ENRICHISSEMENT DE PAIRES DE GÈNES DONT LES
INTERACTIONS CAUSENT LA SCHIZOPHRÉNIE À
L’AIDE DE BASES DE DONNÉES GÉNOMIQUES ET
APPLICATION À UNE ÉTUDE D’ASSOCIATION
CAS-TÉMOINS DE L’EST DU QUÉBEC.
Mémoire
Simon Noël
Maîtrise en neurobiologie - Neurobiologie
Maître ès sciences (M. Sc.)
Québec, Canada
iii
Résumé
Nous essayons de trouver de nouvelles interactions géniques pouvant donner une résistance ou une susceptibilité pour développer la schizophrénie. Nous avons donc fait l’enrichissement de voies en utilisant GSEA et Biofilter. Nous avons ensuite cherché de nouvelles interactions avec la méthode JE et la régression logistique parmi les paires de gènes identifiées. De plus, nous avons obtenu plus de résultats statistiquement significatifs qu’une sélection se basant sur les valeurs d’association marginale. Par ailleurs, les résultats pointent certains candidats intéressants comme le gène NRXN1 qui code pour une protéine d’adhésion cellulaire du système nerveux et qui aurait une interaction causant une susceptibilité avec le gène ROBO1, un gène impliqué dans la guidance des axones, et une autre avec le gène CDH13, un gène jouant le rôle de régulateur négatif dans la croissance des axones. Ces trois gènes sont déjà liés à la schizophrénie dans la littérature et pourraient servir de biomarqueurs.
v
Table des matières
RÉSUMÉ ... III TABLE DES MATIÈRES ... V LISTE DES FIGURES ... IX LISTE DES TABLEAUX ... XI LISTE D’ABRÉVIATIONS ... XIII AVANT-PROPOS ... XV
CHAPITRE 1 ... 1
1.1 INTRODUCTION ... 2
1.2 OBJECTIFS ... 5
1.2.3 Aperçu des étapes ... 5
CHAPITRE 2 ... 7
2.1 MATÉRIEL ET MÉTHODE ... 8
2.1.1 Population/données ... 8
2.1.2 Association marginale ... 9
2.1.3 Interaction gène-gène/Épistasie ... 10
2.1.4 Méthodes de filtrage basées sur les connaissances biologiques ... 11
2.1.5 Logiciel et base de données ... 12
2.1.5.1 GO ...13 2.1.5.2 GEA ...13 2.1.5.3 MSigDB ...14 2.1.5.4 DAVID ...14 2.1.5.5 BIOFILTER ...15 2.1.5.6 GSEA ...16 2.1.5.7 JE ...18 2.1.5.8 R ...20 2.1.5.9 Perl ...20 2.1.5.10 Osprey ...21 CHAPITRE 3 ... 23
vi
3.1 RÉSULTATS ... 24
3.1.1 Résultats de cartographie et filtrage ... 24
3.1.2 Résultats GSEA ... 26
3.1.3 Résultats Biofilter ... 27
3.1.4 Résultats Régression logistique ... 28
3.1.5 Résultats JE ... 28 3.1.6 Résultats retenus ... 30 3.1.7 Résultats Osprey ... 46 CHAPITRE 4 ... 49 4.1 DISCUSSION ... 50 4.1.1 Cartographie et filtrage ... 51 4.1.2 GSEA ... 51 4.1.3 Biofilter ... 52 4.1.4 Régression logistique VS JE ... 53 4.1.5 Enrichissement ... 53 4.1.6 Résultats retenus ... 54 4.1.7 Osprey ... 55
4.1.8 Comparaison avec Jia ... 56
4.1.9 Littérature ... 57
4.1.10 Élagage neuronal ... 58
4.1.11 Autre approche essayée ... 59
4.2 CONCLUSION ... 60
BIBLIOGRAPHIE ... 63
ANNEXE ... 73
A1 : Résumé graphique du projet ... 73
vii
A3 : Information supplémentaire sur les différentes bases de données ... 79
