THÈSE
Pour l'obtention du grade de
DOCTEUR DE L'UNIVERSITÉ DE POITIERS UFR de médecine et de pharmacie
Institut de physiologie et biologie cellulaires - IPBC (Poitiers) (Diplôme National - Arrêté du 25 mai 2016)
École doctorale : Biologie-santé - Bio-santé (Limoges)
Secteur de recherche : Recherche clinique, innovation thérapeutique, santé publique
Présentée par :
Marine Legendre
Le syndrome CHARGE : étude clinique et moléculaire
Directeur(s) de Thèse :
Brigitte Gilbert-Dussardier, Frederic Bilan
Soutenue le 14 décembre 2016 devant le jury
Jury :
Président Damien Sanlaville Professeur et praticien hospitalier, Université de Lyon Rapporteur Damien Sanlaville Professeur et praticien hospitalier, Université de Lyon Rapporteur Didier Lacombe Professeur et praticien hospitalier, Université de Bordeaux Membre Brigitte Gilbert-Dussardier Professeure et praticienne hospitalière, Université de Poitiers
Membre Frederic Bilan Docteur, Université de Poitiers
Membre Tania Attie-Bitach Professeur et praticienne hospitalière, Université Descartes, Paris
Pour citer cette thèse :
Marine Legendre. Le syndrome CHARGE : étude clinique et moléculaire [En ligne]. Thèse Recherche clinique, innovation thérapeutique, santé publique. Poitiers : Université de Poitiers, 2016. Disponible sur Internet <http://theses.univ-poitiers.fr>
Université de Poitiers
Faculté de Médecine et Pharmacie
ANNEE 2016
THESE
En vue de l’obtention du grade de
DOCTEUR DE L’UNIVERSITE DE POITIERS
Ecole Doctorale Biosante
Champ disciplinaire : Biologie, Me decine, Sante
Secteur de Recherche : Recherche clinique, innovation the rapeutique, sante publique soutenue
le 14 De cembre 2016 a Poitiers par Marine LEGENDRE devant la commission d’examen
Le Syndrome CHARGE :
Etude clinique et moléculaire
RAPPORTEURS : Monsieur le Professeur Damien Sanlaville, Universite Claude Bernard Lyon 1
Monsieur le Professeur Didier Lacombe, Universite de Bordeaux
C
OMPOSITION DUJ
URY :Rapporteur : Monsieur le Professeur Damien Sanlaville, Universite Claude Bernard Lyon 1
Examinateurs : Madame le Professeur Tania Attie -Bitach, Universite Paris 5
Madame le Professeur Brigitte Gilbert-Dussardier, Universite de Poitiers Monsieur le Docteur Fre de ric Bilan, Universite de Poitiers
Co- Directeurs de thèse : Madame le Professeur Brigitte Gilbert-Dussardier Monsieur le Docteur Fre de ric Bilan
2
Université de Poitiers
Faculté de Médecine et Pharmacie
ANNEE 2016
THESE
En vue de l’obtention du grade de
DOCTEUR DE L’UNIVERSITE DE POITIERS
Ecole Doctorale Biosante
Champ disciplinaire : Biologie, Me decine, Sante
Secteur de Recherche : Recherche clinique, innovation the rapeutique, sante publique soutenue
le 14 De cembre 2016 a Poitiers par Marine LEGENDRE devant la commission d’examen
Le Syndrome CHARGE :
Etude clinique et moléculaire
RAPPORTEURS : Monsieur le Professeur Damien Sanlaville, Universite Claude Bernard Lyon 1
Monsieur le Professeur Didier Lacombe, Universite de Bordeaux
C
OMPOSITION DUJ
URY :Rapporteur : Monsieur le Professeur Damien Sanlaville, Universite Claude Bernard Lyon 1
Examinateurs : Madame le Professeur Tania Attie -Bitach, Universite Paris 5
Madame le Professeur Brigitte Gilbert-Dussardier, Universite de Poitiers Monsieur le Docteur Fre de ric Bilan, Universite de Poitiers
Co- Directeurs de thèse : Madame le Professeur Brigitte Gilbert-Dussardier
4
R É S U M É
Le syndrome CHARGE est une association malformative rare due a une mutation du ge ne CHD7 dans 60 a 90% des cas. L’objectif de ce travail e tait d’en de crire les e le ments cliniques et mole culaires afin d’optimiser la prise en charge de patients atteints d’un handicap multisensoriel lourd.
Le diagnostic ante natal en est difficile et l’e tude de 40 fœtus a permis d’affiner la description du phe notype, de de crire de nouveaux e le ments cliniques et finalement de proposer un ajustement des crite res diagnostiques chez le fœtus.
L’e tude endocrinienne de 42 patients confirme la pre sence d’un hypogonadisme hypogonadotrope dans 97% des cas. Souvent me connu et non traite il peut avoir des conse quences de le te res sur la qualite de vie. Nous proposons qu’il soit reconnu comme crite re majeur du syndrome.
L’e tude clinique d’une cohorte française de 119 patients a montre que la surdite et l’atteinte des canaux semi circulaires sont les e le ments les plus fre quents du syndrome (suivis de l’atteinte hypophysaire, de l’arhinence phalie et des anomalies de l’oreille externe), et les seuls significativement associe s a la pre sence d’une mutation dans CHD7. Une e tude approfondie des donne es issues de cette e tude est en cours.
Sur le plan mole culaire, alors que la plupart des mutations identifie es sont des mutations tronquantes prive es apparues de novo, nous avons identifie 4 variants au niveau de l’intron 25, re currents pour certains, dont l’interpre tation e tait de licate. Leur e tude in silico puis par une technique de minige ne a permis de mettre en e vidence une configuration particulie re des se quences implique es dans l’e pissage de l’exon 26 (point de branchement distant) et de de montrer leur pathoge nicite .
5
R É S U M É E N A N GL A I S
CHARGE syndrome is a rare disorder of multiple congenital anomalies ascribed to a CHD7 gene mutation in 60% to 90% of cases. The aim of this study was to improve the knowledge regarding molecular and clinical aspects of the syndrome in order to optimize the management of these patients with severe disability.
Antenatal diagnosis remains challenging in many instances and a detailed clinicopathological survey in a series of 40 fetuses allowed us to refine the clinical description of CHARGE syndrome in fetuses, describe some novel features and set up diagnostic criteria.
An endocrinologic study of 42 patients showed a hypogonadotrophic hypogonadism in 97% of cases. For this reason, it should be considered as a major symptom of the syndrome. An early screening should lead to a hormonal replacement therapy which dramatically impacts the condition.
A study of a French cohort of 119 patients found that deafness and semi-circular canals hypoplasia were the most frequent symptoms (followed by hypogonadotrophic hypogonadism, arhinencephaly and external ears anomalies) and the only features statistically associated with a mutation in the CHD7 gene. A detailed study of the data is still going on.
The syndrome is mainly due to de novo and private truncating mutations of the CHD7 gene but we report an intriguing hot spot of intronic mutations located in IVS25. Combining computational in silico analysis and ex vivo minigene assays, we explained this mutation hot spot by a particular genomic context, including a distant branch point, and confirmed the pathogenicity of these mutations.
6
R E M E RC I EM E N TS
Je tiens tout d’abord a exprimer mes since res remerciements a Messieurs les
Professeurs Didier Lacombe
etDamien Sanlaville
pour l’inte re t qu’ils ont porte a ce travail en acceptant d’e tre rapporteurs de cette the se et membre du jury. Ils sont des mode les et leur jugement reve t une importance particulie re pour moi.A Madame le Professeur Gilbert-Dussardier
Je vous remercie since rement d’avoir accepte de codiriger cette the se. Je vous suis reconnaissante de votre investissement dans ce travail qui fut une grande source de motivation et facilita beaucoup de choses. Je ne serais pas la ge ne ticienne que je suis sans vos enseignements, vos encouragements et votre soutien dans mes diffe rents projets professionnels. Je sais ce que je vous dois, merci.
A Monsieur le Docteur Bilan
A toi qui as accepte de codiriger cette the se avec une patience admirable et une disponibilite de chaque instant, j’adresse mes remerciements les plus vifs. Et bien au-dela de cette the se, ces 9 anne es (et oui, quand me me !) d’apprentissage et de collaboration furent riches d’enseignements, de bonne humeur et de beaux projets ; pour tout cela aussi je te remercie.
A Madame le Professeur Attié-Bitach
Une partie des plus passionnantes de mon histoire avec le syndrome CHARGE s’est e crite entre les murs de ton laboratoire. Je te remercie de ton investissement et de ton soutien qui m’auront permis d’emmener « nos fœtus » jusqu'a San Francisco. J’ai beaucoup aime travailler avec toi, j’ai appris e norme ment. Au terme de ce travail, merci de me faire a nouveau le plaisir et l’honneur de ta pre sence.
