HAL Id: dumas-01675326
https://dumas.ccsd.cnrs.fr/dumas-01675326
Submitted on 4 Jan 2018HAL is a multi-disciplinary open access
archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
Syndrome de Sturge-Weber : optimisation du diagnostic
précoce de l’angiome pial et recherche de facteurs
prédictifs électroencéphalographiques du développement
de l’épilepsie
Claire Bar Maillet
To cite this version:
Claire Bar Maillet. Syndrome de Sturge-Weber : optimisation du diagnostic précoce de l’angiome pial et recherche de facteurs prédictifs électroencéphalographiques du développement de l’épilepsie. Médecine humaine et pathologie. 2017. �dumas-01675326�
1
Université de Bordeaux
U.F.R. DES SCIENCES MEDICALES
Année 2017 N° 3206
Thèse pour l’obtention du
DIPLOME d’ETAT de DOCTEUR EN MEDECINE
Présentée et soutenue publiquement
Par Claire BAR MAILLET
Née le 13 octobre 1988 à Nantes
Le 15 novembre 2017
Syndrome de Sturge-Weber : Optimisation du diagnostic
précoce de l'angiome pial et recherche de facteurs prédictifs
électroencéphalographiques du développement de l'épilepsie
Directeur de thèse
Madame le Professeur Rima NABBOUT
Jury
Monsieur le Professeur Jean-François CHATEIL
Président
Monsieur le Professeur Franck BORALEVI
Monsieur le Professeur Dominique GUEHL
Monsieur le Docteur Jean-Michel PEDESPAN
Monsieur le Docteur Frédéric VILLEGA
Madame le Professeur Rima NABBOUT
2
Remerciements
A Monsieur le Professeur Jean-François CHATEIL,
Pour l’honneur que vous me faites en acceptant de présider et de juger cette thèse.
Pour votre disponibilité, vos compétences et votre rigueur que j’ai pu apprécier lors d’un précédent travail réalisé ensemble sur les ischémies médullaires de l’enfant.
A Monsieur le Professeur Franck BORALEVI,
Pour l’honneur que vous me faites en acceptant de juger ce travail. Veuillez trouver ici l’expression de mes remerciements les plus sincères.
A Monsieur le Professeur Dominique GUEHL,
Pour l’honneur que vous me faites en acceptant de juger ce travail et pour m’avoir fait découvrir le monde de l’électrophysiologie et de la neurologie adulte lors de mon stage aux explorations fonctionnelles du système nerveux.
Merci de m’avoir donné le goût des neurosciences par vos explications passionnées, et passionnantes, sur le fonctionnement des ganglions de la base !
A Monsieur le Docteur Jean-Michel PEDESPAN,
Pour avoir accepté d’être le rapporteur de ce travail et pour l’honneur que vous me faites en faisant parti de ce jury.
Pour m’avoir fait découvrir et aimer la neurologie pédiatrique lors de mon passage dans votre service en 3ème semestre. Merci pour votre aide tout au long de mon cursus et pour la confiance que vous me faites en m’accueillant dans votre équipe.
Veuillez trouver ici l’expression de mon profond respect.
A Monsieur le Docteur Frédéric VILLEGA,
Pour ton accompagnement bienveillant tout au long de mon internat, ta disponibilité permanente, tes précieux conseils et pour tout ce que tu m’as appris pendant et après mon passage dans le service, tant sur le plan médical que sur le plan humain. Tes compétences de médecin, ton talent de musicien, ta générosité et ton humilité forcent mon respect et mon admiration. J’ai hâte de travailler à tes côtés.
3
A Madame le Professeur Rima NABBOUT,
Pour l’honneur que tu m’as fait en acceptant de diriger cette thèse et pour m’avoir ensuite accompagné et guidé tout au long de ce travail, et ce malgré la distance. Merci pour ta disponibilité, ton soutien et tes conseils (souvent nocturnes... !).
Je te remercie pour la confiance que tu m’accordes en m’offrant la possibilité de revenir travailler à tes côtés.
Merci à tous ceux qui ont participé à ce travail et notamment au Professeur Nathalie BODDAERT et au Docteur Anna KAMINSKA pour leur aide précieuse, leur disponibilité et leur réactivité à mes fréquentes sollicitations. Les staffs animés et passionnants de neuroradiologie du mardi après-midi et d’épileptologie du jeudi après-midi à Necker resteront comme des grands moments de mon internat de pédiatrie !
4 A tous les médecins qui m’ont accompagné tout au long de mon cursus, depuis mes premiers stages d’externe, qui m’ont transmis leur expérience dans un compagnonnage bienveillant et qui ont fait de moi le médecin que je deviens aujourd’hui…
Je tiens à remercier particulièrement l’équipe de Agen et Claire BRIENNON avec qui j’ai adoré jouer au « duo des Claires » lors de mes premiers pas en pédiatrie ; l’équipe de Bayonne dont Serge RIVEIRA et Leila LAZZARO que je revois toujours avec grand plaisir ; l’équipe du 6èmeC bien sûr, Caroline ESPIL-TARIS et Marie HUSSON que j’ai hâte de retrouver ; l’équipe des explorations fonctionnelles du SNC et particulièrement Imad GHORAYEB qui m’a fait publier mon premier papier ; l’équipe de neuropédiatrie de Necker et particulièrement le Pr Isabelle DESGUERRE pour les traditionnels apéros-transmissions du vendredi soir ; et enfin l’équipe du 3ème A, avec qui je ne pouvais pas mieux terminer mon internat, Cathy, ma référente des internes et chef de clinique préférée, ma marraine Vanessa VAUTIER, Jérôme HARAMBAT, Astrid DUBRASQUET et bien sûr Brigitte LLANAS, notre maman à tous…!
Merci à Pascal BARAT, notre coordinateur du DES avec qui j’ai découvert l’envers du décor de l’hôpital et les réunions à rallonge de l’ARS… Merci pour votre investissement dans notre formation, votre bienveillance et votre disponibilité auprès de tous les internes.
A toutes les équipes paramédicales, si bienveillantes, avec qui j’ai pris plaisir à travailler à chacun de mes stages et sans qui rien ne serait possible…
A Jan-Pieter KONSMAN, alias JPK, pour m’avoir fait découvrir le monde de la recherche fondamentale durant mon année de master 2. Merci de m’avoir poussé au-delà de mes limites, merci pour ta patience, ta pédagogie, ta confiance et ta rigueur scientifique.
Merci au reste de la « Brain Molecular Imaging » team, et notamment à Béa, Aleks et Jérôme BADAUT pour votre indulgence quant à mon anglais !
A mes « co-internes » rencontrés au fil des stages et devenus des ami(e)s, particulièrement à la dream team du 6C, Céline et Barbara, pour avoir été mes mamans lors de mes premiers pas au CHU ; aux 2 Benjamins pour m’avoir chouchouté aux EEG ; à Raph pour m’avoir fait tant progresser en anglais ; à Anne-So, Romain et Roman qui ont fait passer tellement vite ces 6 mois à Paris ; et enfin à Raphaëlle, Audrey, Marie et Soso, véritables coups de cœur de mon dernier stage d’internat...
A la team « Bordal » élargie, Manu et Anne go, Mayo et Pav, Claire et Lolo, Audrey et Arnaud, Nath, Clémence et Christian, Heidi, Nanou et Thibaut, Chloé et Clément, Judith, Damien, Mélo et Adri…sans oublier Simsim, le ptit dernier de la bande ! Merci pour tous ces moments passés ensemble qui me permettent si rapidement d’oublier l’hôpital !
5 A Guitou et Anne-Cha pour les « parenthèses enchantées » à Noirmoutier…
A mes coloc et témoins adorées, Juju, Mag et Nana, auxquelles je rajoute bien sur Claire et Maellou. Merci d’avoir rendu inoubliables ces longues années de fac, pour les moments studieux (ou pas…) rue du général Faidherbe, pour ces amitiés si fortes qui continuent de s’enrichir années après années, et ce malgré la distance...
A Anaïs, ma tite « best » de tous les instants, de tous les moments importants de ma vie. Même si l’insouciance des années « vélo-mobylette » semble bien loin aujourd’hui, d’autres beaux moments à partager nous attendent. En attendant, comme Maud nous le rappelle si bien, je continuerai de sourire avec toi et ma deuxième famille que j’aime tant…
A mes parents, pour m’avoir transmis, au-delà de votre passion pour la médecine, le « goût des autres » à travers les valeurs de générosité, de tolérance et d’empathie que vous incarnez si bien... C’est grâce à vous si j’en suis la aujourd’hui, merci pour tout.
A mes frères et sœurs, Céline, Fabien, et Antoine, pour m’avoir supporté pendant toutes ces années et pour me permettre d’apprendre toujours de nouvelles choses à chacune de nos joyeuses retrouvailles !
A Camille pour tes conseils toujours avisés et, bien sûr, pour Victor et Haydée, les plus merveilleux des neveux et nièces !