A4: Détails sur l’épistasie ... 81
A5 : Résumé MSigDB ... 85
A6 : Résumé des tests pour paramétrer Biofilter ... 87
A7 : Détails des tests pour paramétrer GSEA ... 95
ix
Liste des figures
FIGURE 1 : FORMULE POUR LE CALCUL DE L’ENRICHISSEMENT DE GSEA ... 17
FIGURE 2 : KCNQ1 VS NAV2 ... 31 FIGURE 3 : RELN VS CTNND2 ... 32 FIGURE 4 : GRM3 VS GRM7 ... 33 FIGURE 5 : ADCY8 VS PRDM14 ... 34 FIGURE 6 : ROBO1 VS NRXN1 ... 35 FIGURE 7 : CDH13 VS NRXN1 ... 36 FIGURE 8 : TLK1 VS PDIA6 ... 37 FIGURE 9 : PLCB1 VS PLCL2 ... 38 FIGURE 10 : GPC5 VS PKNOX2 ... 39
FIGURE 11 : FAIM2 VS SHANK2 ... 40
FIGURE 12 : CDH13 VS CYCS ... 41
FIGURE 13 : DLG2 VS RGS7 ... 42
FIGURE 14 : CAMK2D VS KCNQ5 ... 43
xi
Liste des tableaux
TABLEAU 1 : TABLEAU DE FREQUENCE DE GENOTYPE A 2 LOCUS (G ET H) ... 10
TABLEAU 2 : EXEMPLE DE NOTRE FICHIER DE DONNÉES... 19
TABLEAU 3 : RESUME CARTOGRAPHIE ET FILTRE ... 25
TABLEAU 4 : RESULTATS GSEA ... 26
TABLEAU 5 : RESULTATS BIOFILTER ... 28
TABLEAU 6 : RESUME DES RESULTATS DE JE ... 29
TABLEAU 7 : RESUME INTERACTION ... 43
TABLEAU 8 : BIOMARQUEUR POTENTIEL ... 48
TABLEAU 9 : COMPARAISON AVEC JIA ... 56
TABLEAU 10 : MODIFICATION POUR CORRESPONDRE A JIA ... 57
xiii
Liste d’abréviations
ADN : Acide DésoxyriboNucléiqueALIGATOR : Association LIst Go AnnoTatOR BED : Best-Estimate DSM-III-R Diagnosis Ch : Chromosome
CRIUSMQ : Centre de Recherche de l’Institut Universitaire en Santé Mentale de Québec
CRULRG : Centre de Recherche de l’Université Laval affilié à Robert Giffard DAVID : Database for Annotation, Visualization and Integrated Discovery. DIP : The Database of Interacting Proteins
DSM : Diagnostic and Statistical Manual of mental disorders DSM-III : DSM - Troisième révision générale
DSM-III-R : Révision du DSM-III publiée en 1987 DSM-IV : DSM - Quatrième révision générale ES : Enrichment Score
GEA : Gene Expression Atlas GO : Gene Ontology
GSEA : Gene Set Enrichment Analysis
IUSMQ : Institut Universitaire en Santé Mentale de Québec JE : Joint Effect
KEGG : Kyoto Encyclopedia of Genes and Genomes
MAGENTA : Meta-Analysis Gene-set Enrichment of variaNT Associations MSigDB : Molecular Signatures Database
xiv
xv
Avant-propos
Ce mémoire de Maîtrise est le résultat de plusieurs mois de travail et d’innovations. La route ne fut pas toujours facile. De nombreuses embûches se sont présentées. Je dois donc une fière chandelle à une merveilleuse équipe qui m’a supporté tout au long de mon projet.
J’aimerais dans un premier temps remercier mon directeur, M. Alexandre Bureau. Sa grande patience est tout à son honneur. Il a su aussi démontré une grande générosité de son temps et fourni de précieux conseils.
J’aimerais aussi remercier mon codirecteur, M. Simon Hardy. Chacune de nos rencontres fut très enrichissante. Il a su amener de l’eau au moulin chaque fois que je pensais être en période de sécheresse.