7
Je remercie très sincèrement toutes les personnes qui ont participé à
ce travail et l’ont ainsi rendu possible :
Je te moigne toute ma gratitude a l’ensemble des médecins ; Annie Harroche, Christel Thauvin, Christine Coubes, Ge raldine Viot, Brigitte Simon-Bouy, Dominique Martin-Coignard, Marion Ge rard, Bertrand Isidor, Lucille Pinson, Claire Beneteau, Laurence Olivier Faivre, Estelle Colin, Christine Coubes, Sylvie Odent, Jean Chiesa, Patricia Blanchet, Sandrine Marlin, Dominique Gaillard, Joelle Roume, Martine Le Merrer, Jeanne Amiel, Laurent Pasquier, Dominique Boggio, Stanislas Lyonnet, Vale rie Cormier-Daire Annick. Toutain, Patrice. Rodien, Regis Coutant, Sabine Baron Nathalie Magontier, Marc. De Kerdanet, Florence Compain, Richard Mare chaud, Massimiliano Rossi, Ve ronique Abadie, Nicole Revencu, Patricia Blanchet, Fre de ric Brioude, Marie-Ange Delrue, Yassamine Doubaj, Abdelaziz Sefiani, Christine Francannet, Muriel Holder-Espinasse, Pierre-Simon Jouk, Sophie Julia, Judith Melki, Se bastien Mur, Sophie Naudion, Jennifer Fabre-Teste, Tiffany Busa, Sophie Blesson, Chloe Que lin, Me lanie Fradin, Aure lia Jaquette, Sandra Mercier, Didier Lacombe, Se bastien Moutton, Eric Bieth, Alexandre Buffet, MathieuGautier, Nicole Philip, David Genevieve, Pierre Sarda, Sabine Sigaudy, Miche le Mathieu –Dramard, Anne-Marie Guerrot, Catherine Vincent-Delorme, Vale rie Layet, Renaud Touraine, Alice Masurel, Salima El Chehadeh, Bruno Leheup, He le ne Dolfus, Marie Gonzales , Pauline Parisot Marie-Jose e Perez, Maryse Bonnie re, Bettina Bessie res, Jelena Martinovic, Anne Lise Delezoide, Fre derique Jossic, Catherine Fallet-Bianco, Martine Bucourt, Julia Tantau, Philippe Loget, Laurence Loeuillet, Nicole Laurent, Brigitte Leroy, Houria Salhi, Nicole Bigi, Caroline Rouleau, Fabien Guimiot, Anne Bazin, Caroline Alby, Yves Ville , Ferechte Razavi, Michel Vekemans qui nous ont adresse s les patients.
Montserrat Rodriguez – Ballesteros
Un immense merci a toi pour ta patience d’ange, tes explications mais bien plus que cela pour l’e norme quantite de travail que tu as fourni pour obtenir des re sultats et pour ton investissement (et ce n’est pas peu dire) dans ce travail de the se.
Je n’oublie pas de remercier Géraldine Goudefroye, Amale Ichkou, Nadia Elkhartoufi, Roselyne Gesny et Barbara Manière pour leur aide, leur sympathique accueil et le travail qu’elles effectuent quotidiennement pour les fœtus, les patients et les familles. Je remercie Marie Laure Rodes qui a collecte la majeure partie des donne es endocrinologiques.
Je remercie since rement madame Stéphanie Ragot qui s’est montre tre s investie et tre s re active dans notre inte ressante collaboration. Sa participation fut d’une grande aide.
8 Je remercie Chantal Buet et Nathalie Klein qui ont eu la charge de la partie du travail qui n’e tait pas la plus simple. De l’art d’harceler les PU de France et de Navarre avec tact a celui de de busquer les erreurs dans des masses de donne es ; je leur tire mon chapeau ! Guylène Page
Merci beaucoup pour tes pre cieux conseils. Ton investissement pour la Recherche est contagieux !
Merci a Priscilla HAMIAUX pour son travail sur le versant neuropsychologique du syndrome CHARGE, la combinaison de nos travaux devrait apporter d’inte ressantes perspectives.
Xavier Piguel
Je te remercie vivement pour tous les e claircissements que tu as pu m’apporter sur le versant endocrinologique de cette the se et pour ton implication. Travailler avec toi fut un plaisir !
Ce travail a e te supporte par le financement du Projet Hospitalier de Recherche Clinique « Etude clinique mole culaire et neuro-comportementale du syndrome CHARGE »
Je tiens à remercier tout(e) celles et ceux qui ont fait partie de ces
années « CHARGEES »
Alain Kitzis
Je vous remercie de m’avoir accueillie dans votre laboratoire et de m’avoir soutenue durant ces riches anne es de formations.
Je remercie Dominique Couet pour sa disponibilite , sa gentillesse et son enseignement lors de mon passage dans son service de cytoge ne tique.
Merci a toute l’équipe du laboratoire de génétique de Poitiers et de l’EA 3808 pour leur accueil et leur bonne humeur. Le travail avec eux au quotidien est un bonheur ! Je remercie e galement toute l’e quipe de l’unite INSERM U781 pour leur accueil et leurs enseignements.
Merci a toute ma bulle professionnelle au sein du service de ge ne tique clinique :
Aurore qui subit de longue date mes soupirs derrie re le chaos qui re gne sur mon bureau et qui sait, a l’oreille, de tecter le moment ou un carre de chocolat serait le bienvenu ! Karine dont j’ai souvent use des talents quand la pression montait ! Me laisser contaminer par sa joie de vivre fut la meilleure prescription. Et puis paraitrait que mon cas n’est pas comple tement de sespe re , ouf !
9 Audrey avec qui j’ai partage la prise en charge des familles. Les pauses autour d’un cafe (ou d’une bassine de the en ce qui me concerne) sont, dans notre sous-sol, de vrais rayons de soleil.
Amandine, Lydie et Céline, qui n’ont jamais sourcille quand je leur demandais pour la nie me fois de me sortir un dossier qui e tait (forcement) de ja dans mon bureau…
Victoria, qui fait chanter un petit morceau d’Ame rique du sud dans le service.
Gwenaël, nos quelques grammes de testoste rone dans un monde de filles ! Acolyte momentane ment en exil (sur une plane te lointaine ?) : reviens vite, c’est a ton tour de refiler a quelqu’un d’autre le remplissage de CIROCO !
Manon et bien qu’elle m’ait abandonne e dans les derniers me tres (sous le pre texte tout a fait honteux de soit-disant « vacances »…) : tu as pris le pli a la vitesse de l’e clair et m’as quasi remplace e quand le devoir m’appelait ailleurs, c’est rien de dire que je te remercie !
Merci e galement a Xavier d’avoir pris le relais au pied leve , contraint de faire son bapte me du feu en solitaire et a Julien pour les petits coups de main qui font toute la diffe rence.
Bien su r je n’oublie pas le passe et celles et ceux qui, en leur temps, eurent toute leur importance : Bénédicte ; un amour de colle gue, la rocambolesque Chantal ; notre maman a toutes, Virginie ; ma p’tite sœur (il manque un lama pre t de moi depuis que tu bosses chez Renault !), Christelle ; la premie re coloc’, Anaelle et sa bonne humeur, Radu qui n’a pas re siste a l’appel des pistes, Marie avec qui j’ai pu partager mon inte re t (souvent perçu comme un peu bizarre) pour les fœtus, Pierre et ses pommes, Clara, Charlotte, …
10
Et parce que je ne serai rien sans eux, je les remercie :
Je remercie ma famille pour son soutien inde fectible et la ressource qu’ils repre sentent pour moi.
Je remercie surtout mon « petit » frère, de sormais « tonton Oignon », parce que nos discussions me font sentir plus le ge re ; elles ont toujours un parfum de madeleine (… de Proust, sous forme de malabars bi-gout a l’arrie re de la BX) !
Un infini merci a mes parents, toujours la lorsque je jetais un œil par-dessus mon e paule au moment de passer les portes qui se sont pre sente es devant moi. Ils n’avaient peut-e tre pas pre vu que j’aurais encore une carte d’e tudiant a 34 ans ! Quand on aime on ne compte pas mais, cette fois ci, (a priori…) c’est fini !
Je remercie mes amis, ceux de toujours et les suivants et surtout Aurélie, Bapt, Geoffrey et Fabiola, Groulty, Cathy, Fabien, Raph, parce qu’il n’y a pas que le syndrome CHARGE dans la vie!
Je remercie Raphaëlla et Lucile, mes acolytes du M6, pour le souvenir de mon anne e parisienne, M6 forever !
A nouveau, ce travail je le de die a Vincent, infaillible Porte-Bonheur de mes travaux et de mes jours, sauveur attitre de cette the se pour son ro le dans la mise en page et motivateur inconditionnel. Je ne sais pas ce que je serais sans toi… et c’est parfait comme ca !
