce point du cycle cellulaire (Hoffmann et al., 1993). De plus Cdc25C déphosphoryle uniquement le complexe CDK1-Cycline B1 (Gabrielli et al., 1997). Cependant des études plus récentes ont permis de montrer que Cdc25C jouerait également un rôle lors la transition G1/S (figure 9). En effet, la micro-injection d’ARN antisens dirigés contre l’ARNm codant pour Cdc25C ainsi que la transfection de cellules HeLa par des siRNA anti-Cdc25C provoque une inhibition de la réplication et un arrêt des cellules en phase S (Turowski et al., 2003). Enfin seule la surexpression de Cdc25B conduit à une entrée précoce des cellules en mitose, même si la réplication de l’ADN n’a pas été correctement accomplie, alors qu’il faut co-exprimer en même temps la Cycline B avec Cdc25C pour qu’une accélération de l’entrée en mitose soit observée (Gabrielli et al., 1996; Karlsson et al., 1999). Le modèle actuel présente donc Cdc25B et Cdc25A comme les activateurs initiaux du complexe CDK1-Cycline B1, tandis que Cdc25C permet la progression de la cellule en mitose, après son activation par CDK1-Cycline B1 (Lindqvist et al., 2005).
Des souris invalidées pour les gènes de cdc25b (Lincoln et al., 2002) et cdc25c (Chen et al., 2001) ont été générés. Il est intéressant de noter que ces souris sont viables, ont des cycles cellulaires normaux et répondent correctement aux points de contrôles (en réponse aux cassures double brin ou aux arrêts de la réplication). Seule les souris femelles cdc25b
sont stériles, leurs ovocytes sont bloqués en prophase de méiose I par leur incapacité à activer le MPF (Chen et al., 2001; Lincoln et al., 2002). Plus récemment des souris invalidées pour les deux gènes cdc25b et cdc25c ont été obtenues, de la même manière ces souris sont viables, répondent correctement aux points de contrôle et Cdc25A est régulée normalement (Ferguson et al., 2005). Ces résultats suggèrent que Cdc25B et Cdc25C ne sont pas requises pour le développement des souris ou lors de l’entrée en mitose, suggérant que peut être Cdc25A ou d’autres phosphatases sont capables de compenser fonctionnellement la perte de cdc25b et cdc25c chez les souris.
I-3-B-b : Régulation des phosphatases Cdc25
En tant qu’acteurs majeurs du cycle cellulaire, les phosphatases Cdc25 sont finement régulées au cours du cycle. Elles sont ainsi régulées par différents mécanismes qui contrôlent notamment leur localisation, leur activité catalytique et leur dégradation. Nous verrons dans la partie suivante de l’introduction générale que ces mécanismes de régulation sont mis à profit dans l’activation des points de contrôle lors de la réponse cellulaire aux dommages sur l’ADN.
(i) Cdc25A
L’activité et l’abondance de Cdc25A est régulée par des évènements de phosphorylation réversibles, des interactions protéine-protéine, ainsi qu’une protéolyse ubiquitine-dépendante (figure10) (Bernardi et al., 2000; Hoffmann et al., 1994; Mailand et al., 2002; Molinari et al., 2000) (Conklin et al., 1995).
La régulation subcellulaire de Cdc25A au cours de cycle cellulaire est contrôlée par un processus dynamique de navettes entre le noyau et le cytoplasme (Kallstrom et al., 2005). Cette dynamique est dépendante de la présence d’une NES (Nuclear Export Signal) qui a la caractéristique de ne pas se lier au facteur d’export nucléaire, l’exportine-1, et d’une NLS (Nuclear Localisation Signal) bipartite canonique KRX10-12KRRK (Kallstrom et al., 2005). A l’origine
Cdc25A était considérée comme une protéine strictement nucléaire, ce qui était en accord avec son activité d’activation des complexes nucléaires CDK2-Cycline A et CDK2-Cycline E en G1/S (Blomberg and Hoffmann, 1999). Cependant la mise en évidence de sa localisation également cytoplasmique montre un rôle de Cdc25A en mitose où elle déphosphorylerait les complexes CDK1-Cycline B1 (Lindqvist et al., 2005).