To Margaret, my official proofreader, I’m looking forward to meeting you!
A tout le reste de ma famille et notamment mes grands-parents pour leur tendresse et leur affection et à mon arrière-arrière-grand-père, le professeur Paul BAR, inventeur du célèbre « Clamp de Bar » (ce qui me permet de l’écrire correctement ici !).
A ma belle-famille, Kiki et Annie, Marie, Bastien et Zoé, Charly, pour votre accueil chaleureux au royaume de Montjean, pour votre simplicité et votre générosité à chacune de nos retrouvailles !
A Alex, surtout.
Pour ton soutien infaillible depuis maintenant 5 ans, ta présence à mes côtés qui me rend plus forte, pour toutes tes petites attentions du quotidien qui rendent la vie avec toi si douce et pour avoir assumé une grande partie de la « charge mentale » depuis plusieurs mois, toujours avec amour et bienveillance… ! Merci de me suivre dans l’aventure parisienne, malgré les contraintes. Merci pour tout. Totem.
Enfin, merci à tous les enfants et leur famille rencontrés au cours de mon internat et auprès de qui j’ai le plus appris. Je leur dédie cette thèse.
6
« Celui à qui la souffrance est épargnée doit se sentir appelé à soulager celle des autres. »
7
Table des matières
1. ABREVIATIONS ...8
2. LE SYNDROME DE STURGE-WEBER...9
2.1. Introduction ...9
2.2. Histoire du syndrome de Sturge-Weber ... 10
2.3. Définition ... 11
2.4. Epidémiologie ... 12
2.5. Données de génétique et physiopathologie ... 12
2.6. Présentation clinique ... 14 2.6.1. Atteinte cutanée ... 14 2.6.2. Atteinte ophtalmologique ... 15 2.6.3. Complications neurologiques ... 16 2.7. Données de neuro-imagerie ... 17 2.7.1. Diagnostic radiologique ... 17
2.7.2. Apports des autres techniques de neuro-imagerie ... 17
2.8. Traitements ... 19
3. PROBLEMATIQUES ... 21
3.1. Diagnostic précoce de l’angiome leptoméningé ... 21
3.2. Identification des enfants à risque de développer une épilepsie ... 22
4. ARTICLE 1 : Improving early MRI detection of presymptomatic leptomeningeal angioma in Sturge-Weber syndrome. ... 23
4.1. Abstract ... 25
4.2. Introduction ... 26
4.3. Patients and Methods ... 27
4.4. Results ... 30
4.5. Discussion ... 32
4.6. Conclusion ... 35
4.7. References ... 36
4.8. Tables and Figures ... 39
5. ARTICLE 2 : Electroencephalographic anomalies preceding the onset of epilepsy in children with Sturge Weber syndrome. ... 45
5.1. Abstract ... 46
5.2. Introduction ... 47
5.3. Patients and Methods ... 48
5.4. Results ... 49
5.5. Discussion ... 50
5.6. Conclusion ... 52
5.7. References ... 53
5.8. Tables and figures ... 55
6. CONCLUSION ET PERSPECTIVES... 57
7. REFERENCES ... 58
8
1. ABREVIATIONS
DTI : Diffusion Tensor Imaging EEG : Electroencéphalogramme
FLAIR : Fluid-Attenuated Inversion-Recovery
GNAQ : Guanine Nucleotide-binding protein Alpha-Q IRM : Imagerie par Résonance Magnétique
MRI : Magnetic Resonance Imaging mTOR : Mechanistic Target Of Rapamycin NEM : Necker-Enfants Malades
PET : Positron Emission Tomography PWS : Port-Wine Stain
SPECT : Single Photon Emission Computed Tomography SWI : Susceptibility Weighted Imaging
SWS : Sturge Weber Syndrome
9
2. LE SYNDROME DE STURGE-WEBER
2.1. Introduction
Van der Hoeve créa en 1923 le mot " phacomatose " à partir de deux termes grecs : " phakos " (tache) et " oma " (tumeur) pour désigner un groupe d’affections associant des lésions du système nerveux à des lésions cutanées. Ces différents syndromes résultent généralement d’une anomalie précoce du développement embryonnaire. L’implication des dérivés ectodermiques a donné naissance à d’autres dénominations telles que "syndromes neurocutanés", "dysplasies neuroectodermiques" ou "neuroectodermoses". Finalement moins restrictif, le terme de « phacomatose » reste le plus approprié puisqu’il inclut également les affections dérivant des autres feuillets embryonnaires, notamment du mésoblaste.
Trois phacomatoses possédant un caractère familial et des tumeurs multiples ont été initialement décrites : La Neurofibromatose de type 1, La Sclérose Tubéreuse de Bourneville et la maladie de Von Hippel-Lindau. Des phacomatoses à traduction essentiellement vasculaire (angiomatoses cérébrales comprenant le syndrome de Sturge-Weber) et d'autres à traduction essentiellement cutanée (phacomatoses pigmentaires) ont ensuite été ajoutées dans ce groupe de pathologies.
Les progrès accomplis dans le domaine de la génétique ont permis, dans certains cas, la découverte, la localisation et le clonage des gènes en cause, permettant de mieux comprendre la pathogenèse de ces phacomatoses, d’envisager un véritable conseil génétique et le développement de perspectives thérapeutiques.
10
2.2. Histoire du syndrome de Sturge-Weber
En 1879, William Allen Sturge rapporta l’histoire d’une enfant de 6 ans présentant des secousses de l’hémicorps gauche et porteuse d’une « marque maternelle » étendue, touchant la partie droite du visage, bien délimitée sur la ligne médiane, associée à un glaucome congénital de l’œil droit. Il nomma cette marque une tâche « lie-de-vin » (« port-wine stain » en anglais) et suggéra qu’une malformation sur la surface ipsilatérale du cerveau puisse être à l’origine du déficit neurologique. Selon lui, une lésion du parenchyme cérébral lui-même aurait dû entrainer une apparition plus précoce de l’épilepsie (1). Son hypothèse sera confirmée 18 ans plus tard, en 1901, par Siegfried Kalischer sur une autopsie d’une enfant de 18 mois (2).
En 1922, Parkes Weber rapporta la présence d’une lésion calcifiée et festonnée à la surface de l’hémisphère gauche sur des radiographies du crâne d’une jeune patiente présentant une tâche « lie-de-vin » étendue et un glaucome congénital gauche (Figure 1) (3).
Krabbe montra en 1934 que ces calcifications étaient situées dans le parenchyme cérébral plutôt que dans les méninges. En 1935, l’association d’une tâche « lie-de vin » du visage, d’un
Figure 1. Patiente porteuse d’un angiome plan étendu, prédominant à gauche, et sa radiographie du crâne révélant un liseré calcifié à la surface de l'hémisphère gauche.
11 glaucome congénital et d’une malformation vasculaire leptoméningée fut regroupée sous le syndrome de « Sturge-Weber » par le professeur Hilding Bergstrand (Figure 2) (2).
2.3. Définition
Le syndrome de Sturge-Weber est une phacomatose neuro-cutanée et oculaire congénitale et sporadique. Dans sa forme complète, elle associe une malformation capillaire faciale (nommée « tâche lie-de-vin » ou « angiome plan »), une malformation vasculaire leptoméningée et une atteinte oculaire (glaucome, angiome choroïdien).
Roach (4) a classé le syndrome de Sturge-Weber en 3 types :
- Type 1 associant angiome de la face et angiome leptoméningé avec atteinte oculaire inconstante.
- Type 2 avec angiome de la face sans atteinte du système nerveux central et atteinte oculaire inconstante.
- Type 3 avec angiome leptoméningé isolé, sans atteinte cutanée.
Ce travail porte sur le type 1 de cette classification, forme classique du syndrome de Sturge-Weber et dont le pronostic est le plus sévère.
Figure 2. William Allen Sturge (1850-1919) et Frederick Parkes Weber (1863-1962)
12
2.4. Epidémiologie
L’angiome plan ou « tâche lie-de-vin » est une malformation vasculaire fréquente à la naissance, présente chez 0,3% des nouveau-nés avec un sexe ratio de 1 :1 (5). Seuls les enfants avec un angiome plan touchant la partie supérieure du visage, front ou paupière supérieure, sont à risque de présenter une atteinte cérébrale et/ou oculaire associée. Ce risque est évalué entre 10 et 20% selon les études (6–8) et pourrait atteindre 35% lorsque l’atteinte cutanée est bilatérale (9). En l’absence de données épidémiologiques basées sur une étude de population, la prévalence du syndrome de Sturge-Weber est estimée entre 1 pour 20 000 à 50 000 naissances vivantes (10).