A la plus passionnante de mes « collaborations » qui a vu naitre, un vendredi 13, le plus parfait des ouvrages…
11
S O M M A I R E
RÉSUMÉ ... 4
RÉSUMÉ EN ANGLAIS ... 5
SOMMAIRE ... 11
LISTE DES TABLEAUX ... 15
LISTE DES FIGURES ... 16
ABREVIATIONS ... 17
INTRODUCTION... 20
I. CLINIQUE DU SYNDROME CHARGE ... 21
1. Historique ... 21
2. Epidémiologie ... 21
3. Signes cliniques du syndrome CHARGE ... 21
4. Diagnostics différentiels ... 25
5. Critères diagnostiques du syndrome CHARGE ... 28
II. GENETIQUE DU SYNDROME CHARGE... 29
1. Historique ... 29
2. Le gène CHD7 ... 31
3. La protéine CHD7 ... 31
OBJECTIF DE LA THESE ... 33
PARTIE 1 ... 37
ANTENATAL SPECTRUM OF CHARGE SYNDROME IN 40 FETUSES WITH CHD7 MUTATIONS ... 38
I. ABSTRACT ... 40
II. INTRODUCTION ... 40
III. MATERIAL AND METHODS ... 41
1. Fetuses ... 41
2. CHD7 sequence analysis ... 42
IV. RESULTS ... 42
1. CHD7 mutations ... 46
12
V. DISCUSSION ... 51
1. Molecular analysis ... 51
2. Clinical signs ... 52
3. Male to female ratio ... 54
4. Fetuses without CHD7 mutation/deletion ... 55
5. Major features of CHARGE syndrome at fetal or neonatal autopsy ... 58
PARTIE 2 ... 61
HYPOGONADOTROPHIC HYPOGONADISM AS A MAJOR SIGN IN CHARGE SYNDROME? AN ENDOCRINOLOGICAL STUDY OF 42 PATIENTS ... 62
I. ABSTRACT ... 64
II. INTRODUCTION ... 64
III. METHODS AND PATIENTS ... 65
IV. RESULTS ... 67
V. DISCUSSION ... 69
VI. CONCLUSION ... 73
PARTIE 3 ... 74
CHARGE SYNDROME: AN INTRIGUING RECURRENT HOTSPOT OF MUTATIONS IN CHD7 IVS25 ANALYZED BY BIO-INFORMATICS TOOLS AND MINIGENE ASSAYS. ... 75
I. ABSTRACT ... 77
II. INTRODUCTION ... 78
III. MATERIAL AND METHOD ... 79
1. Patients ... 79
2. Software prediction tools ... 79
3. Generation of minigene reporter ... 83
4. Lariat RT-PCR to determine the branch point ... 87
5. Minigene product semi-quantitative analysis using fluorescent liquid chromatography ... 88
IV. RESULTS ... 89
1. Clinical findings ... 89
1. In silico splice site prediction ... 95
2. Minigene product splicing assays ... 97
13
4. CHD7 IVS25 belongs to the “AG independent” intron class... 106
V. DISCUSSION ... 108
1. Pathogenic effects of variants ... 108
2. CHD7 IVS25 harbors a distant branch point ... 109
3. “AG” introducing process is probably the main mechanism of CHD7 IVS25 hot spot of mutations ... 110
4. Phenotypic variability ... 110
PARTIE 4 ... 113
ETUDE CLINIQUE ET MOLECULAIRE DU SYNDROME CHARGE – DONNEES ISSUES DU PHRC ... 114
I. INTRODUCTION... 114
II. MATERIEL ET METHODES ... 114
1. Objectifs de l’étude ... 114
2. Patients ... 115
3. Etude statistique ... 117
4. Elaboration du questionnaire en ligne (e-crf) ... 117
5. Etude moléculaire ... 119
6. Classification des mutations selon leur impact sur la protéine CHD7 ... 120
III. RESULTATS ... 120
1. Etude Clinique de la cohorte de patients présentant un syndrome CHARGE ... 120
2. Etude moléculaire de la cohorte de patients atteints de syndrome CHARGE (Tableau 21, p129) ... 138
DISCUSSION ET PERSPECTIVES ... 144
DISCUSSION ... 145
I. LE DIAGNOSTIC ANTENATAL DE SYNDROME CHARGE ... 145
II. ETUDE DES CRITERES DIAGNOSTIQUES ... 148
1. Etude descriptive des éléments phénotypiques ... 148
2. Comparaison des patients atteints de formes typiques vs atypiques ... 150
3. Leçon des patients ne remplissant pas les critères diagnostiques du syndrome (patients exclus) ... 152
4. Evaluation de la pertinence des critères diagnostiques ... 153
14
1. Etude phénotypique selon le sexe (Tableau 26, p154) ... 156
2. Recherche de facteurs pronostiques... 157
IV. ETUDE MOLECULAIRE ET CORRELATION GENOTYPE/PHENOTYPE ... 158
1. Etude des mutations de CHD7 ... 158
2. Comparaison des patients avec et sans mutation dans CHD7 ... 159
3. Comparaison phénotypique des patients porteurs d’une mutation tronquante ou non tronquante ... 160
4. Les formes familiales ... 161
5. EFTUD2 ... 163
V. LIMITES DE L’ETUDE... 164
PERSPECTIVES ... 166
I. ETUDE PAR EXOME DES PATIENTS SANS CAUSE MOLECULAIRE IDENTIFIEE ... 166
II. ETUDE PAR MINIGENE DES MUTATIONS POTENTIELLEMENT IMPLIQUEES DANS L’EPISSAGE. .... 167
CONCLUSION ... 168
REFERENCES ... 172
ANNEXES ... 186
ANNEXE 1 - SURVEILLANCE DES PATIENTS ATTEINTS DE SYNDROME CHARGE D’APRES BERGMAN ET AL.(6) ... 187
ANNEXE 2 - LISTE DES PUBLICATIONS ... 190
Publications ... 190
Communications orales ... 190
Posters avec comité de révision ... 191 ANNEXE3 - CAHIER D’OBSERVATION DU PHRC ... ERREUR!SIGNET NON DEFINI.
ANNEXE4 - ANTENATAL SPECTRUM OF CHARGE SYNDROME IN 40 FETUSES WITH CHD7 MUTATIONS
15
L I ST E D E S TA B L EAUX
TABLEAU 1 :CRITERES DIAGNOSTIQUES CLINIQUES DU SYNDROME CHARGE SELON BLAKE ET SELON VERLOES (46). ... 29
TABLEAU 2:SUMMARY OF THE CLINICAL AND GENETICS DATA OF THE 40CHARGE FETUSES WITH CHD7 MUTATION ... 43
TABLEAU 3:DESCRIPTION OF HEART DEFECTS IN THE 40 FETAL/NEW BORNS CASES WITH CHD7 MUTATION ... 48
TABLEAU 4:DESCRIPTION OF CENTRAL NERVOUS SYSTEM ANOMALIES IN THE 40 FETAL/NEW BORN CASES WITH CHD7 MUTATION ... 49
TABLEAU 5:COMPARISON OF THE FREQUENCY OF POOR PROGNOSIS FACTORS IN MALES AND FEMALES ... 55
TABLEAU 6:SUMMARY OF THE CLINICAL DATA OF THE 20 FETUSES WITHOUT CHD7 MUTATION ... 56
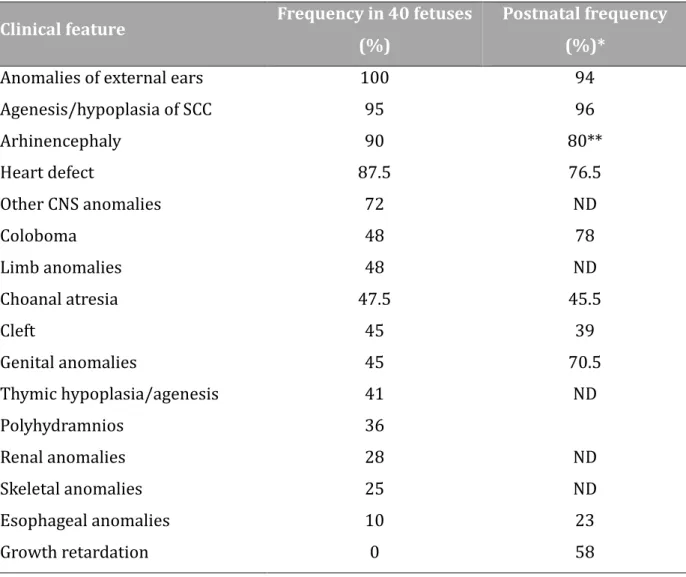
TABLEAU 7:CLINICAL FEATURES IN CHD7 MUTATED FETUSES AS COMPARED WITH POSTNATAL CASES ... 58
TABLEAU 8:DIAGNOSTIC CRITERIA FOR CHARGE SYNDROME AT FETAL OR NEONATAL AUTOPSY ... 59
TABLEAU 9:VERLOES’ DIAGNOSTIC CRITERIA SYSTEM ... 66
TABLEAU 10:FREQUENCY OF PITUITARY DEFECTS IN CHARGE SYNDROME. ... 71
TABLEAU 11:BIOINFORMATIC ON LINE TOOLS USED FOR THIS STUDY (THEIR UNIFORM RESOURCE LOCATOR AND THEIR REFERENCE) ... 80
TABLEAU 12:DNA SEQUENCE OF OLIGONUCLEOTIDES USED FOR THIS STUDY ... 87
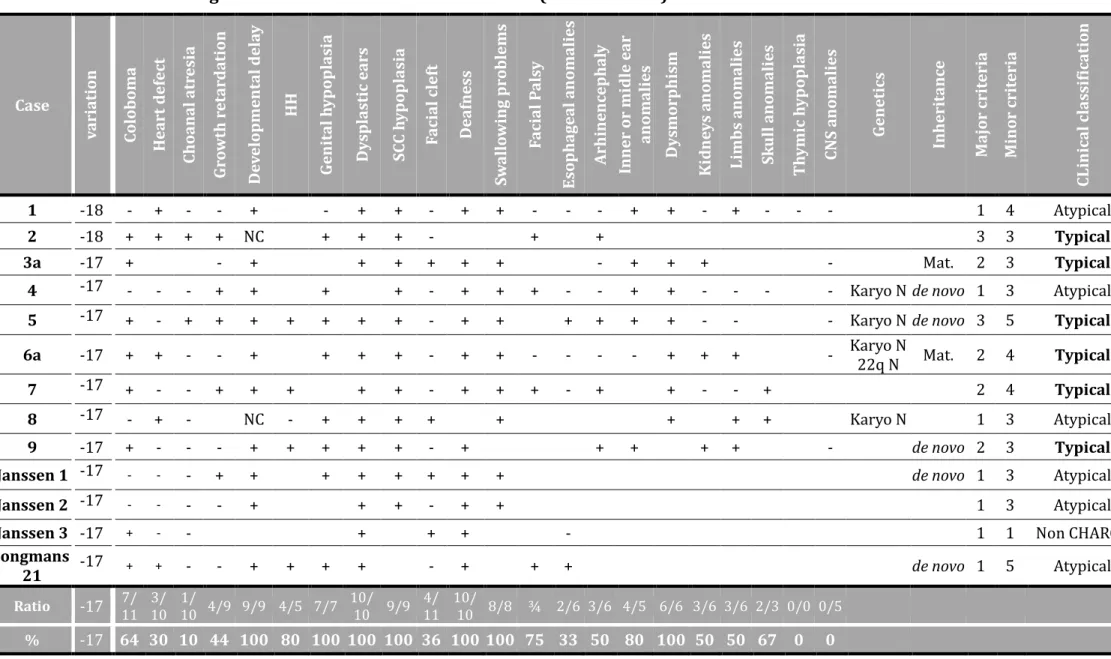
TABLEAU 13:CLINICAL AND GENETIC DATA OF INDEX CASES IN OUR SERIES (CASES 1A TO 20) AND IN LITERATURE. ... 91
TABLEAU 14:CLINICAL DATA OF FAMILIAL CASES. ... 94
TABLEAU 15:IVS25 ACCEPTOR AND DONOR SPLICE SITE ANALYSIS USING DIFFERENT PREDICTION SOFTWARE.EVALUATION OF C.5405-2A>G, C.5405-7G>A, C.5405-13G>A, C.5405-17G>A AND C.5405-18C>A MUTATIONS IMPACT UPON SPLICING. ... 96