La phosphorylation de Cdc25A par le complexe CDK2-Cycline E au moment de l’entrée en phase S conduit à une augmentation de son activité phosphatase (Hoffmann et al., 1994). Cette phosphorylation de Cdc25A par son propre substrat crée une boucle de rétrocontrôle positif qui conduit à l’augmentation de l’activité des complexes CDK-Cyclines lors de la transition G1/S. Mais la phosphorylation par CDK2-Cycline E et/ou CDK2-Cycline A pourrait également avoir un contrôle négatif sur la phosphatase Cdc25A (Ducruet and Lazo, 2003). De plus au début de la mitose, CDK1-Cycline B phosphoryle Cdc25A sur les résidus S18 et S116, conduisant à une
37 stabilisation de la protéine par un mécanisme inconnu qui empêcherait son ubiquitinylation (Mailand et al., 2002).
Au cours du cycle cellulaire Cdc25A est dégradée par l’intermédiaire de deux ubiquitine-ligases différentes. A la sortie de mitose Cdc25A est ubiquitinylée par le cyclosome ou APC (Anaphase Promoting Complex) en association avec la protéine Cdh1. Cdh1 reconnaît le motif KEN de Cdc25A, alors que la mutation de ces résidus inhibe l’ubiquitinylation par l’APC/C-Cdh1. Par contre en interphase, Cdc25A est ubiquitinylée indépendamment du motif KEN par le complexe SCF (Skp1/Cullin/F-box) (Donzelli et al., 2002). La dégradation en phases S et G2 de Cdc25A est dépendante de la phosphorylation par la kinase Chk1. En effet en absence de dommages sur l’ADN, Chk1 phosphoryle les résidus : S124, S178, S279 et S293 (Sorensen et al., 2003; Zhao et al., 2002). Il semblerait qu’elle soit aussi responsable de la phosphorylation du résidu S76, alors que la protéine kinase responsable de la phosphorylation des acides-aminés S82 et S88 du motif DSG est encore inconnue. Ces phosphorylations sont nécessaires à la liaison de βTrCP permettant ensuite le recrutement du complexe ubiquitine-ligase SCF (Busino et al., 2004; Busino et al., 2003; Jin et al., 2003).
De plus il semblerait que Chk1 soit également responsable de la liaison aux protéines 14-3-3, impliquées dans la régulation et la localisation de Cdc25A au cours du cycle cellulaire. Cependant ces études sont controversées. Chk1 phosphoryle la thréonine 504 de xCdc25A et la thréonine 507 de hCdc25A, qui sont situées dans une région conservée en fin du domaine carboxy-terminal (Chen et al., 2003; Uto et al., 2004). Cette phosphorylation est responsable de la liaison aux protéines 14-3-3, cependant il apparaît que la liaison aux 14-3-3 ne provoque pas directement la diminution de l’activité de Cdc25A observée après la phosphorylation par Chk1 (Uto et al., 2004). La fin du domaine carboxy-terminal de Cdc25A et Cdc25B a été décrite comme le site de liaison aux substrats (Powers et al., 2000; Wilborn et al., 2001). Ainsi la phosphorylation de la thréonine 507 empêcherait la liaison de la phosphatase à ses substrats, conduisant à la diminution de son activité catalytique observée après la phosphorylation par Chk1 (Uto et al., 2004).
Figure 11 : Les principaux variants d’épissage de Cdc25B
Les trois isoformes de Cdc25B diffèrent par la présence ou l’absence des séquences codantes pour les domaines A et B de 14 et 41 acides aminés respectivement.
Figure 10 : Régulation de la phosphatase Cdc25A
Les phosphorylations de Cdc25A par Chk1 conduisent aux liaisons avec les 14-3-3. Cdc25A est stabilisée en mitose par des phosphorylations de CDK1. La dégradation de Cdc25A est dépendante de SCF/βTrCP qui induit son ubiquitinylation et de manière dépendante de la boite KEN, le complexe APC/C conduit également à la dégradation de Cdc25A à la suite de son ubiquitinylation en fin de mitose.
39
(ii) Cdc25B
Parmi les cinq variants d’épissage de Cdc25B dont les séquences sont accessibles dans les bases de données, trois variants ont été relativement bien décrits : Cdc25B1, Cdc25B2 et Cdc25B3 (Baldin et al., 1997b). Ils sont différenciés par la présence de deux domaines situés dans la région amino-terminale régulatrice : le domaine A qui est composé de 14 acides aminés et le domaine B de 43 acides aminés (figure 11). Ainsi le variant Cdc25B1 ne possède pas le domaine A, le variant B2 ne possède pas le domaine B et le variant B3 contient les deux domaines A et B. Ces trois variants ont été exprimés dans la levure à fission et sont capables de complémenter la souche de levure Cdc25-22TS
. C’est le variant Cdc25B2 qui présente la plus forte activité inductrice de mitose (Baldin et al., 1997b).