2.5. Données de génétique et physiopathologie
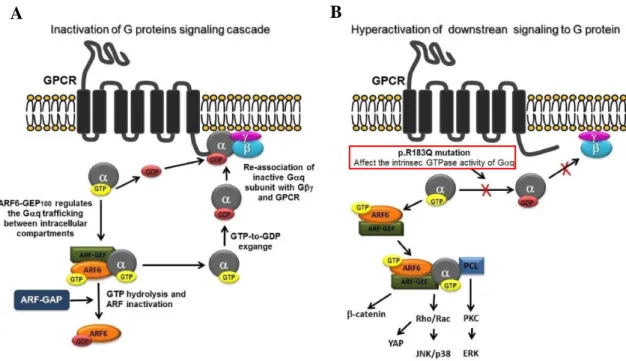
En 2013, Shirley et al. ont mis en évidence une mutation somatique dans le gène GNAQ, (Guanine Nucleotide-binding protein Alpha-Q) dans 88% d’échantillons de tissus de patients présentant un syndrome de Sturge-Weber (peau et cerveau), mais également dans 92% d’échantillons de tissus issus de tâches « lie-de-vin » non syndromiques (11). Ce gène GNAQ code pour la sous unité Gαq, jouant un rôle important dans la transduction du signal entre certains récepteurs couplés aux protéine G, comme ceux de l’endothéline, et leurs effecteurs en aval. La mutation en cause (c.548G > A; p.R183Q), retrouvée en mosaïque dans les tissus affectés, entrainerait une réduction de l’activité intrinsèque GTPase de Gαq à l’origine d’une hyperactivation de plusieurs cascades de signalisation en aval (11,12) (Figure 3).
13 Figure 3. Représentation schématique du cycle d'activation et d'inactivation des protéines G par les récepteurs couplés aux protéines G. A) Après transduction du signal, l'inactivation de la cascade de signalisation des protéines G se produit suite à la conversion du GTP en GDP grâce à l'activité intrinsèque GTPase des sous-unités Gα. B) La mutation activatrice sur le gène GNAQ (p.R183Q) entrainerait des modifications conformationnelles de la sous-unité Gαq affectant son activité intrinsèque GTPase. L’absence d’inactivation de Gαq conduirait alors à l’hyperactivation de plusieurs voies de signalisation en aval. (D’après Martins et al. J Mol Graph Model. sept 2017;76:429‑40.)
Ces récepteurs couplés aux protéines G et liés à Gαq jouent un rôle essentiel dans la prolifération cellulaire, notamment pour le développement des vaisseaux sanguins (13). Des études récentes datant de 2016 et 2017 ont isolé cette mutation GNAQ dans les cellules endothéliales des malformations capillaires cutanées et cérébrales (14,15). Le dysfonctionnement de ces cellules endothéliales pourrait expliquer l’aspect pathologique des structures vasculaires leptoméningées du syndrome de Sturge-Weber, décrites comme tortueuses et dilatées en histologie (16).
Par ailleurs, il a été montré que le facteur de croissance vasculaire VEGF, régulé par la cascade de signalisation Ras-Raf-MEK-ERK-mTOR, est surexprimé dans les cellules endothéliales des vaisseaux leptoméningés anormaux. L’hyperactivation de cette voie de signalisation secondaire à la mutation GNAQ pourrait donc participer au remodelage vasculaire dans le syndrome de Sturge-Weber (17).
14 Enfin, la variabilité phénotypique associée à cette mutation GNAQ pourrait être liée au moment de survenue de cette mutation durant la vie fœtale. Sa survenue précoce durant le développement embryonnaire pourrait être responsable du syndrome de Sturge-Weber par l’atteinte d’un progéniteur commun aux différents tissus affectés. En effet, les leptoméninges, l’œil et le tissu cutané de la partie supérieure du visage possèdent une origine embryologique commune, dérivant tous de cellules de la crête neurale céphalique provenant du prosencéphale. La même mutation survenant plus tardivement dans des cellules endothéliales vasculaires pourrait être responsable des angiomes plan du visage non syndromiques (11).
La découverte des bases génétiques du syndrome de Sturge-Weber ouvre de nouvelles perspectives sur la compréhension des mécanismes physiopathologiques sous-jacents, et pourrait permettre dans l’avenir d’identifier des cibles thérapeutiques susceptibles de ralentir la progression de l’atteinte cérébrale (13).
2.6. Présentation clinique
2.6.1. Atteinte cutanée
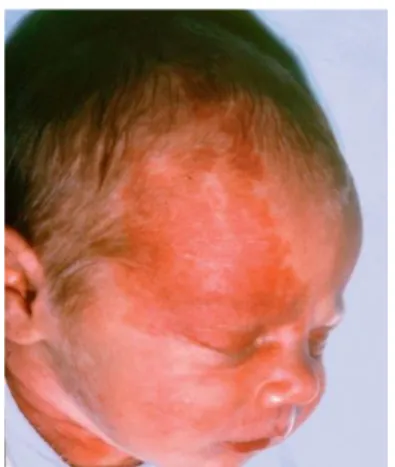
Le diagnostic de syndrome de Sturge-Weber est évoqué à la naissance devant un angiome plan couleur lie-de-vin, affectant la partie supérieure du visage (Figure 3).
Figure 3. Tâche lie-de-vin (angiome plan).
(D’après Nabbout et al. Handb Clin Neurol. 2013;111:315‑21.)
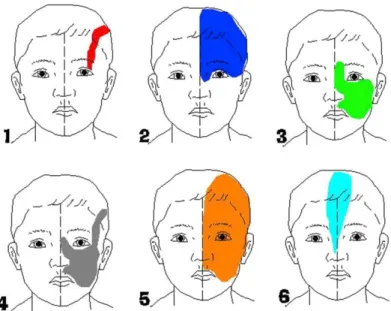
15 Historiquement, l’angiome plan du visage était décrit selon son extension sur les territoires d’innervation des branches du nerf trijumeau (V). Le risque d’angiome leptoméningé était alors associé à une atteinte cutanée sensée correspondre au territoire de la branche ophtalmique V1. La découverte récente des mécanismes génétiques suggère plutôt une distribution de l’atteinte cutanée liée au mosaïcisme génétique, en rapport avec le développement embryologique du système vasculaire. Dutkiewicz et al. ont ainsi identifié deux patterns d’atteinte cutanée associés au risque d’angiome leptoméningé sous-jacent (Figure 4, patterns 5 et 6) (7).
2.6.2. Atteinte ophtalmologique
La prévalence du glaucome chez les patients atteint du syndrome de Sturge-Weber varie de 30% à 60% (18,19). Il existe trois pics d’apparition du glaucome : 40 % durant la première année, 23 % entre 5 et 9 ans et 20 % après l’âge de 20 ans (18). L’autre atteinte ophtalmologique fréquente est l’hémangiome choroïdien, souvent asymptomatique et donnant un aspect rouge diffus du pôle postérieur au fond d’œil. Cet hémangiome peut se compliquer d’un décollement de rétine ou d’un glaucome néovasculaire (20).
Figure 4. Représentation schématique des différents patterns de distribution de l'angiome plan dans une population de 66 enfants. Seuls les patterns 5 et 6 étaient associés à un risque significatif de syndrome de Sturge-Weber. (D’après Dutkiewicz et al. J Am Acad Dermatol. mars 2015;72(3):473‑80.)
16
2.6.3. Complications neurologiques
L’examen clinique de ces enfants est généralement normal à la naissance mais la survenue précoce de complications neurologiques fait toute la gravité de cette pathologie. L'épilepsie est souvent la première manifestation, touchant environ 80% des patients (21,22). Le début des crises survient avant l’âge de 1 an dans 75% des cas, avant l’âge de 2 ans dans 86% des cas et dans 95% des cas avant l’âge de 5 ans (18). Une susceptibilité des crises à l’hyperthermie est retrouvée chez la plupart de ces patients (22). Les états de mal convulsifs sont fréquents et souvent inauguraux. Les crises sont généralement motrices et à début focal, en rapport avec l'angiome leptoméningé localisé chez la plupart des patients dans les régions occipitales (22). L’atteinte cognitive est fréquente dans le syndrome de Sturge-Weber mais présente à des degrés variables. Une étude rétrospective portant sur 171 patients avec un syndrome de Sturge-Weber rapportait un retard de développement psychomoteur chez 58% des patients. Plus récemment, dans une série de 40 patients suivis de manière prospective, seulement 30% d’entre eux avaient un quotient intellectuel normal, supérieur à 90. Le début précoce et la sévérité de l’épilepsie sont associés à un pronostic cognitif péjoratif (18,21,23).
Des céphalées récurrentes à l’origine d’une altération de la qualité de vie sont retrouvées chez environ 60% des patients (18), répondant aux critères de migraines dans 28% des cas (24). Enfin, des déficits neurologiques focaux à type d’hémiplégie/hémiparésie spastique ou d’hémianopsie peuvent apparaitre au cours du temps, soit de manière transitoire suite à une crise prolongée ou associés à une migraine, soit définitivement en rapport avec des phénomènes ischémiques de type « stroke-like » (25). Dans une cohorte de 77 patients avec un syndrome de Sturge-Weber, l'apparition précoce des crises avant l’âge de 6 mois était associée à un déficit moteur plus sévère (26).