TABLEAU 16:BRANCH POINT ANALYSIS USING SVM PREDICTION TOOL DEPENDING ON THE IVS25 SEQUENCE ANALYZED.106 TABLEAU 17:CRITERES D’INCLUSION ET DE NON INCLUSION A L’ETUDE ... 115
TABLEAU 18:CRITERES DIAGNOSTIQUES DE SYNDROME CHARGE RETENUS POUR L'ETUDE. ... 116
TABLEAU 19:FREQUENCE DES CRITERES DIAGNOSTIQUES CHEZ LES PATIENTS ATTEINTS DE SYNDROME CHARGE ... 129
TABLEAU 20:FREQUENCE COMPAREE DES ELEMENTS CLINIQUES EN FONCTION DE LA PRESENCE OU NON D'UNE MUTATION IDENTIFIEE DANS CHD7 ... 130
TABLEAU 21:PHENOTYPE DES PATIENTS ATTEINTS DE SYNDROME CHARGE AVEC UNE MUTATION DANS LE GENE CHD7 131 TABLEAU 22:PHENOTYPE DES PATIENTS ATTEINTS DE SYNDROME CHARGE SANS ANOMALIES IDENTIFIEE DANS LE GENE CHD7 ... 135
TABLEAU 23:FREQUENCE COMPAREE DES ELEMENTS CLINIQUES EN FONCTION DE LA CLASSIFICATION DU PHENOTYPE ... 136
TABLEAU 24:MUTATIONS EXCLUES DE LA CLASSIFICATION TRONQUANTE /NON TRONQUANTE... 141
TABLEAU 25:MUTATIONS IDENTIFIEES COMME NON TRONQUANTES ... 142
TABLEAU 26:FREQUENCE COMPAREE DES ELEMENTS CLINIQUES EN FONCTION DU SEXE DES PATIENTS ... 156
TABLEAU 27:FREQUENCE COMPAREE DES ELEMENTS CLINIQUES EN FONCTION DU CARACTERE TRONQUANT OU NON DE LA MUTATION ... 161
16
L I ST E D E S F I G U R E S
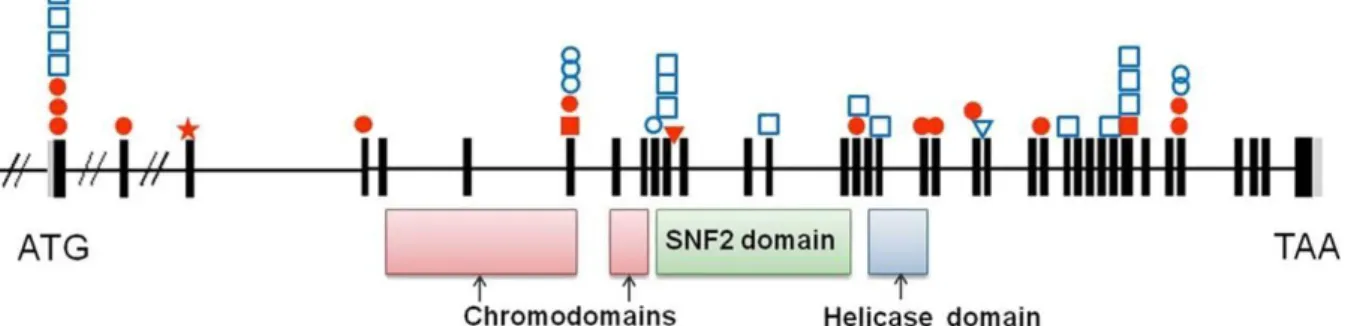
FIGURE 1:REPRESENTATION OF THE CHD7 MUTATIONS IDENTIFIED IN OUR SERIES. ... 46
FIGURE 2:TYPICAL CLINICAL FEATURES IN FETAL CHARGE SYNDROME. ... 51
FIGURE 3:THE DIFFERENT STEPS OF MINIGENE GENERATION. ... 86
FIGURE 4:CHD7IVS25 RECURRENT MUTATION ANALYSIS BY MINIGENE ASSAYS. ... 99
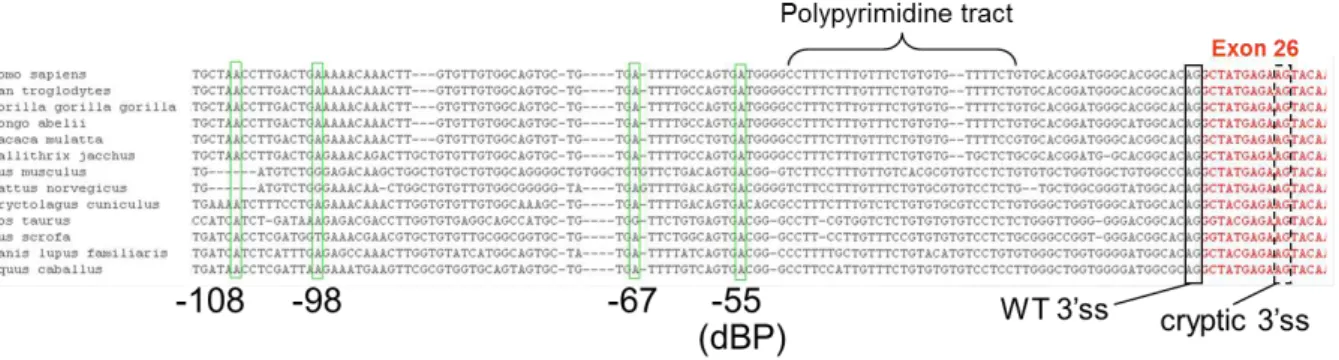
FIGURE 5:GENOMIC SEQUENCE ALIGNMENT OF CHD7IVS25 ACROSS DIFFERENT MAMMALIAN SPECIES.NUCLEOTIDE NUMBERING IS BASED UPON THE HUMAN SEQUENCE. ... 100
FIGURE 6:CHD7IVS25 BRANCH POINT DETERMINATION BY LARIAT RT-PCR AND MINIGENE ASSAYS. ... 105
FIGURE 7:TARGETED MUTAGENESIS OF CRITICAL SITE OF CHD7IVS25 MAY EXPLAIN THE MUTATIONAL HOT SPOT. ... 107
FIGURE 8:FAMILY TREE OF CASES 2A AND 2B ON THE LEFT -5A,5B,5C AND 5D ON THE RIGHT ... 111
FIGURE 9 :ELEMENTS MORPHOLOGIQUES A DIFFERENTS AGES DES PATIENTS TYPIQUES, PORTEURS D'UNE MUTATION DANS CHD7. ... 127
FIGURE 10:ELEMENTS MORPHOLOGIQUES DU PATIENT PORTEUR D'UNE TRANSLOCATION DESEQUILIBREE T(3 ;14)... 127
FIGURE 11:ELEMENTS MORPHOLOGIQUES DES PATIENTS DONT L'ORIGINE MOLECULAIRE DU SYNDROME CHARGE N'EST PAS CONNU.LES PATIENTS 29001 ET 1009 PRESENTENT UNE INCISIVE MEDIANE UNIQUE. ... 128
FIGURE 12:REPRESENTATION DU TAUX DE PATIENTS MUTES DANS LE GENE CHD7 EN FONCTION DE LEUR PHENOTYPE .... 137
FIGURE 13:ELEMENTS MORPHOLOGIQUES DE PATIENTS PORTEUR D'UNE FORME ATYPIQUE DE SYNDROME CHARGE A DIFFERENTS AGES. ... 137
FIGURE 14:REPRESENTATION DES MUTATIONS DE CHD7 IDENTIFIEES DANS NOTRE COHORTE. ... 140
FIGURE 15:REPRESENTATION DU TYPE DE MUTATION DANS LE GENE CHD7 EN FONCTION DU PHENOTYPE TYPIQUE OU ATYPIQUE DES PATIENTS ... 143
17
A B R E V I AT I O N S
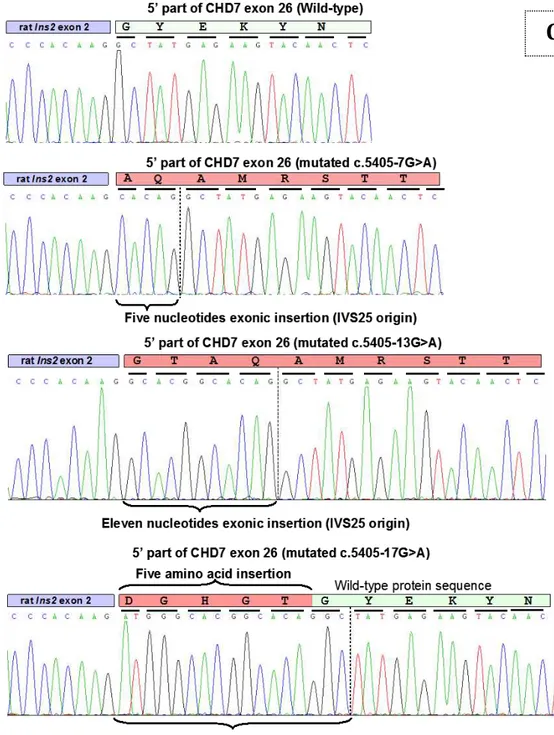
-13 C.5405-13G>A -17 C.5405-17G>A 22q FISH 22q11.2 -7 C.5405-7G>AACTH Adre no Cortico Trophic Hormone
AGEZ Ag Exclusion Zone An Anamnios
ASSP Alternative Splice Site Predictor
BP Branch Point
bp Base Pair
BS Branch Site
CAV Canal Atrio-Ventriculaire
CdP Cellules De Purkinje
CESSA Centre d’Education Spe cialise Pour Enfants Sourd Aveugles Et Sourds Malvoyants
cDNA Complementary Deoxyribonucleic Acid
CGH Comparative Genomic Hybridization
CHD7 Chromodomain Helicase DNA Binding Protein 7
CIA Communication Inter Auriculaire
CIV Communication Inter Ventriculaire
CLAD Centres Labellise s Anomalies Du De veloppement
CNS Central Nervous System
CS CHARGE Syndrome
CSC Canaux Semi Circulaires
dBP Distant Branch Point
dbsnp Single Nucleotide Polymorphism Database
dHPLC Denaturing High Performance Liquid Chromatography
DAN Diagnostic Ante natal
DMF-GA Dysostose Mandibulo-Faciale De Type Guion-Almeida
DNA Deoxyribonucleic Acid
DORV Double Outlet Right Ventricle
DS Deviation Standard
e-crf Electronic Case Report Form
EFTUD2 Elongation Factor Tu Gtp Binding Domain Containing 2 ERG E lectrore tinogramme
ESEs Exonic Splicing Enhancers ESSs Exonic Splicing Silencers ExAC Exome Aggregation Consortium F Fe minin
18
FO Fond D’œil GH Growth Hormone
HH Hypogonadisme Hypogonadotrope / Hypogonadotropic Hypogonadism
HSF Human Splicing Finder
F Fe minin / Female
IGF 1 Insulin-Like Growth Factor 1
IRM Imagerie Par Re sonnance Magne tique
IHH Hypogonadisme Hypogonadotrophique Idiopathique Idiopatic Hypogonadotropic Hypogonadism
IMC Indice De Masse Corporel
IMG Interruption Me dicale De Grossesse
ISEs Intronic Splicing Enhancers ISSs Intronic Splicing Silencers IUGR Intrauterine Growth Restriction
IVS Intervening Sequence
KS Kallmann Syndrome
LH Luteinizing Hormone
LHRH Luteinizing Hormone Releasing Hormone
M Masculin / Male
Mat Maternelle
MLPA Multiplex Ligation-Dependent Probe Amplification
MRI Magnetic Resonance Imaging
N Normal
NC Non Concerne ND Not Determined
NMD Nonsense Mediated Decay
niHH Non Idiopatic Hypogonadotropic Hypogonadism NR Non Renseigne / not relevant
OGE Organes Ge nitaux Externes
ORL Oto-Rhino-Laryngologiste
Patho. Pathologique
PC Pe rime tre Cranien
PCR Polymerase Chain Reaction PEA Potentiels Evoque s Auditifs
PEV Potentiels Evoque s Visuels
PPT Polypyrimidine Tract PF Paralysie Faciale
PHRC Programme Hospitalier De Recherche Clinique Prot. Prote ique
RCIU Retard De Croissance Intra Ute rin
19
RT PCR Reverse Transcription Polymerase Chain Reaction RVU Reflux Ve sico - Ure te ral
SA Semaines D’ame norrhe e
SC Syndrome CHARGE
SCC Semicircular Canals
SD Standard Deviation
SK Syndrome De KALLMANN
SNC Syste me Nerveux Central
SNP Single-Nucleotide Polymorphism
SROOGLE Splicing Regulation On Line Graphical Engine SS Site D’e pissage (Splicing Site)
SVM Support Vector Machine TC Tronc Ce re bral
TPO-Abs Thyroperoxidase (Tpo) Antibodies
TSH Thyreostimulating Hormone
VDDI Ventricule Droit A Double Issue
WG Weeks Of Gestation.