Cdc25B fait la navette entre le noyau et le cytoplasme au cours du cycle cellulaire. Cette variation de localisation s’explique par la présence d’une NES et d’une NLS bipartite située dans la région amino-terminale régulatrice de Cdc25B (Davezac et al., 2000; Lindqvist et al., 2004; Uchida et al., 2004) (figure 12). Ces signaux de localisation sont présents dans les différents variants de Cdc25B, cependant il semblerait que Cdc25B1 soit plutôt localisée dans le noyau où elle est active lors de l’entrée en mitose (Baldin et al., 2002), alors que Cdc25B3 doit être cytoplasmique pour entraîner les cellules en mitose (Lindqvist et al., 2004), suggérant ainsi une régulation de la localisation différente pour chaque variant d’épissage.
Trois sites de liaison aux protéines 14-3-3 sont présents dans le domaine régulateur amino-terminal de Cdc25B et sont impliqués dans la régulation de sa localisation et de son activité : RFQpS151MP, RPSpS230AP et RSPpS323MP (Giles et al., 2003; Uchida et al., 2004). La mutation
en alanine de la sérine 323 conduit à une inhibition de la liaison aux protéines 14-3-3 et entraîne une localisation strictement nucléaire de Cdc25B (Davezac et al., 2000), alors que les simples et doubles mutations en alanine des sérines 151 et 230 entraînent une augmentation de la localisation cytoplasmique de la phosphatase. La proximité de ces résidus par rapport aux séquences NES et NLS pourrait ainsi permettre aux protéines 14-3-3 de réguler la localisation intracellulaire de Cdc25B en bloquant la liaison de Crm1 ou des Importines à leur séquence de reconnaissance respective, NES ou NLS (Giles et al., 2003). Il semblerait que la liaison aux protéines 14-3-3 joue également un rôle dans la modulation de l’activité de Cdc25B en bloquant l’accès de Cdc25B à son substrat CDK1-Cycline B1 (Forrest and Gabrielli, 2001; Giles et al., 2003).
Il a d’autre part été montré que Cdc25B jouait un rôle dans l’activation initiale du complexe CDK1-Cycline B1 dans le cytoplasme des cellules, au niveau des centrosomes (Jackman et al., 2003; Lindqvist et al., 2005). De récentes études ont permis de montrer des événements de régulation positifs et négatifs de Cdc25B aux centrosomes. Tout d’abord la protéine Chk1 régule négativement la transition G2/M du cycle cellulaire en conduisant à une inhibition de l’activité des complexes CDK1-Cyclines B aux centrosomes (Kramer et al., 2004). Des travaux de notre équipe ont montré que Cdc25B3 était phosphorylée par Chk1 in vivo sur son résidu thréonine 563 (homologue du résidu T507 sur Cdc25A) et sur la sérine 230 aux centrosomes (homologue de la S178 de Cdc25A) (Schmitt et al., 2006). Les acides aminés S151 et S323 sont également phosphorylés in vitro par la kinase Chk1 (Schmitt et al., 2006) et Cdc25B3 portant une phosphorylation sur la S323 est localisée aux centrosomes (Lemaire et al., 2006). La T563 de Cdc25B est localisée dans le domaine carboxy-terminal, qui est bien conservé avec Cdc25A, et permet la liaison aux substrats. Ainsi la phosphorylation par Chk1 de la T563 conduit à une inhibition de la liaison de Cdc25B avec les cyclines A1, B1 et E1 (Uto et al., 2004). La protéine kinase mitotique Aurora A phosphoryle et active Cdc25B3 sur le résidu sérine 353 au niveau des centrosomes, ce qui a pour conséquence le recrutement et l’activation des complexes CDK1-Cycline B lors de l’entrée des cellules en mitose (Dutertre et al., 2004) (Hirota et al., 2003). Enfin, la protéine kinase Eg3 est responsable de la phosphorylation de la S169 de Cdc25B3 qui est située dans le domaine B (Mirey et al., 2005). Cette phosphorylation a été détectée au niveau des centrosomes, elle n’a pas d’effet sur l’activité catalytique de Cdc25B in vitro et son mécanisme d’action au sein de la cellule reste encore à déterminer. Cependant la co-expression des deux protéines dans les cellules humaines montre qu’Eg3 a un effet négatif sur l’entrée des cellules en mitose et que cet effet est antagonisé par Cdc25B (Davezac et al., 2002), ainsi Eg3 pourrait agir comme un régulateur négatif de Cdc25B.