17
2.7. Données de neuro-imagerie
2.7.1. Diagnostic radiologique
Le diagnostic de certitude du syndrome de Sturge-Weber repose sur l’imagerie par résonance magnétique (IRM) cérébrale avec injection de produit de contraste permettant de confirmer la présence d’une malformation vasculaire leptoméningée (angiome leptoméningé ou angiome pial). Celle-ci peut être visible directement sous la forme d’une prise de contraste épousant le contour des sillons et associée à une visibilité augmentée des vaisseaux sous-corticaux. D’autres anomalies neuroradiologiques décrites dans la littérature pourraient constituer des signes indirects de l’angiome leptoméningé : une hypertrophie homolatérale du plexus choroïde, une inversion du signal de la substance blanche, une atrophie cérébrale ou des calcifications en regard de l’angiome.
Cependant, la sensibilité de l’IRM cérébrale durant les premiers mois de vie est considérée comme faible dans la littérature, de sorte qu’il n’existe pas de véritable consensus sur son délai de réalisation chez un nouveau-né présentant une tâche lie-de-vin du visage (27). Il est actuellement admis que si l’enfant a un examen clinique et un développement neurologique normal, sans antécédent d’épilepsie et avec une IRM cérébrale injectée normale après l’âge de 1 an, le diagnostic de syndrome de Sturge-Weber peut être raisonnablement écarté (25).
2.7.2. Apports des autres techniques de neuro-imagerie
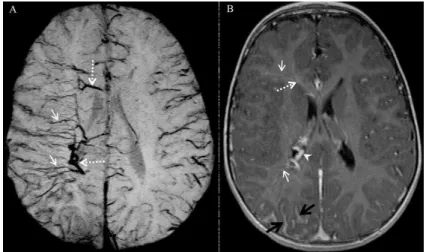
La séquence FLAIR basée sur la technique d'inversion-récupération et l’imagerie de susceptibilité magnétique (SWI) basée sur les différences de susceptibilité magnétique entre les tissus pourraient être plus sensibles pour détecter l'angiome leptoméningé et l’élargissement de veines transmédullaires profondes (Figure 5) (28,29).
18 Figure 5. Syndrome de Sturge-Weber chez un enfant de 2 ans. Réseau
de veines transmédullaires (flèches pleines) et périventriculaires (flèches pointillées) mieux visible sur la séquence de susceptibilité magnétique SWI (A) par rapport à la séquence T1 avec injection de produit de contraste (B). (D’après Hu J et al. J Magn Reson Imaging. août 2008;28(2):300‑7.)
L'imagerie de diffusion met en évidence une restriction de la diffusion associée à une augmentation du coefficient apparent de diffusion dans la substance blanche adjacente à l’angiome (30). L’imagerie de perfusion révèle des modifications dynamiques du flux sanguin cérébral à différents stades d’évolution de la maladie. Une augmentation transitoire de la perfusion cérébrale est retrouvée chez les patients les plus jeunes alors qu’une diminution de la perfusion cérébrale est retrouvée chez les patients plus âgés, corrélée à la durée d’évolution de l’épilepsie, sa sévérité et à l’atrophie cérébrale sous-jacente (31).
L’imagerie cérébrale fonctionnelle, d’introduction plus récente, apporte des informations intéressantes pour la compréhension des mécanismes physiopathologiques sous-jacents à l’angiome leptoméningé. L’étude du métabolisme du glucose par PET et de la perfusion cérébrale par SPECT ont mis en évidence à un stade précoce de la maladie un hypermétabolisme cortical transitoire en regard de l’angiome, probablement en rapport avec l’hyperperfusion décrite ci-dessus (32,33). A un stade plus avancé de la maladie, on note un hypométabolisme régional s’étendant au-delà des anomalies structurelles apparentes, associé à une diminution de la perfusion cérébrale et corrélé à la gravité de l’épilepsie (30,34).
19 Ces techniques d’imagerie fonctionnelle, actuellement difficilement réalisables en routine, ont surtout une indication dans l’évaluation pré-chirurgicale des enfants avec une épilepsie réfractaire.
Enfin, l’augmentation de la concentration en glutamate dans l’hémisphère atteint par l’angiome leptoméningé pourrait jouer un rôle dans l’épileptogènese du syndrome de Sturge-Weber, comme démontré dans une étude récente utilisant la spectroscopie par résonnance magnétique (35).
2.8. Traitements
Peu de données sont disponibles dans la littérature sur l’évaluation de la prise en charge thérapeutique des patients atteints d’un syndrome de Sturge-Weber. Les recommandations reposent principalement sur l’expérience des cliniciens, à travers le traitement symptomatique des différentes complications neurologiques et ophtalmologiques (17).
Un avis dermatologique doit être pris précocement pour discuter de l’indication du traitement de l’angiome cutané par laser (17). La mise en jeu du pronostic visuel impose un suivi ophtalmologique régulier, dès les premiers jours de vie. Il inclut, outre l’examen de la fonction visuelle adapté à l’âge, la réalisation d’un fond d’œil et la mesure de la pression intra-oculaire. Un traitement par collyre anti-hypertenseur est instauré en cas d’élévation de la pression intra-oculaire. En cas d’échec, une prise en charge chirurgicale est nécessaire (36).
Le pronostic neurologique de ces patients étant corrélé à la gravité de l’épilepsie, une prise en charge précoce et agressive est recommandée dès la survenue de la première crise (25). Dès la première consultation avec le neuropédiatre, les parents sont éduqués à la reconnaissance des crises et à l’administration du traitement par Diazépam intra-rectal en cas de crise. Après la survenue d’une première crise, un traitement de fond antiépileptique doit être instauré rapidement. Les crises étant le plus souvent focales, le traitement de 1ère intention repose sur le
20 valproate de sodium, la carbamazépine ou l’oxcarbamazépine (37). Environ 30% à 50% des patients auront une épilepsie pharmaco-résistante (19,22). Chez ces patients, un traitement chirurgical peut être envisagé selon différentes méthodes (lésionectomie, hémisphérotomie, callosotomie ou hémisphérectomie). Si la chirurgie semble avoir une bonne efficacité sur le contrôle des crises dans le syndrome de Sturge-Weber, le moment optimal pour sa réalisation reste discuté dans la littérature (17).
Connaissant le rôle délétère des crises précoces sur la progression de la maladie, un traitement préventif antiépileptique pourrait permettre de retarder l’apparition des crises et d’améliorer le pronostic de ces enfants (38). Ville et al. ont montré un bénéfice de l’utilisation préventive du phénobarbital sur l’évolution cognitive d’enfants avec un syndrome de Sturge-Weber (39). Cependant, en l’absence de données suffisantes pour valider cette approche, le traitement antiépileptique préventif n’est pas recommandé en pratique courante.
Par ailleurs, un traitement par aspirine à faibles doses a été étudié dans quelques études, dans le but de prévenir la survenue de phénomènes ischémiques participant à la détérioration cognitive de ces enfants (40,41). Si ce traitement pourrait permettre de diminuer à la fois la fréquence des phénomènes de stroke-like et des crises d’épilepsies, des études prospectives sont nécessaires pour valider l’efficacité et la sécurité de cette approche thérapeutique (17).
Enfin, la découverte récente des bases génétiques du syndrome de Sturge-Weber et des mécanismes moléculaires sous-jacents permet d’envisager de nouvelles perspectives thérapeutiques. Un essai clinique interventionnel de phase II-III est ainsi en cours, portant sur l’évaluation du Sirolimus, un inhibiteur de la voie mTOR, dans le traitement du syndrome de Sturge-Weber (https://clinicaltrials.gov/ct2/show/NCT03047980).
21
3. PROBLEMATIQUES
Depuis 2009, un protocole de suivi prospectif des nourrissons suspect de syndrome de Sturge-Weber a été mis en place à l’Hôpital Necker-Enfants Malades (NEM) à Paris. Celui-ci inclut tous les nouveau-nés de moins de 3 mois vus en consultation de dermatologie pédiatrique de l’hôpital NEM pour un angiome plan du visage incluant au moins une partie du front. Ces enfants sont ensuite orientés rapidement vers un neuropédiatre de l’hôpital NEM (RN) qui explique aux parents les risques liés à la présence d’un éventuel angiome leptoméningé et les éduque à la reconnaissance des crises ainsi qu’à l’utilisation d’un traitement par Diazépam intrarectal en cas de crise. Après accord des parents, un électroencéphalogramme (EEG) est programmé à l’unité de neurophysiologie clinique de l’hôpital NEM ainsi que deux IRM cérébrales réalisées selon le protocole de routine du service de radiologie de l’hôpital NEM, la première avant l’âge de 3 mois et la deuxième après 9 mois.
Lors de mon stage hors subdivision en neurologie pédiatrique à l’hôpital NEM à Paris, j’ai eu l’opportunité de travailler sur les données recueillies dans le cadre de ce protocole entre 2009 et 2017 afin d’essayer de répondre à 2 problématiques majeures du syndrome de Sturge-Weber.