20
21
I N T RO D U CT I O N
I.
C
LINIQUE DU SYNDROMECHARGE
1. Historique
Le syndrome CHARGE (OMIM 214800) est une affection ge ne tique rare qui fut de crite pour la premie re fois par deux e quipes inde pendantes en 1979. A l’e poque, l’e quipe de Hall (1) de crivait une cohorte de 17 patients pre sentant l’association re currente d’une atre sie des choanes et de multiples anomalies quand Hittner et al. (2) rapportaient chez neuf cas index un nouveau syndrome constitue d’une microphtalmie colobomateuse, d’une cardiopathie, d’une paralysie faciale, de troubles fonctionnels pharynge s, d’une surdite et d’un de ficit intellectuel.
Par la suite, Pagon et al. (3) pre cise rent la description clinique et propose rent l’acronyme anglais C.H.A.R.G.E afin de rappeler les diffe rents signes de l’association a savoir un Colobome (C), une cardiopathie (Heart), une Atre sie des choanes (A), un Retard de croissance ou de de veloppement (R) et des anomalies Ge nitales (G) et de l’oreille externe et/ou une surdite (Ear).
2. Epidémiologie
L’incidence du syndrome est estime e entre 1/10.000 et 1/17.000 (4,5). Quelques rares cas familiaux et l’a ge paternel e leve sugge raient une he re dite dominante.
3. Signes cliniques du syndrome CHARGE
L’acronyme CHARGE
Un colobome uni ou bilate ral est retrouve dans 75 a 81% des cas (6). Lorsque le colobome implique la re tine, il peut s’e tendre au nerf optique et impacter la vision. Dans ce cas il pre dispose au de collement de re tine du fait de l’amincissement de la zone d’adhe sion a l’e pithe lium pigmente de la re tine.
Une asyme trie de taille entre les deux yeux est fre quente. En ge ne ral, une microphtalmie (mode re e a importante) est associe e a une atteinte de la macula, compromettant la vision.
22 Une cardiopathie est pre sente chez 76 a 77% des individus atteints de syndrome CHARGE (6). Ce sont des cardiopathies en ge ne ral complexes et tre s variables. Les anomalies conotroncales (te tralogie de Fallot, interruption de l’arche aortique, communication inter ventriculaire (CIV) infundibulaire, ventricule droit a double issue (VDDI) ou tronc arte riel commun), le canal atrioventriculaire (CAV) et les autres anomalies de l’arche aortique sont fre quents. Communication inter auriculaire (CIA), CIV non conotroncale et persistance du canal arte riel ont e galement e te de crites.
Une atrésie des choanes bilate rale est responsable d’une de tresse respiratoire ne cessitant des manœuvres de re animation imme diates a la naissance tandis que les formes unilate rales passent parfois inaperçues jusqu'a ce que l’enfant pre sente une rhinorrhe e unilate rale persistante.
Les enfants atteints de syndrome CHARGE naissent en ge ne ral avec une biome trie (taille et poids) normale. Le retard de croissance apparait plus tard au cours de la vie. Dans une e tude portant sur 25 enfants ayant un statut nutritionnel normal par ailleurs, Pinto et al. (7), de crivent une se cre tion normale d’hormone de croissance pour 22 d’entre eux et un de ficit en GH chez les 3 autres.
Le retard de développement est a la fois moteur et intellectuel dans le syndrome CHARGE.
Les enfants pre sentent en ge ne ral un grand retard dans les acquisitions motrices. Les hospitalisations prolonge es, l’hypotonie de l’axe et la laxite ligamentaire, les de ficits visuels et auditifs ainsi que l’atteinte vestibulaire participent aux difficulte s. Tellier et al. (8) rapportent un a ge moyen de la tenue de te te a 5 mois, de la tenue assise a 14.8 mois et de la marche sans aide a 33 mois (8).
Le de veloppement du langage est souvent retarde en raison de la perte auditive et ce, d’autant plus que la vision est e galement alte re e ce qui porte atteinte a la lecture labiale d’une part et la perception des indices du langage corporel d’autre part.
De s lors, l’e valuation des capacités cognitives est difficile en raison du manque d'outils standardise s pour e valuer les personnes ayant une de ficience a la fois visuelle et auditive. Raqbi et al. (9) ont montre que les performances intellectuelles des personnes atteintes du syndrome CHARGE e taient variables, allant des troubles majeurs d'apprentissage sans aucun langage jusqu’a des acquisitions presque normales. Selon cette e tude, la microce phalie, les malformations ce re brales et le colobome bilate ral e tendu (impactant la vision) sont les seuls facteurs pre dictifs d’un mauvais
23 de veloppement intellectuel. Les re sultats sugge rent que pour environ la moitie des enfants atteints du syndrome CHARGE, les retards moteur et du langage sont dus principalement aux de ficits sensoriels multiples et non a un dysfonctionnement du syste me nerveux central (SNC).
50% a 60% des patients de sexe masculin pre sentent une hypoplasie génitale a type de micrope nis ou de cryptorchidie. Wheeler et al (10) ont sugge re que l’hypogonadisme hypogonadotrope serait responsable non seulement de l’hypoplasie ge nitale chez le garçon mais aussi du de faut de de veloppement pubertaire dans les deux sexes. Dans cette e tude, les 9 patients e tudie s pre sentaient des taux anormalement bas de FSH et LH. La surdité est l’une des atteintes les plus fre quentes du syndrome. Elle peut e tre mode re e a profonde et il est parfois difficile de la quantifier, ne cessitant la re alisation ite rative de potentiels e voque s auditifs du tronc ce re bral. La composante perceptive de la surdite est souvent associe e a une malformation de Mondini de la cochle e et l’hypoplasie du nerf auditif a e galement e te de crite. La composante transmissionnelle de la surdite pourrait quant a elle re sulter d’une atteinte ossiculaire variable (11,12) et fluctuer notamment du fait d’otites chroniques fre quentes.
Les autres éléments cliniques du syndrome CHARGE
Au-dela des e le ments constitutifs de l’acronyme « CHARGE », plusieurs autres signes fre quents dans cette association ont e te rapporte s et certains sont de sormais tre s discriminants pour le diagnostic.