D’autre part la localisation cellulaire de Cdc25B est également régulée par la phosphorylation de la sérine 353 par la protéine kinase PKB/Akt. En effet, la surexpression d’une forme constitutivement active de PKB/Akt dans les cellules humaines conduit à une relocalisation cytoplasmique de Cdc25B dépendante du facteur d’export nucléaire Crm1 (Baldin et al., 2003). La protéine kinase CK2 (Caséine Kinase 2) qui interagit avec Cdc25B par sa sous unité β, phosphoryle les résidus S186 et S187, ce qui se traduit par une augmentation de l’activité phosphatase de Cdc25B observée in vitro mais aussi in vivo (Theis-Febvre et al., 2003). Des boucles d’amplifications positives et négatives impliquant Cdc25B et les complexes CDK-Cyclines ont été décrites. CDK1-Cycline A phosphoryle Cdc25B1 et induit sa dégradation de
41 manière dépendante du protéasome en phase G2 du cycle cellulaire (Baldin et al., 1997a). Le complexe CDK1-Cycline B1 phosphoryle, lui aussi, Cdc25B1 sur le résidu sérine 146, situé dans le domaine B (sérine 160 de Cdc25B3), ce qui entraîne une inhibition de son export nucléaire mais n’a pas d’effet sur l’activité phosphatase de Cdc25B1 (Baldin et al., 2002). Il a d’autre part été montré au laboratoire que CDK1-Cycline B1 phosphoryle in vitro de nombreux sites dans la région régulatrice amino-terminale de Cdc25B (Bouché JP, en préparation). Parmi ces sites, la phosphorylation in vivo du résidu thréonine 69, situé dans le domaine A de Cdc25B et donc absente de Cdc25B1, contribuerait à l’augmentation de l’activité de Cdc25B2 et Cdc25B3 en mitose (Izumi and Maller, 1993; Margolis et al., 2006).
Figure 12 : Régulation de la phosphatase Cdc25B
Cdc25B est régulée négativement par Chk1 et Eg3. La phosphorylation sur les S151, S230 et S323 par Chk1 conduit à la liaison avec 14-3-3 et une diminution de l’activité phosphatase. Aurora A régule positivement Cdc25B à l’entrée en mitose et il semblerait que Cdc25B soit également phosphorylée par CDK1.
Enfin Cdc25B est une protéine instable qui est dégradée au cours de la mitose par le protéasome (Baldin et al., 1997a). Des études récentes réalisées dans notre laboratoire ont permis de montrer que les différents variants de Cdc25B étaient dégradés de manière différentielle dépendant du domaine B. Ainsi les variants Cdc25B1 et Cdc25B3 sont dégradés suivant un mécanisme dépendant du complexe SCF/βTrCP entre la transition métaphase-anaphase de la mitose (Kieffer et al., 2007).