3.1. Diagnostic précoce de l’angiome leptoméningé
Parmi tous les enfants avec une tâche lie-de-vin du visage incluant le front, le risque d’angiome leptoméningé associé est estimé entre 10 et 20% selon les études (6–8). Si le diagnostic de syndrome de Sturge-Weber est donc facilement évoqué dès la naissance, sa confirmation chez un nouveau-né asymptomatique est un enjeu diagnostique majeur. Le diagnostic présymptomatique permettrait de délivrer précocement une information pronostique aux familles, d’organiser un suivi médical adapté et de discuter la mise en place de thérapeutiques préventives. Cependant, il existe très peu de données sur l’imagerie cérébrale de ces enfants
22 dans les premiers mois de vie, considérée comme peu sensible dans la littérature. L’âge médian de survenue des premières crises étant de 6 mois, le diagnostic est souvent fait suite à l’apparition de l’épilepsie.
Le 1er objectif de ce travail était d’évaluer la performance diagnostique de l’IRM cérébrale précoce dans la détection d’un éventuel angiome leptoméningé chez des nourrissons asymptomatiques porteurs d’une tâche lie-de-vin du visage.
3.2. Identification des enfants à risque de développer une épilepsie
L’épilepsie est la complication la plus fréquente du syndrome de Sturge-Weber et son apparition précoce semble être le principal déterminant du pronostic neurologique à long-terme. La mise en route de traitements préventifs, antiépileptiques ou aspirine, reste pourtant discutée dans la littérature en raison de la difficulté à réaliser un diagnostic présymptomatique, des effets secondaires non négligeables de ces traitements et de la grande variabilité phénotypique du syndrome de Sturge-Weber. L’identification d’un facteur prédictif du développement de l’épilepsie pourrait permettre de cibler la population susceptible de bénéficier de ces thérapeutiques neuroprotectrices.
Le 2ème objectif de ce travail était d’analyser les EEG réalisés avant l’apparition des crises dans une population d’enfants avec un diagnostic présymptomatique de syndrome de Sturge-Weber, à la recherche d’anomalies EEG prédictives du développement de l’épilepsie.
23
4. ARTICLE 1 : Improving early MRI detection of presymptomatic
leptomeningeal angioma in Sturge-Weber syndrome.
Claire Bara, MD, Jean-Michel Pedespana, MD, Olivia Boccarab, MD, Raphael Levyc, MD, David Gréventc, MD, Nathalie Boddaert*c,d, MD, PhD, Rima Nabbout*d,e, MD, PhD
Affiliations:
aDepartment of Pediatric Neurology, Hôpital des enfants, CHU Bordeaux, France;
bDepartment of Pediatric Dermatology, Hôpital Necker-Enfants Malades, Paris, France; cDepartment of Pediatric Radiology, Hôpital Necker-Enfants Malades and IMAGINE Institute, INSERM UMR 1163 and INSERM U1000,
dDepartment of Pediatric Neurology, Hôpital Necker-Enfants Malades, reference center for rare epilepsies, INSERM U1129, Paris, France
eParis Descartes University, Sorbonne Paris Cité, Paris, France *These authors equally contributed to the paper
Address correspondence to:
Rima Nabbout, Reference center for rare epilepsies, Necker-Enfants Malades hospital, Department of Pediatric Neurology, 149 rue de Sèvres, 75015 Paris, France
E-mail: rima.nabbout@aphp.fr
Short title: Sensitivity of Early MRI in Detecting Pial Angioma Funding Source: None
Financial Disclosure: The authors have no financial relationships relevant to this article to
disclose.
Conflict of Interest: The authors have no conflicts of interest relevant to this article to disclose. Abbreviations: SWS, Sturge Weber Syndrome, MRI, Magnetic resonance imaging, FLAIR,
Fluid-Attenuated Inversion-Recovery, SWI, Susceptibility Weighted Imaging, DTI, Diffusion Tensor Imaging, SPECT, Single Photon Emission Computed Tomography, PET Positron Emission Tomography
24
Table of Contents Summary
Specific MRI markers can provide an early and pre-symptomatic diagnosis of the leptomeningeal angioma in Sturge-Weber syndrome during the first months of life.
What’s Known on This Subject
The diagnosis of Sturge-Weber syndrome is easily identified at birth because of upper facial port-wine stain. However, the diagnosis of the leptomeningeal angioma is often delayed after one year due to the difficulty detecting it on early MRI.
What This Study Adds
We propose a new MRI protocol for early diagnosis of Sturge-Weber syndrome based on specific MRI findings to confirm the diagnosis of leptomeningeal angioma before the onset of epilepsy or other neurological signs.
25
4.1. Abstract
Objective
Early diagnosis of Sturge–Weber syndrome (SWS), associating a facial port-wine stain with cerebral and ocular vascular malformations, is thought to be complicated by the low sensitivity of magnetic resonance imaging (MRI) in neonates. In this study, we assessed the diagnostic accuracy of a routine MRI protocol in detecting leptomeningeal angioma in neonates with facial port-wine stain.
Methods
Children with a facial port-wine stain suspected of SWS underwent an MRI before 3 months old and a control after 9 months. Leptomeningeal enhancement on T1-weighted imaging, direct sign of the leptomeningeal angioma, and four indirect signs (choroid plexus enlargement, focal cerebral atrophy and a signal inversion of the white matter with T2 hyposignal and T1 hypersignal) were screened on the first MRI and correlated to a positive diagnosis of SWS on the second MRI considered as the gold standard.
Results
Thirty children were included in the study, 19 prospectively in 2009-2016 and 11 retrospectively in 2001-2009. Thirteen patients had a diagnosis of SWS confirmed by the second MRI. Eleven of them had a leptomeningeal enhancement on the first MRI of whom 10 presented indirect signs. The presence of direct or indirect signs of the leptomeningeal angioma on the first MRI was associated with a sensitivity of 100% and a specificity of 94%.
Conclusion
Early diagnosis of SWS is possible on a contrast-enhanced MRI performed in asymptomatic neonates with a facial port-wine stain. Although underestimating the extension of the angioma, this early diagnosis paves the way for clinical trials assessing neuro-protective strategies.
26
4.2. Introduction
Sturge-Weber syndrome (SWS) is a congenital neurocutaneous syndrome defined by the association of a facial capillary malformation named a port-wine stain (PWS) with an ipsilateral leptomeningeal angioma and an inconstant ipsilateral glaucoma. Isolated PWS and SWS have a common genetic etiology with a somatic mosaic mutation that has been recently identified in the guanine nucleotide-binding protein alpha-q (GNAQ) gene.1–3
Isolated PWS is a congenital cutaneous lesion, occurring in 3 per 1000 live births.4 Only patients with a facial PWS on the forehead and/or the upper eyelid are at risk of SWS.5 Although no epidemiologic data are available, this risk is estimated between 10% and 20%.6 In the historical definition of the SWS, this territory was thought to correspond with the area of innervation of the ophthalmic division of the trigeminal nerve.5,7,8 However, the recent discovery of an underlying genetic mechanism suggests a somatic mosaicism in the vascular embryological distribution.9,10
Neurological examination of children with SWS is usually normal at birth and during the first months, making a pre-symptomatic diagnosis of the leptomeningeal angioma challenging. The prognosis of SWS is, however, mainly related to the neurological complications that often develop early during the first year of life.11 Epilepsy is typically the first manifestation related to the leptomeningeal angioma.12 Seizures occur in 75 to 85 % of patients with SWS with an onset before 1 year of age in 75% of them.5,13,14 The cognitive outcome of SWS patients is highly variable, but seems to correlate with the onset of early seizures and the severity and intractability of epilepsy.13,15–17
The leptomeningeal angioma can also be responsible for contralateral neurological deficits such as hemiparesis or hemianopsia that often develop progressively during the course of the disease.13,18 Chronic brain tissue hypoperfusion due to abnormal venous drainage leads to progressive cerebral atrophy worsened by seizures.19,20 In addition to neurological
27 complications, 40 to 60% of SWS patients will develop glaucoma.5,13 In 60 % of them, it appears before the age of one year and may lead to early visual impairment.13,21 Therefore, early diagnosis of SWS is needed to accurately identify the infants at risk, to educate parents to properly identify and manage seizures and to organize consequent medical follow-up.