Ainsi, les examens d’imagerie approprie s permettent d’objectiver des anomalies des canaux semi-circulaires (CSC) chez 94 a 98% des individus atteints (6,8,11,13–16). L’absence ou l’hypoplasie des CSC est a l’origine de troubles de l’e quilibre, en particulier lorsqu'elle est associe e a un de ficit visuel, contribuant au de calage des acquisitions motrices.
Les anomalies cérébrales commune ment rapporte es comprennent l’arhinence phalie, l’age ne sie du corps calleux et des anomalies de la fosse poste rieure (8). Les anomalies des voies et des bulbes olfactifs variant de l’hypoplasie mode re e a l’aplasie comple te a
24 l’origine d’une hyposmie voire d’une anosmie ont e te signale es dans de nombreuses e tudes (7,17) et sont assez e vocatrices du diagnostic de syndrome CHARGE.
Une atrésie de l'œsophage ou une fistule trachéo-œsophagienne est pre sente chez environ 19% a 29% des nourrissons atteints du syndrome (6) et peut exacerber des difficulte s d'alimentation ou une de tresse respiratoire dans les premiers jours de vie. Un diagnostic pre coce et une prise en charge approprie e ame liorent conside rablement la survie.
Une paralysie faciale unilate rale ou bilate rale est pre sente dans 39 a 66% cas (6). Les formes bilate rales sont a l’origine d’une amimie pouvant retentir sur la communication interpersonnelle.
Les anomalies du tractus urinaire telles qu’un rein unique, une hydrone phrose ou une hypoplasie re nale sont pre sentes chez 25 a 40% des enfants atteints du syndrome CHARGE (18,19).
Des anomalies des membres sont observe es chez plus d'un tiers des individus atteints du syndrome CHARGE (20). Les anomalies les plus fre quemment rapporte es sont les hypoplasies ungue ales, les clinodactylies (du cinquie me doigt et du deuxie me orteil), la polydactylie, les contractures, la brachydactylie, les pieds bots, les anomalies tibiales et l’hyperlaxite articulaire.
Des luxations de hanche, des co tes manquantes et des anomalies verte brales ont aussi e te de crites. Ces dernie res peuvent e tre responsables de scolioses qui peuvent par ailleurs e tre d’origine neuromusculaire dans l’enfance.
Les anomalies dentaires comprennent les anomalies de l’articule e dentaire, un re trognatisme, une hypodontie affectant la dentition de finitive et une mauvaise mine ralisation de l'e mail (21).
Un déficit immunitaire peut e tre parfois observe dans le syndrome CHARGE sous la forme d’une se quence de DiGeorge a l’origine d’une lymphope nie le ge re a se ve re des cellules (22).
25 4. Diagnostics différentiels
Du fait de l’atteinte multisyste mique et de la grande variabilite phe notypique du syndrome CHARGE, les diagnostics diffe rentiels sont nombreux et de pendent du mode d’entre e dans le syndrome et des signes au premier plan (colobome, atre sie de l’œsophage, cardiopathie, surdite etc.…).
Les génopathies
La fente palatine
La microdélétion 22q11.2 est caracte rise e par une cardiopathie conge nitale, le plus souvent de type conotroncal, des anomalies du palais (insuffisance ve lo-pharynge e, fente palatine), des traits morphologiques caracte ristiques et des difficulte s d'apprentissage. S’y associent parfois un de ficit immunitaire, une hypocalce mie, des proble mes d’alimentation, des anomalies re nales, une perte auditive (transmission et/ou perception), des anomalies laryngo-trache o-œsophagiennes, un de ficit en hormone de croissance, des atteintes auto-immunes, des crises convulsives (en dehors d’e pisode d’hypocalce mie) et des anomalies squelettiques.
Bien que plusieurs caracte ristiques cliniques de la microde le tion 22q11.2 soient chevauchantes avec le syndrome CHARGE, les dysmorphies sont distinctes. Les anomalies des canaux semi-circulaires, l’atre sie des choanes ou le colobome oculaire qui sont fre quentes dans le syndrome CHARGE sont rares dans la microde le tion 22q11.2 et les difficulte s concernant l’alimentation durent ge ne ralement plus longtemps chez les enfants atteints du syndrome CHARGE (23). La difficulte du diagnostic clinique repose sur le fait que la se quence de DiGeorge existe en dehors de la pre sence d’une microde le tion 22q11.2 et peut e tre retrouve e chez des patients atteints du syndrome CHARGE (23).
L’existence chez des patients atteints de SC, de signes caracte ristiques de syndrome de DiGeorge (hypo/age ne sie thymique, hypocalce mie, de ficit immunitaire cellulaire) fait entrer ces patients dans une cate gorie nomme e syndrome CHARGE/DiGeorge (24,25).
L’atre sie des choanes
Dans sa forme syndromique l’atre sie des choanes peut faire e voquer une anomalie chromosomique, une atteinte du spectre des craniosténoses syndromiques ou le syndrome de Treacher Collins.
26 L’association Hypogonadisme / Arhinence phalie
Elle fait e voquer un syndrome de Kallmann. Bien que des mutations dans le ge ne CHD7 aient e te rapporte es chez des individus atteints du syndrome de Kallmann (hypogonadisme hypogonadotrophique et anosmie ou hyposmie)(26,27), on retrouve en ge ne ral chez ces individus des e le ments cliniques supple mentaires caracte ristiques du syndrome CHARGE et finalement ce diagnostic devrait e tre retenu dans ces cas. Le syndrome de Kallmann est ge ne tiquement he te roge ne avec 5 ge nes identifie s a ce jour. De manie re inte ressante, le chevauchement avec le syndrome CHARGE est particulie rement remarquable au locus KAL2 pour lequel une atre sie des choanes, une surdite , un retard de croissance, des fentes faciales, des anomalies des oreilles externes et des extre mite s et des colobomes sont possibles chez les patients mute s pour FGFR1 (28–32).
L’atteinte œsophagienne
L’association VACTERL est une combinaison d’anomalies verte brales (V), d’une atre sie anale (A), d’anomalies cardiaques (C), d’une fistule trache o-œsophagienne ou d’atre sie de l'œsophage (T) et d’anomalies re nales (R) et des membres (L pour limb). Le syndrome CHARGE partage avec l’association VACTERL les anomalies verte brales, les atteintes œsophagiennes, cardiaques et re nales mais s’en distingue par la pre sence de colobome, de l’anomalie caracte ristique de l’oreille externe et de l’atre sie choanale. L'anomalie de l’os temporal fre quemment observe dans le syndrome CHARGE est rarement rapporte e dans le VACTERL (33). L’association VACTERL survient habituellement de façon sporadique et la cause en est inconnue.
Les syndromes de Feingold et AEG (Anophthalmia-Esophageal-Genital) par mutation de SOX2 peuvent e galement e tre e voque s dans le cadre de ces malformations œsophagiennes syndromiques (34).
Plus re cemment la dysostose mandibulo-faciale de type Guion-Almeida, due a une mutation du ge ne EFTUD2, a e te de crite comme partageant de nombreux e le ments cliniques avec le syndrome CHARGE. Une surdite , une hypoplasie des canaux semi-circulaires, une atre sie des choanes ou une fente, des anomalies de l’oreille externe, une asyme trie de la face, une de ficience intellectuelle, une cardiopathie et une atteinte de l’appareil uroge nital ou une age ne sie des bulbes peuvent e tre retrouve es chez les patients atteints de ce type de dysostose mandibulo-faciale. En revanche il n’est pas
27 de crit de colobome chez ces patients qui pre sentent par ailleurs fre quemment une microce phalie, un retard de croissance intra-ute rin et une dysmorphie spe cifique dans tous les cas (35).
Les anomalies ophtalmologiques – le colobome oculaire
Des chevauchements entre le syndrome CHARGE et le syndrome de Kabuki tels que la pre sence d’une fente palatine, d’une cardiopathie, d’anomalies squelettiques mineures ou d’un colobome ont e te mis en e vidence (36,37). Ce sont les dysmorphies bien distinctes de chaque syndrome (longues fentes palpe brales avec e version de tiers late ral des paupie res infe rieures, sourcils clairseme s et grandes oreilles proe minentes) et la persistance de l’aspect fœtal, en baguette de tambour, des doigts dans le syndrome de Kabuki qui distinguent les deux entite s. Une mutation dans les ge nes KMT2D ou KDM6A, d’autres membres de la machinerie de modification de la chromatine, est identifie e chez plus de 56% - 76% des individus ayant un diagnostic clinique de syndrome de Kabuki (38–41).
Le syndrome rein - colobome cause par des mutations dans le ge ne PAX2, se caracte rise par des colobomes re tiniens ou du nerf optique, des anomalies re nales et une perte auditive inconstante mais sans les autres atteintes conge nitales multiples que l’on retrouve dans le syndrome CHARGE. Aucune mutation dans le ge ne PAX2 n’a pu e tre identifie e chez des individus atteints, d’un point de vue clinique, du syndrome CHARGE (42).
Le syndrome du « cat eye », caracte rise par la combinaison d’un colobome irien, d’une atre sie ou fistule anale, d’anomalies pre -auriculaires (sinus ou tubercules pre tragiens) et la survenue fre quente de malformations cardiaques et re nales, est cause par la pre sence d'un petit chromosome surnume raire de rive du chromosome 22 qui signe le diagnostic. Ce marqueur, le plus souvent dicentrique, porteur d’ADN satellites et en mosaï que, correspond a la duplication inverse e de la partie proximale d'un chromosome 22.
Les anomalies des canaux semi-circulaires
Enfin, l’association de malformations de l’oreille externe, d’hypoplasie du canal semi-circulaire late ral et d’anomalies re nales peut amener a conside rer le diagnostic de syndrome BOR (Branchio-Oto-Rénal, du a une mutation dans le ge ne EYA1) mais
28 l’absence de fistule et de kyste branchiaux et la pre sence d’autres crite res diagnostiques du syndrome CHARGE redresseront le diagnostic.