(iii) Cdc25C
L’activité de Cdc25C doit être finement régulée afin de permettre la progression des cellules en mitose de manière rapide et irréversible. La présence d’une NES dépendante du facteur d’export nucléaire Crm1 et d’une NLS bipartite canonique au niveau de la région amino-régulatrice de Cdc25C permettent à la phosphatase de réaliser une navette continue entre le cytoplasme et le noyau de la cellule (Graves et al., 2001; Yang and Kornbluth, 1999). En réalité, en interphase Cdc25C est phosphorylée sur la sérine 216 et est majoritairement cytoplasmique et devient majoritairement nucléaire lors de l’entrée en prophase (Dalal et al., 1999). Cette phosphorylation de la S216 crée un site de liaison aux protéines 14-3-3 qui cache probablement la séquence de localisation nucléaire et maintient la phosphatase sous une forme inactive dans le cytoplasme (Dalal et al., 1999; Graves et al., 2001; Peng et al., 1997). Cette interaction entre 14-3-3 et Cdc25C, cruciale pour le contrôle de l’entrée en mitose, est dépendante de la phosphorylation de la S216. En effet, la mutation en alanine de la S216 empêche la liaison avec 14-3-3 et conduit à l’observation de cellules présentant une condensation prématurée de la chromatine (Graves et al., 2001). En absence de dommages sur l’ADN la protéine kinase C-TAK1 (Cdc Twenty-five C Associated Kinase 1) est responsable de la phosphorylation de ce résidu (figure13) (Peng et al., 1998). Cependant il est aussi possible que Chk1 soit impliquée dans cet événement de phosphorylation en raison de son rôle joué lors de la régulation du cycle cellulaire en absence de dommages sur l’ADN. Lors de l’entrée en mitose, la dissociation de la liaison entre Cdc25C et 14-3-3 permet l’import nucléaire de la phosphatase où elle peut activer pleinement les complexes CDK1-Cycline B1. Cdc25C est alors activée par CDK1-Cycline B1 à travers une boucle de retro-contrôle positif (Bulavin et al., 2003a; Bulavin et al., 2003b). Ainsi CDK1 phosphoryle le résidu sérine 214 très proche de la sérine 216 et empêche la phosphorylation de la S216. Des études réalisées chez le xénope ont permis de montrer que Cdc25C et 14-3-3 sont
43 dissociées après la phosphorylation de la T138. Le site S285 (S214 de hCdc25C) est alors accessible à la phosphorylation par la kinase CDK1, accélérant considérablement le recrutement de la phosphatase PP1 qui induit la déphosphorylation de la S216 et entraîne l’entrée des cellules en mitose (Margolis et al., 2006; Margolis et al., 2003). Cdc25C est également régulée par un processus d’isomérisation dépendant de la protéine Pin1 (Stukenberg and Kirschner, 2001). Pin1 est une peptidyl-proline isomérase qui interagit par son domaine WW avec Cdc25C phosphorylée par CDK1 au niveau des motifs pS/pTP (Crenshaw et al., 1998; Shen et al., 1998) (Zhou et al., 2000). La résultante de la conversion peptidique des liaisons S/TP induit un changement conformationnel de la phosphatase accompagné de la déphosphorylation par PP2A de motifs phosphorylés par CDK1, inhibant ainsi Cdc25C (Zhou et al., 2000).
Enfin d’autres phosphorylations sont impliquées dans la régulation de Cdc25C. Ainsi la protéine kinase CK2 qui phosphoryle le résidu thréonine 236 très proche de la NLS, inhibe la liaison aux importines α/β et prévient l’import nucléaire de Cdc25C (Schwindling et al., 2004). La protéine kinase CaMKII (Calcium calmoduline-dependent protein Kinase II), dont les sites de phosphorylation ne sont pas encore identifiés, permet l’augmentation très nette de l’activité phosphatase de Cdc25C (Patel et al., 1999). De la même manière la phosphorylation en amino-terminal de Cdc25C par Pim1 entraîne une augmentation de son activité lors de la progression G2/M (Bachmann et al., 2006). Cdc25C est aussi phosphorylée par les membres de la famille Plk (Polo kinase). En effet la sérine 198 est phosphorylée in vitro par Plk1 et Plk3 (Toyoshima-Morimoto et al., 2002) et la sérine 191 est elle aussi phosphorylée par Plk3 (Bahassi el et al., 2004). Ces études montrent que les phosphorylations engendrées par les kinases Plk conduisent à une accumulation nucléaire de Cdc25C suggérant un rôle important des Plk lors du contrôle de la translocation nucléaire de Cdc25C lors de l’entrée de la cellule en mitose (Bahassi el et al., 2004) (Toyoshima-Morimoto et al., 2002).
Il a aussi été démontré chez l’ovocyte de xénope que les kinases Erk/MAPK étaient impliquées dans la phosphorylation de trois résidus de Cdc25C lors de la transition G2/M, associées à une augmentation de son activité (Wang et al., 2007). Ces résidus sont hautement conservés chez la phosphatase humaine : T48, T67 et S168, et ont été montrés, en plus des résidus S122 et T130, comme étant phosphorylés in vivo lors de l’entrée en mitose (Izumi and Maller, 1993; Strausfeld et al., 1994). Il semblerait que ces sites soient phosphorylés par CDK1 lors de la boucle d’auto-amplification entre Cdc25C et CDK1-Cycline B1 à l’entrée en mitose.
Enfin une étude récente a permis de montrer qu’un événement de phosphorylation était impliqué dans la dégradation de Cdc25C. La sérine 216, phosphorylée par la kinase p90ARF