Moreover, preventive treatments with antiepileptic drugs (AEDs) and low-dose aspirin might prevent or delay the onset of seizures and cerebral atrophy.22–24 Hence, criteria helping to establish pre-symptomatic diagnosis are essential for introducing early intervention and developing clinical trials that target the use of neuroprotective strategies.25 Although SWS is easily suspected at birth in the presence of an upper facial PWS, the diagnosis is confirmed by Magnetic Resonance Imaging (MRI) that reveals an ipsilateral leptomeningeal enhancement on T1-weighted imaging.6 This direct sign of the leptomeningeal angioma can be, however, absent on the early MRI, and other radiological signs are thought to provide indirect evidence of the leptomeningeal angioma: an ipsilateral choroid plexus enlargement, cerebral atrophy, and a signal inversion of the white matter (hyposignal on T2 and hypersignal on T1-weighted imaging).26,27 The sensitivity of these abnormalities on early MRI is still debated and was not assessed in pre-symptomatic neonates and infants.20 Therefore, the diagnosis is often made on the MRI after the age of one and often following the occurrence of the neurological symptoms.5 The aim of this study is to evaluate the diagnostic accuracy of early MRI in asymptomatic neonates and infants with facial PWS. Our goal was to establish MRI diagnostic criteria at an early stage before the onset of these neurological complications.
4.3. Patients and Methods
4.3.1. Study design and participants
We performed a longitudinal study at the University Hospital of Paris Necker-Enfants Malades from January 2009 to February 2016. This protocol was performed within the best clinical care
28 recommendations. Consecutive children aged under 3 months old and presenting at pediatric dermatology clinic for a facial PWS covering at least one part of the forehead and/or eyelid were eligible for the study. They were referred to the child neurologist, who explained the potential risk of leptomeningeal angioma and its complications, and educated the family on identifying seizures and administering rescue medication. The family also received an emergency certificate explaining the risk of developing seizures and detailing how to administer emergency care if a seizure were to occur. Patients were enrolled in a prospective protocol with a clinical follow-up in addition to an early MRI performed before the child was 3 months of age and a confirmatory MRI after 9 months of age. The study protocol was approved by the ethical board of our hospital as part of the good care practice, and informed consent was obtained from parents.
4.3.2. Data collection
For each patient included, we collected information on age, sex, PWS distribution, clinical and ocular findings at the first consultation, prophylactic treatment, along with follow-up data regarding neurological and ocular complications. For patients seen several times during the follow-up, data from the first and last visit were recorded. Patients were excluded from the protocol if the MRI revealed a pathology different from SWS. To increase the power of our statistical analysis, we carried out a retrospective review among children seen at the University Hospital of Paris Necker-Enfants Malades between 2001 and 2009 for a PWS on the forehead. We included patients who fulfilled the same inclusion criteria as those for the prospective part and collected their data through medical files.
29
4.3.3. Test methods
The early MRI performed before 3 months of age was considered as the index test and was compared to the second MRI conducted after 9 months of age as the reference standard. All MRI were performed using a routine protocol including a 3DT1-weighted sagittal and axial images, T2-weighted axial, and coronal images, and a post-gadolinium 3DT1-weighted acquisition. In addition, the prospective protocol included a cube post-gadolinium T1-weighted acquisition with fat saturation. When necessary to avoid movement artifacts, children were sedated using intrarectal pentobarbital (5mg per kilogram of the child’s weight <20 Kgs), as is the protocol of our radiology department. The same pediatric neuroradiologist (NB) reviewed the brain images of each child, blinded to clinical information except for the side of the PWS. The early MRI was evaluated for the presence of a leptomeningeal enhancement on post-contrast T1-weighted imaging as a direct sign of leptomeningeal angioma. Results of each MRI were classified as either a positive or negative diagnosis of SWS based on whether or not this direct sign was present. We additionally screened for associated signs already described in the literature: white matter abnormalities with a T1 hypersignal or a T2 hyposignal, an asymmetry of choroid plexus or a localized cerebral atrophy.27–29 The diagnosis of SWS was then confirmed or ruled out on the second MRI performed after the child reached 9 months, as the gold standard, by the direct visualization of the leptomeningeal enhancement.
4.3.4. Analysis
Descriptive statistics were used to analyze the characteristics of the study population. To determine test performance characteristics, the results of the early MRI (the index test) along with those of the second MRI (the reference standard) were illustrated in a cross tabulation. We estimated sensitivity, the specificity, and post-test probability using standard methods, along with corresponding confidence intervals of 95%.
30
4.4. Results
Thirty patients were included in this study. Twenty-one were eligible for the prospective part of the study, but two patients were excluded because of a final diagnosis of megalencephaly capillary malformation (MCAP). Eleven patients were added with the retrospective review fulfilling the inclusion criteria.
4.4.1. Baseline demographic and clinical characteristics of participants
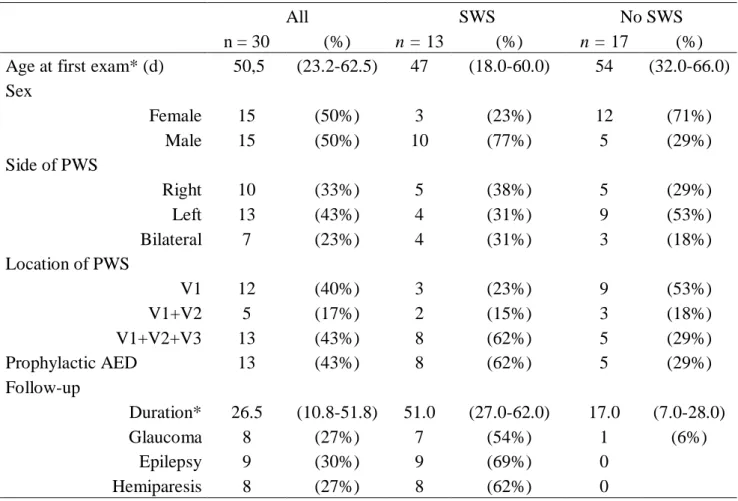
The diagnosis of SWS was confirmed in 13 out of 30 patients by the visualization of a leptomeningeal angioma on the second MRI (7 out of 19 from the prospective and 6 out of 11 from the retrospective study). Main clinical and follow-up data of the study population are detailed in Table 1.
Thirteen patients received a prophylactic AED, of whom 8 had a confirmed diagnosis of SWS on the second MRI. Nine patients developed epilepsy during the follow-up period, among whom 5 had received prophylactic AEDs. The first seizure occurred in all patients before the second MRI at a median age of 6 months (range 2-8). All children presenting epilepsy had an associated leptomeningeal angioma confirmed by the second MRI. The first seizures were reported as focal in all cases, contralateral to the leptomeningeal angioma. Three patients presented as status epilepticus, while another 3 patients developed status epilepticus at respectively 6 months, 13 months and 5 years old. Three patients underwent surgery for refractory epilepsy, 2 for a hemispherotomy at 15 months old, and one for a lobectomy at 17 months old. At the last visit, all 9 patients were seizure free. Five patients received monotherapy, 3 patients received bitherapy and one stopped AEDs.
31
4.4.2. MRI results
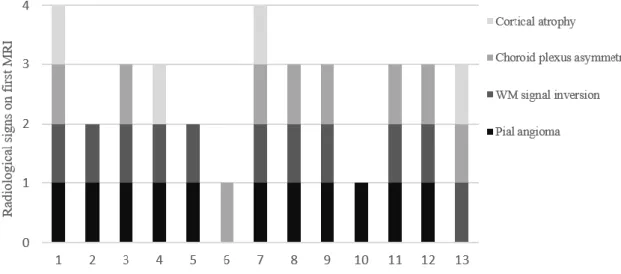
The median age at the first MRI was 2 months (range 0-3) and 12 months at the time of the second MRI (range 9-36). Thirteen patients had a diagnosis of SWS confirmed on the second MRI by the enhancement of the angioma with gadolinium (Figure 1, D). Out of these 13 patients, 11 already had a leptomeningeal contrast enhancement on the first MRI, which was better seen on images with fat saturation (Figure 1, A). Ten of them also had associated indirect signs of leptomeningeal angioma on the first MRI (Figure 2). A signal inversion of the white matter with T1 hypersignal and T2 hyposignal were the most often reported (Figure 1, B-C). The two remaining patients did not have visible leptomeningeal contrast enhancement, but presented at least one indirect sign of leptomeningeal angioma: one with all four indirect signs and the other with an isolated choroid plexus asymmetry (Figure 3). The diagnosis of SWS was ruled out by the second MRI in 17 patients. The first MRI of 16 patients came out negative for direct and indirect signs of leptomeningeal angioma, and in only one case was there an isolated direct sign (suspicion of leptomeningeal enhancement) (Figure 4). The second MRI of 3 patients confirmed the presence of bilateral leptomeningeal angioma. Given the visibility of a leptomeningeal contrast enhancement contralateral on the facial PWS, the results had already been suggested on the first MRI. Despite the good predictability of the early MRI, the extension of leptomeningeal angioma was underestimated in 9 out of 13 patients. The leptomeningeal contrast enhancement that was often restricted to the occipital lobe on the first MRI extended to more lobes on the second MRI perhaps on account of the angioma increasing in size with age or its better visibility. Progressive cortical atrophy was present on the second MRI of all 9 patients with epilepsy, but only in 1 out of 4 patients with no history of seizures.