L’exposition aux tératogènes
L’embryopathie à l’acide rétinoïque est due a l’effet te ratoge ne du Roaccutane® (isotre tinoï ne) au cours du premier trimestre de grossesse, a l’origine de malformations associe es a une anomalie de la migration des cellules de la cre te neurale (microtie / anotie, micrognathie, fente palatine, malformations cardiaques conotroncales et anomalies de l’arche aortique, atteintes thymiques, re tiniennes ou du nerf optique et malformations du syste me nerveux central) (43). Bien que certains chevauchements existent, les nourrissons atteints d’embryopathie re tinoï que ne re pondent pas aux crite res diagnostiques du syndrome CHARGE.
L’embryopathie alcoolique partage avec le syndrome CHARGE l’atteinte cardiaque, oculaire et neurologique mais le RCIU, la microce phalie, la dysmorphie et l’absence d’anomalies de l’oreille externe et des CSC orientent le diagnostic lorsque la preuve d’une exposition a l’alcool ne peut e tre faite.
5. Critères diagnostiques du syndrome CHARGE
Initialement, le diagnostic du syndrome CHARGE reposait sur les e le ments mne motechniques de l’acronyme ; la pre sence chez un individu de quatre caracte ristiques parmi les six signait le diagnostic. Une meilleure connaissance du syndrome a peu a peu mis a jour la ne cessite d’affiner les crite res diagnostiques propose s par Pagon et al. (3), puis Mitchell et al. en 1985 (44). En 1998, un groupe d’experts a e tabli des crite res diagnostiques majeurs et mineurs pour le syndrome CHARGE (18).
Les crite res diagnostiques furent plusieurs fois rede finis (45,46) et aujourd’hui, les e tudes se re fe rent ge ne ralement aux crite res majeurs et mineurs de Verloes (46) ou Blake ((18) re vise s en 2006 (47)) (Tableau 1, p29).
29 Tableau 1 : Critères diagnostiques cliniques du syndrome CHARGE selon Blake et selon Verloes (46).
CRITERES MAJEURS CRITERES MINEURS CLASSIFICATION
D ’a pr ès V erloes 1. Colobome oculaire 2. Atre sie des choanes 3. Hypoplasie des canaux
semi-circulaires
1. Dysfonction
rhombence phalique (anomalies du tronc ce re bral ou des paires cra niennes III a XII incluant la surdite neurosensorielle) 2. Dysfonction hypothalamo-hypophysaire 3. Malformation de l’oreille (interne ou externe) 4. Malformation des organes me diastinaux (cœur, œsophage) 5. De ficience intellectuelle CHARGE typique : 3 crite res majeurs
OU 2 majeurs + 2 mineurs CHARGE partiel : 2 majeurs et 1 mineur CHARGE atypique : 2 majeurs seuls OU 1 majeur + 2 mineurs D ’a pr ès Bl ak e 1. Colobome oculaire - microphtalmie
2. Atre sie des choanes ou fente labiale et /ou palatine*
3. Anomalies
caracte ristiques de l’oreille (quel que soit le segment concerne – dont l’hypoplasie des CSC) – surdite mixte
4. Atteinte des paires cra niennes
1. Hypoplasie ge nitale ou retard pubertaire 2. Retard des acquisitions,
de ficience intellectuelle 3. Cardiopathie
4. Retard de croissance 5. Fente labiale et/ou
palatine 6. Atteinte trache o-œsophagienne 7. Dysmorphie e vocatrice CHARGE : 4 majeurs ou 3 majeurs + 3 mineurs
* : d’apre s l’actualisation de Blake
II.
G
ENETIQUE DU SYNDROMECHARGE
1. Historique
Bien qu’aucune preuve n’ait jamais pu e tre apporte e en ce sens, une origine te ratoge ne avait initialement e te e voque e pour l’association CHARGE.
La plupart des cas de syndrome CHARGE sont sporadiques au sein de familles par ailleurs sans ante ce dent mais parfois la transmission du syndrome d’un parent a un
30 enfant ou une re currence au sein d’une fratrie (18) a pu e tre observe e, faisant e voquer une origine ge ne tique au syndrome. Par ailleurs, il existe une concordance entre les jumeaux monozygotes, une discordance chez les dizygotes et un lien statistique entre l’a ge paternel avance et la survenue de cas sporadiques de CHARGE (a ge supe rieur ou e gal a 34 ans dans 43% des cas. (8,18) Tous ces e le ments e taient en faveur du fait que la plupart des patients devaient e tre porteurs d’une mutation de novo ou d’une microde le tion chromosomique impliquant un ge ne inconnu.
En 1991 Hurst et al. (48) de crivaient l’association d’une te tralogie de Fallot, d’un colobome, d’une atre sie choanale, d’une malformation de l’oreille externe, d’une surdite , d’une paralysie faciale et d’une fistule trache o-œsophagienne chez une patiente porteuse d’une translocation en apparence e quilibre e t(6;8)(6p8p;6q8q) de novo puis en 2001, Martin et al. (49) de crivirent le cas d’un individu porteur d’une translocation de novo t(2;7)(p14;q21.11) et d’une association CHARGE avec atre sie des choanes, hypoplasie des CSC et de la cochle e, sugge rant de potentiels loci implique s dans le syndrome CHARGE.
En paralle le, plusieurs the ories incluant une anomalie du de veloppement des cellules des cre tes neurales (50), une perturbation de l’interaction entre le me soderme et les cellules des cre tes neurales (51) ou une perturbation de l’interaction e pithe lium-me senchylium-me (52) ont e te propose es afin d’expliquer le lium-me canislium-me physiopathologique du syndrome.
Finalement c’est l’utilisation de la CGH-array qui a permis en 2004 la de couverte par Vissers et al. (53) du ge ne CHD7 comme responsable de la majorite des cas de syndrome CHARGE gra ce a l’e tude de 2 patients (dont le patient de crit par Hurst et al. en 1991 (48)) pre sentant 2 de le tions chevauchantes impliquant cette re gion.
Cependant, encore aujourd’hui, l’analyse du ge ne par les techniques standard (recherche de mutations ou de remaniements) ne retrouve pas de mutation chez 10% des patients pre sentant une forme typique du syndrome selon les crite res diagnostiques (54). Le taux de patients sans mutation identifie e dans CHD7 pourrait atteindre 40% en conside rant les formes dites « atypiques » du syndrome (55). La variabilite clinique du syndrome et le nombre important de patients non mute s dans le ge ne CHD7 sugge rent une he te roge ne ite ge ne tique mais l’identification des autres ge nes qui pourraient e tre implique s est difficile du fait du caracte re sporadique de la maladie avec d’exceptionnelles formes familiales. On ne peut exclure l’implication de se quences non codantes du ge ne, non encore identifie es a ce jour (mutations introniques profondes par exemple).
31 Un autre ge ne SEMA3A a e te sugge re comme pouvant e tre implique dans le syndrome mais seuls deux patients ont e te rapporte s (56) (dont un est porteur d’une variation de novo identifie e a 2 reprises dans le navigateur de l’Exome Aggregation Consortium (ExAC)) et aucune autre e tude n’a confirme l’implication de ce ge ne dans la maladie. Par contre, un cas familial de syndrome de Kallmann impliquant ce ge ne a e te rapporte (57). En 2014 Lehalle et al. de crivaient une se rie de 36 patients porteurs d’une mutation dans le ge ne EFTUD2. Parmi eux, 14 cas avaient initialement e te adresse s pour syndrome CHARGE (35). Ce ge ne est probablement responsable d’un pourcentage de cas de syndrome CHARGE, qui reste a de terminer, notamment lorsqu’il existe une atre sie de l’œsophage chez les patients.
2. Le gène CHD7
Le ge ne CHD7 (MIM# 608892) est situe sur le chromosome 8 (8q12) en position 61,59-61,78 Me ga bases (hg 19). Il s’e tend sur 188 kilobases et comprend 38 exons (le premier est non codant). Ce ge ne est tre s conserve a travers les espe ces et des orthologues ont e te identifie s chez le xe nope, le poisson ze bre, la souris et le poulet (58–60). Ceci, combine au fait que les souris homozygotes pour une mutation de chd7 meurent a un stade pre coce du de veloppement embryonnaire sugge re une forte pression de se lection et l’importance fonctionnelle de la prote ine (61,62).
Le motif d’expression du ge ne CHD7 a e te e tudie chez l’embryon humain, murin et gallinace entre autres et a chaque fois, on retrouve la corre lation entre le tissu ou le ge ne s’exprime et les anomalies du de veloppement observe es dans le syndrome CHARGE (55,58,60,63). L’expression de CHD7 varie selon le tissu et le stade de de veloppement ; on la retrouve notamment dans les cellules de rive es des cre tes neurales quelle que soit l’espe ce e tudie e (55,58–61,63), dans certaines re gions du cerveau et du cœur en de veloppement et dans les bourgeons otiques et optiques (55,58,60,61,63–66).
3. La protéine CHD7
CHD7 est une prote ine de 2.997 acides amine s que l’on retrouve dans le nucle ole et le nucle oplasme cellulaire (67). Elle appartient a une famille de prote ines implique es dans la re gulation transcriptionnelle via la modification de l’organisation chromatinienne. Toutes les prote ines CHD posse dent deux chromodomaines (domaines de modification de l’organisation de la chromatine) en position N-terminale et un motif he licase central (« sucrose non fermenting (SNF) like helicase motif »). Chez l’Homme, cette famille de
32 neuf prote ines est subdivise e en 3 groupes selon leurs structures et leurs se quences (68,69). La prote ine CHD7 appartient a la sous-famille III caracte rise e par les domaines apparie s C-terminaux Brahma and Kismet (BRK) et par un domaine «Switching-defective protein 3, Adaptor 2, Nuclear receptor corepressor, Transcription factor IIIB (SANT)-like » (68,69).