Cross tabulation of the early MRI results with the results of the later MRI is represented in Table 2. Table 3 shows the diagnostic accuracy of the early MRI in predicting leptomeningeal angioma. In our population, having one direct sign with at least one indirect sign of
32 leptomeningeal angioma on the first MRI performed in patients younger than 3 months could confirm the presence of the leptomeningeal angioma. An isolated direct sign should be carefully considered, as it might represent a false negative, unlike indirect signs that are more indicative of a positive diagnosis even in the absence of gadolinium enhancement.
4.5. Discussion
This is the first study to evaluate the diagnostic performance of early cerebral MRI in the detection of leptomeningeal angioma before the age of 3 months. A pre-symptomatic diagnosis of SWS remains a major challenge with regards to establishing a prognosis for the families, identifying patients at risk of seizures, and proposing possible preventive therapeutic strategies. Our data show that cerebral MRI with gadolinium enhancement has an excellent sensitivity and specificity for the early detection of leptomeningeal angioma in patients under the age of 3 months. The presence of leptomeningeal enhancement on T1-weighted imaging led to the correct diagnosis in up to 85% of SWS children on the early MRI with a sensitivity and specificity of 85 and 94%, respectively. Screening of indirect signs increased the sensibility of the early MRI up to 100% and permitted, along with the presence of the direct sign, detecting all patients with leptomeningeal angioma confirmed by later MRI.
Found in almost 80% of cases, white matter abnormalities were the most frequent indirect signs associated with the leptomeningeal angioma (T1 hypersignal and T2 hyposignal). Described for the first time by Jacoby in 1987,27 these abnormalities are thought to be related to an accelerated myelination process as suggested by the hyperperfusion visualized in the same area on SPECT imaging29 and the increase of the fraction of anisotropy with a decrease of the apparent diffusion coefficient in diffusion-tensor imaging.30 However, these abnormalities are rarely reported, as most imaging series involve older children whose MRI pattern changes after neurological complications.19,24,31
33 Enlargement of the choroid plexus is a well-recognized feature of Sturge-Weber syndrome,32– 34 first described on Computed Tomography scans.28,33 However, this sign is not constant since it was found in 9 out of 13 patients, emphasizing the superiority of the MRI already reported in previous studies.28,34 Its pathophysiology remains unclear, but could be related to a vascular engorgement of the choroid plexus upstream from the leptomeningeal angioma.32 More common in later stages of the disease because of chronic hypoperfusion and hypoxia,19,35 cortical atrophy was rare on the early MRI, but was highly specific (100% associated with an underlying leptomeningeal angioma).
The gradient Echo T2* sequence should be added in the MRI protocols of these children for the detection of early microcalcifications, historically the first radiological diagnostic sign on skull radiographs.28 Gadolinium-enhanced fluid-attenuated inversion-recovery (FLAIR) imaging may increase the sensitivity for detecting leptomeningeal disease when compared with routine contrast-enhanced T1-weighted imaging.36,37 This could be partly due to the suppression of signal intensity from normal vascular structures on the surface of the brain, allowing easier visualization of abnormal leptomeninges.37 Contrast-enhanced FLAIR imaging is not currently performed in routine pediatric MRI protocols even though it should probably be the case for suspected SWS.
More recent MRI techniques such as susceptibility weighted imaging (SWI) or diffusion tensor imaging (DTI) could identify enlarged trans medullary collaterals veins and microstructural white matter damage associated with the leptomeningeal vascular malformation.26,38 Diagnostic accuracy of these sequences has not yet been assessed in pre-symptomatic children. Functional imaging based on glucose metabolism (FDG-PET) or cerebral perfusion (SPECT) provide complementary information to the conventional MRI.19,38–42 These techniques use radioactive tracers, are available in specialized centers and often reserved for the presurgical workup ahead of an epilepsy surgery. New techniques like arterial spinal labeling which quantifies cerebral
34 blood flow without the need for contrast injection could provide information on the leptomeningeal angioma diagnosis and the progression of the disease.43,44
A newborn with a facial PWS on at least one part of the forehead and/or upper eyelid should have an early contrast-enhanced MRI during the first months of life. The radiologist should be aware of the lateralization of the facial PWS to carefully look for an ipsilateral leptomeningeal enhancement. Rather than routine post-gadolinium T1-weighted imaging, post-gadolinium FLAIR and T1-weighted acquisition with fat saturation should be performed first, as they appear to be more sensitive to enhancement detection. Screening for ipsilateral indirect signs is also essential for raising the diagnostic suspicion if the leptomeningeal angioma is not seen and to reinforce the suspicion in case of visible leptomeningeal enhancement. Finally, the extension of the angioma should be carefully stated on the first MRI since it was often underestimated. Having direct or indirect signs on an early MRI increases the probability of a patient having SWS from around 15% in the literature5,9 to over 75% in our study. In the absence of the direct and indirect signs of leptomeningeal angioma, the diagnosis of SWS can be reasonably ruled out, reassuring the family and eliminating the need for preventative care. A proposal for a diagnostic approach is summarized in Figure 5.
Our study shows some limitations. The number of children included in the prospective cohort was limited by the low prevalence of the disease. The addition of retrospective data increased the total number and constituted the largest cohort of infants under the age of one year with a SWS diagnosis. MRI images were analyzed by a neuroradiologist of a tertiary medical center, who could have overestimated the presence of the indirect signs due to the frequently associated visualization of the leptomeningeal angioma.
35
4.6. Conclusion
We showed that early diagnosis of SWS is possible on a contrast-enhanced MRI performed in asymptomatic neonates with facial PWS through reliable radiological markers for the detection of the leptomeningeal angioma. Recognition of pre-symptomatic brain involvement is essential to identify population at risk of neurological complications, to help families with the prognosis and organizing subsequent medical follow-ups. This early pre-symptomatic diagnosis might facilitate the development of clinical trials to evaluate the safety and efficacy of current and future neuroprotective strategies for SWS.
36
4.7. References
1. Shirley MD, Tang H, Gallione CJ, et al. Sturge–Weber Syndrome and Port-Wine Stains Caused by Somatic Mutation in GNAQ. N Engl J Med. 2013;368(21):1971-1979.
2. Nakashima M, Miyajima M, Sugano H, et al. The somatic GNAQ mutation c.548G>A (p.R183Q) is consistently found in Sturge-Weber syndrome. J Hum Genet. 2014;59(12):691-693.
3. Huang L, Couto JA, Pinto A, et al. Somatic GNAQ Mutation is Enriched in Brain Endothelial Cells in Sturge-Weber Syndrome. Pediatr Neurol. 2017;67:59-63.
4. Kanada KN, Merin MR, Munden A, Friedlander SF. A prospective study of cutaneous findings in newborns in the United States: correlation with race, ethnicity, and gestational status using updated classification and nomenclature. J Pediatr. 2012;161(2):240-245.
5. Piram M, Lorette G, Sirinelli D, Herbreteau D, Giraudeau B, Maruani A. Sturge-Weber syndrome in patients with facial port-wine stain. Pediatr Dermatol. 2012;29(1):32-37.
6. Comi AM. Presentation, diagnosis, pathophysiology, and treatment of the neurological features of Sturge-Weber syndrome. The Neurologist. 2011;17(4):179-184.
7. Enjolras O, Riche MC, Merland JJ. Facial port-wine stains and Sturge-Weber syndrome.
Pediatrics. 1985;76(1):48-51.
8. Ch’ng S, Tan ST. Facial port-wine stains - clinical stratification and risks of neuro-ocular involvement. J Plast Reconstr Aesthetic Surg JPRAS. 2008;61(8):889-893.
9. Dutkiewicz A-S, Ezzedine K, Mazereeuw-Hautier J, et al. A prospective study of risk for Sturge-Weber syndrome in children with upper facial port-wine stain. J Am Acad Dermatol. 2015;72(3):473-480.
10. Waelchli R, Aylett S, Robinson K, Chong W, Martinez A, Kinsler V. New vascular classification of port-wine stains: improving prediction of Sturge–Weber risk. Br J Dermatol. 2014;171(4):861-867.
11. Nabbout R, Juhász C. Sturge-Weber syndrome. Handb Clin Neurol. 2013;111:315-321. 12. Pinto A, Sahin M, Pearl PL. Epileptogenesis in neurocutaneous disorders with focus in Sturge Weber syndrome. F1000Research. 2016;5. Retrieved February 6, 2017, from http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4805158/.
13. Sujansky E, Conradi S. Sturge-Weber syndrome: age of onset of seizures and glaucoma and the prognosis for affected children. J Child Neurol. 1995;10(1):49-58.
14. Pascual-Castroviejo I, Pascual-Pascual S-I, Velazquez-Fragua R, Viaño J. Sturge-Weber syndrome: study of 55 patients. Can J Neurol Sci J Can Sci Neurol. 2008;35(3):301-307. 15. Bosnyák E, Behen ME, Guy WC, Asano E, Chugani HT, Juhász C. Predictors of Cognitive Functions in Children With Sturge-Weber Syndrome: A Longitudinal Study. Pediatr Neurol. 2016;61:38-45.