Re cemment, des e tudes concernant la fonction de la prote ine CHD7 ont sugge re un ro le dans le contro le de la programmation de l’expression des ge nes par un remodelage ATP-de pendant ATP-de la chromatine dans les cellules souches embryonnaires et dans d’autres types cellulaires (59,67).
CHD7 interviendrait a deux niveaux :
- D’une part, par liaison aux histones H3 lysine 4 dans les re gions promotrices de certains ge nes, en fonction du stade de de veloppement et du type cellulaire ; elle jouerait un ro le activateur de transcription (67).
- D’autre part, elle serait implique e dans la re gulation positive de la synthe se des ARNr dans le nucle ole de façon tissu spe cifique (70).
Les e tudes re centes sur la fonction de la prote ine CHD7 me nent a la conclusion que l’haplo-insuffisance du ge ne alte re la transcription, normalement re gie par CHD7 (ou par des complexes prote iques dans lesquels CHD7 est implique e), de ge nes cibles spe cifiques (67,70–72).
L’effet de la prote ine CHD7 varie en fonction du tissu et du stade de de veloppement car les sites de liaisons, les complexes prote iques et les ge nes cibles implique s sont diffe rents (67,70). L’hypothe se actuelle pour expliquer la grande variabilite phe notypique du syndrome CHARGE serait celle d’une alte ration fine du niveau d’expression de CHD7 dans le temps et l’espace (5). Ainsi les organes les plus fre quemment atteints, comme l’oreille interne, seraient plus sensibles au dosage de CHD7 que par exemple le palais, dont l’atteinte est moins constante (70,73). Toutefois, les me canismes mole culaires et les voies de signalisation communs dans lesquels CHD7 est implique e commencent juste a e merger (73,74). D’autres e tudes seront ne cessaires afin de de terminer les facteurs ge ne tiques, e pige ne tiques et environnementaux qui interviennent dans la variabilite phe notypique du syndrome.
33
34
O B J EC T I F DE L A T H ES E
La pre sence historique de centres de prise en charge de patients atteints de surdi-ce cite est a l’origine d’un nombre important de patients atteints du syndrome CHARGE dans l’ancienne re gion Poitou-Charentes, ce qui a incite l’e quipe de ge ne tique du CHU de Poitiers a s’investir dans la recherche sur ce syndrome. La premie re e tape a e te la mise en place du diagnostic mole culaire du syndrome dans notre laboratoire, le deuxie me apre s celui de l’ho pital Necker Enfants-Malades a proposer l’analyse en France. J’ai ainsi participe a la mise en place de la technique de Chromatographie liquide a haute performance en condition de naturante utilise e en routine a ce moment-la dans le laboratoire (75).
La prise en charge de ces patients atteints de handicaps sensoriels multiples et d’anomalies variables du de veloppement est complexe et les me thodes e ducatives sont imparfaites du fait de la varie te de la symptomatologie et du caracte re intrique du de ficit sensoriel et d’une e ventuelle de ficience intellectuelle primitive.
Cette the se a pour propos de faire la synthe se des travaux que nous avons effectue s et qui avaient pour but :
L’amélioration la prise en charge et du suivi des patients : o Mieux connaï tre le phe notype associe au syndrome
o Donner les cle s d’un diagnostic pre coce voir ante natal du syndrome afin d’ame liorer la prise en charge des grossesses et des enfants a la naissance o Identifier des facteurs influençant les pronostics vitaux et de veloppementaux
des patients
La possibilité de proposer un conseil génétique adapté aux familles et patients concernés par le syndrome :
o Ame liorer les techniques d’e tude du ge ne CHD7
o Reconnaitre les diagnostics diffe rentiels (dont les mutations du ge ne EFTUD2) o Mettre en e vidence une ou plusieurs autres cause(s) mole culaire(s)
35 Elle se compose des travaux suivants :
PARTIE 1 : Etude fœtopathologique et moléculaire de 40 cas
Un travail a e te spe cifiquement re alise sur une se rie de fœtus dont l’ADN avait e te adresse dans l’un des 2 laboratoires français re alisant l’e tude du ge ne CHD7. L’ensemble des e le ments des examens fœtopathologiques a e te e tudie , ainsi que les re sultats des analyses mole culaires.
Ceci a permis de conforter l’hypothe se d’un phe notype plus se ve re du syndrome chez les individus de sexe masculin, de proposer le retard de croissance intra-ute rin comme un crite re d’exclusion du diagnostic et de confirmer la fre quence plus e leve e des mutations tronquantes chez les fœtus qui repre sentent l’extre mite la plus se ve re de spectre phe notypique.
PARTIE 2 : Etude du retentissement hypophysaire du syndrome CHARGE
Il a e te re alise une e tude de l’atteinte hypophysaire de 42 patients atteints du SC. L’hypogonadisme hypogonadotrope est pre sent chez 97% des patients. Alors me me qu’il impacte le pronostic statural et la qualite de vie des patients, il n’est souvent diagnostique que tardivement, surtout chez les filles, empe chant une substitution hormonale optimale. En raison de sa fre quence tre s e leve e dans le syndrome, nous proposons que l’hypogonadisme hypogonadotrophique soit conside re comme un crite re majeur du diagnostic. Ce travail a e galement permis une description pre cise de l’atteinte hypophysaire de ces patients.
PARTIE 3 : Etude clinique et moléculaire par minigène de 4 variants introniques de CHD7
Notre expe rience d’e tude diagnostique de CHD7 a mis en e vidence une accumulation de mutations, dont certaines sont re currentes au niveau de l’intron 25 du ge ne CHD7, ce qui nous a incite s a mettre en place une technique simple d’aide a l’interpre tation de variants alte rant potentiellement l’e pissage : le minige ne. Il s’agissait de 4 variants identifie s chez 22 patients dont 2 avaient de ja e te rapporte s. Nous avons ainsi de montre leur pathoge nicite et, en combinant les analyses in silico et ex vivo, nous avons mis en e vidence une configuration particulie re des se quences implique es dans l’e pissage de l’intron 25, dont notamment un point de branchement distant.
36 PARTIE 4 : Etude clinique et moléculaire du syndrome CHARGE – données issues du PHRC
Une e tude soutenue par un Projet Hospitalier de Recherche Clinique (PHRC) national a permis de de crire le phe notype d’une cohorte française de 119 patients pre sentant les crite res diagnostiques du syndrome CHARGE, depuis la pe riode pre natale jusque chez l’adulte. Une mutation dans le ge ne CHD7 a e te identifie e chez 77% des patients et nous pre voyons un se quençage d’exome chez les patients pour qui aucune cause mole culaire n’a pu e tre identifie e.
37
38
A N T ENATAL
S P EC T RU M
O F
C H A RGE
SY ND RO M E I N 4 0 F E T U S E S WI T H C H D 7
M U TAT I O N S
Legendre M, Gonzales M, Goudefroye G, Bilan F, Parisot P, Perez MJ, Bonnie re M, Bessie res B, Martinovic J, Delezoide AL, Jossic F, Fallet-Bianco C, Bucourt M, Tantau J, Loget P, Loeuillet L, Laurent N, Leroy B, Salhi H, Bigi N, Rouleau C, Guimiot F, Que lin C,
Bazin A, Alby C, Ichkou A, Gesny R, Kitzis A, Ville Y, Lyonnet S, Razavi F, Gilbert-Dussardier B, Vekemans M, Attie -Bitach T.
J Med Genet. 2012 Nov;49(11):698-707 (Voir p Erreur ! Signet non défini.) OBJECTIFS DE L’ETUDE
Le syndrome CHARGE est une association malformative rare mais de sormais connue chez l’enfant et l’adulte. Cependant, seule une se rie de 10 fœtus atteints de ce syndrome a e te publie e jusqu'a pre sent et le diagnostic ante natal reste parfois difficile.
La pre sentation fœtale du syndrome CHARGE conduisant a une interruption me dicale de grossesse (IMG) constitue probablement l’extre mite la plus se ve re de son spectre phe notypique. Les crite res diagnostiques de Verloes utilise s chez l’enfant ou l’adulte ne sont pas applicables aux fœtus car un certain nombre d’entre eux, notamment ceux concernant les atteintes fonctionnelles (surdite , hypogonadisme, paralysie faciale et de ficience intellectuelle) ne sont pas accessibles tandis que d’autres sont difficiles a appre hender a l’e chographie obste tricale (colobome, atre sie des choanes). Le diagnostic e tant de sormais bien connu des fœtopathologistes et e chographistes, le syndrome est fre quemment e voque devant une association malformative re unissant une cardiopathie, des anomalies de l’oreille externe, une fente ou une hypoplasie vermienne qui incitent a rechercher des signes associe s et notamment l’arhinence phalie, l’hypoplasie ou age ne sie des CSC, l’atre sie des choanes ou une anomalie oculaire.
METHODES
Nous avons e largi l’e tude pre ce dente a 40 fœtus/nouveau-ne s pour lesquels un examen fœtopathologique de taille a e te re alise et une mutation dans le ge ne CHD7 identifie e.
39 RESULTATS
Nous retiendrons notamment de l’e tude de notre se rie le taux e leve de garçons par rapport aux filles (2,6:1), sugge rant des formes plus se ve res chez le garçon. Les signes les plus constants sont l’anomalie des oreilles externes, suivie de l’age ne sie des canaux semi-circulaires et de l’arhinence phalie. En revanche aucun fœtus ne pre sentait de retard de croissance intra-ute rin. Enfin, a l’exception d’un cas, toutes les mutations identifie es dans notre se rie e taient des mutations tronquantes sugge rant une corre lation phe notype/ge notype.
CONCLUSION
L’analyse clinique de notre se rie nous a permis d’affiner la description du phe notype du syndrome CHARGE chez le fœtus, de de crire de nouveaux e le ments cliniques et finalement de proposer un ajustement des crite res diagnostiques chez le fœtus afin d’aider au diagnostic de syndrome CHARGE au de cours d’une interruption me dicale de grossesse pour syndrome malformatif se ve re.