16. Jagtap S, Srinivas G, Harsha KJ, Radhakrishnan N, Radhakrishnan A. Sturge-Weber syndrome: clinical spectrum, disease course, and outcome of 30 patients. J Child Neurol. 2013;28(6):725-731.
17. Udani V, Pujar S, Munot P, Maheshwari S, Mehta N. Natural history and magnetic resonance imaging follow-up in 9 Sturge-Weber Syndrome patients and clinical correlation. J
Child Neurol. 2007;22(4):479-483.
18. Kossoff EH, Ferenc L, Comi AM. An infantile-onset, severe, yet sporadic seizure pattern is common in Sturge-Weber syndrome. Epilepsia. 2009;50(9):2154-2157.
19. Miao Y, Juhász C, Wu J, et al. Clinical correlates of white matter perfusion changes in Sturge-Weber syndrome: A dynamic MR perfusion-weighted imaging study. Ajnr Am J
37 20. Sudarsanam A, Ardern-Holmes SL. Sturge-Weber syndrome: from the past to the present.
Eur J Paediatr Neurol EJPN Off J Eur Paediatr Neurol Soc. 2014;18(3):257-266.
21. Mantelli F, Bruscolini A, La Cava M, Abdolrahimzadeh S, Lambiase A. Ocular manifestations of Sturge–Weber syndrome: pathogenesis, diagnosis, and management. Clin
Ophthalmol Auckl NZ. 2016;10:871-878.
22. Ville D, Enjolras O, Chiron C, Dulac O. Prophylactic antiepileptic treatment in Sturge-Weber disease. Seizure. 2002;11(3):145-150.
23. Bay MJ, Kossoff EH, Lehmann CU, Zabel TA, Comi AM. Survey of aspirin use in Sturge-Weber syndrome. J Child Neurol. 2011;26(6):692-702.
24. Maria BL, Neufeld JA, Rosainz LC, et al. Central nervous system structure and function in Sturge-Weber syndrome: evidence of neurologic and radiologic progression. J Child Neurol. 1998;13(12):606-618.
25. Comi A. Current Therapeutic Options in Sturge-Weber Syndrome. Semin Pediatr Neurol. 2015;22(4):295-301.
26. Hu J, Yu Y, Juhasz C, et al. MR Susceptibility Weighted Imaging (SWI) Complements Conventional Contrast Enhanced T1 Weighted MRI in Characterizing Brain Abnormalities of Sturge-Weber Syndrome. J Magn Reson Imaging JMRI. 2008;28(2):300-307.
27. Jacoby CG, Yuh WT, Afifi AK, Bell WE, Schelper RL, Sato Y. Accelerated myelination in early Sturge-Weber syndrome demonstrated by MR imaging. J Comput Assist Tomogr. 1987;11(2):226-231.
28. Griffiths PD. Sturge-Weber syndrome revisited: the role of neuroradiology.
Neuropediatrics. 1996;27(6):284-294.
29. Adamsbaum C, Pinton F, Rolland Y, Chiron C, Dulac O, Kalifa G. Accelerated myelination in early Sturge-Weber syndrome: MRI-SPECT correlations. Pediatr Radiol. 1996;26(11):759-762.
30. Moritani T, Kim J, Sato Y, Bonthius D, Smoker WRK. Abnormal hypermyelination in a neonate with Sturge-Weber syndrome demonstrated on diffusion-tensor imaging. J Magn
Reson Imaging JMRI. 2008;27(3):617-620.
31. Chiron C, Raynaud C, Tzourio N, et al. Regional cerebral blood flow by SPECT imaging in Sturge-Weber disease: an aid for diagnosis. J Neurol Neurosurg Psychiatry. 1989;52(12):1402-1409.
32. Griffiths PD, Blaser S, Boodram MB, Armstrong D, Harwood-Nash D. Choroid plexus size in young children with Sturge-Weber syndrome. AJNR Am J Neuroradiol. 1996;17(1):175-180.
33. Stimac GK, Solomon MA, Newton TH. CT and MR of angiomatous malformations of the choroid plexus in patients with Sturge-Weber disease. AJNR Am J Neuroradiol. 1986;7(4):623-627.
34. Benedikt RA, Brown DC, Walker R, Ghaed VN, Mitchell M, Geyer CA. Sturge-Weber syndrome: cranial MR imaging with Gd-DTPA. AJNR Am J Neuroradiol. 1993;14(2):409-415. 35. Pinto AL, Chen L, Friedman R, et al. Sturge-Weber Syndrome: Brain Magnetic Resonance Imaging and Neuropathology Findings. Pediatr Neurol. 2016;58:25-30.
36. Griffiths PD, Coley SC, Romanowski CAJ, Hodgson T, Wilkinson ID. Contrast-Enhanced Fluid-Attenuated Inversion Recovery Imaging for Leptomeningeal Disease in Children. Am J
Neuroradiol. 2003;24(4):719-723.
37. Mathews VP, Caldemeyer KS, Lowe MJ, Greenspan SL, Weber DM, Ulmer JL. Brain: Gadolinium-enhanced Fast Fluid-attenuated Inversion-Recovery MR Imaging. Radiology. 1999;211(1):257-263.
38. Juhász C, Hu J, Xuan Y, Chugani HT. Imaging increased glutamate in children with Sturge-Weber syndrome: Association with epilepsy severity. Epilepsy Res. 2016;122:66-72.
38 39. Juhász C, Haacke EM, Hu J, et al. Multimodality Imaging of Cortical and White Matter Abnormalities in Sturge-Weber Syndrome. Am J Neuroradiol. 2007;28(5):900-906.
40. Alkonyi B, Miao Y, Wu J, et al. A perfusion-metabolic mismatch in Sturge-Weber syndrome: a multimodality imaging study. Brain Dev. 2012;34(7):553-562.
41. Lin DDM, Barker PB, Hatfield LA, Comi AM. Dynamic MR perfusion and proton MR spectroscopic imaging in Sturge-Weber syndrome: correlation with neurological symptoms. J
Magn Reson Imaging JMRI. 2006;24(2):274-281.
42. Evans AL, Widjaja E, Connolly DJA, Griffiths PD. Cerebral perfusion abnormalities in children with Sturge-Weber syndrome shown by dynamic contrast bolus magnetic resonance perfusion imaging. Pediatrics. 2006;117(6):2119-2125.
43. Blauwblomme T, Naggara O, Brunelle F, et al. Arterial spin labeling magnetic resonance imaging: toward noninvasive diagnosis and follow-up of pediatric brain arteriovenous malformations. J Neurosurg Pediatr. 2015;15(4):451-458.
44. Wang J, Licht DJ, Jahng G-H, et al. Pediatric perfusion imaging using pulsed arterial spin labeling. J Magn Reson Imaging JMRI. 2003;18(4):404-413.
39
4.8. Tables and Figures
Table 1. Demographic, clinical and follow-up data of the population according to the presence or
absence of leptomeningeal angioma.
All SWS No SWS
n = 30 (%) n = 13 (%) n = 17 (%)
Age at first exam* (d) 50,5 (23.2-62.5) 47 (18.0-60.0) 54 (32.0-66.0)
Sex Female 15 (50%) 3 (23%) 12 (71%) Male 15 (50%) 10 (77%) 5 (29%) Side of PWS Right 10 (33%) 5 (38%) 5 (29%) Left 13 (43%) 4 (31%) 9 (53%) Bilateral 7 (23%) 4 (31%) 3 (18%) Location of PWS V1 12 (40%) 3 (23%) 9 (53%) V1+V2 5 (17%) 2 (15%) 3 (18%) V1+V2+V3 13 (43%) 8 (62%) 5 (29%) Prophylactic AED 13 (43%) 8 (62%) 5 (29%) Follow-up Duration* 26.5 (10.8-51.8) 51.0 (27.0-62.0) 17.0 (7.0-28.0) Glaucoma 8 (27%) 7 (54%) 1 (6%) Epilepsy 9 (30%) 9 (69%) 0 Hemiparesis 8 (27%) 8 (62%) 0
40
Table 2. Cross tabulation of MRI results.
MRI ≤ 3 months old MRI ≥ 9 months old
SWS No SWS n=13 % n=17 % Negative 0 16 Direct sign 11 (85%) 1 (8%) Indirect sign T1 hypersignal 10 (77%) 0 T2 hyposignal 10 (77%) 0
choroid plexus asymmetry 9 (69%) 0
cortical atrophy 4 (31%) 0
Table 3. Diagnostic accuracy of early MRI according to the presence of radiological
markers (95% confidence interval).
Early MRI Sensitivity Specificity Posttest Probability
Presence of the direct sign 85 (55-98) 94 (71-100) 68
Direct or indirect sign 100 (75-100) 94 (71-100) 72
Direct and ≥ 1 indirect sign 77 (46-95) 100 (80-100) -