Je voudrais également remercier du fond du cœur une équipe du tonnerre du CRIUSMQ. Un merci tout spécial à M. Jordie Croteau, M. Molière Nguilé Makao , M. David Dubé St-Hilaire et M. Thomas Paccalet. Vous avez été les meilleurs alliés que l’on peut espérer avoir pour relever les divers défis que ma Maîtrise a offerts.
Une Maîtrise ne se fait pas sans un certain financement. J’aimerais donc remercier l’Institut canadien de recherche en santé (IRSC, subvention MOP-106448) et le Fonds de recherche du Québec – Santé (FRSQ).
Je dois par ailleurs souligner l’immense contribution du Dr Michel Maziade pour l’accès aux données de l’échantillon de patients schizophrènes et de témoins.
En terminant, j’aimerais remercier famille et amis. Vous avez été un support moral essentiel tout au long de ma Maîtrise. Derrière chaque grand homme, il y a une très grande femme. J’aimerais terminer avec un merci tout spécial à une super
xvi
grande femme. Merci Julie, ma tendre amour(e). Tu as été pour moi tel un phare dans la nuit. Ta présence, autant dans les bons que dans les mauvais moments, a été pour moi une source d’inspiration.
1
2
1.1 Introduction
Depuis la nuit des temps, l’homme a été confronté à la maladie mentale. De par le passé, l’absence de connaissance scientifique et le désir de l’Homme de tout comprendre ont amené de fausses croyances comme la sorcellerie, la possession démoniaque, le vaudou, etc. Par la suite, les connaissances ont évolué et le surnaturel fut remplacé par la maladie. Face à l’incompréhension, nous utilisions la lobotomie ou les électrochocs. Avec l’avancement des connaissances, on en est venu à la médication. Cette méthode n’est malgré tout pas parfaite. La science continue toujours d’avancer et aujourd’hui, on étudie les mécanismes mêmes des maladies mentales par diverses techniques telles que le séquençage et l’identification de variants rares. Une meilleure connaissance des mécanismes d’une maladie permet l’identification de biomarqueurs pour diagnostiquer plus tôt la maladie et permet des thérapies pharmaceutiques beaucoup plus ciblées et efficaces. Certaines maladies comme l’épilepsie sont maintenant considérées comme étant le symptôme d’autres maladies. La science avance, mais pour la schizophrénie, la route est encore longue. Le projet de ma maîtrise consiste à étudier cette maladie au niveau génétique par le biais de l’analyse des interactions géniques dans le cadre d’une étude d’association. La schizophrénie échappe encore à notre compréhension, car elle est une maladie très complexe. Nous pouvons définir la schizophrénie comme étant « une psychose grave survenant chez l'adulte jeune, habituellement chronique, cliniquement caractérisée par des signes de dissociation mentale, de discordance affective et d'activité délirante incohérente, entraînant généralement une rupture de contact avec le monde extérieur et parfois un repli autistique. Schizophrénie vient du grec skizein : fendre et de phren : pensée.» [Infirmiers.com, 2010]. Aux facteurs environnementaux s’ajoute un arrière-plan multigénique très complexe. De plus, les psychiatres de l’équipe du CRULRG ainsi que ceux un peu partout dans le monde s’accordent à dire qu’il existe plusieurs types de schizophrénie. Leur désaccord est cependant très grand quand vient le temps de les définir. Le problème déjà complexe peut même être empiré par ce manque de classification. C'est comme si nous en étions
3
à comparer des pommes à des oranges tout en tentant de trouver les causes de la maladie.
Le côté multigénique de la maladie la rend très complexe à analyser, mais cette tâche n’est pas impossible. En effet, d’autres maladies multigéniques complexes comme la maladie de Hirschsprung [Cantrell, 2004] [Owens, 2005] [De Pontual, 2009] [Tam, 2009] ou de Bardet-Biedl [Badano, 2005] ont maintenant plusieurs de leurs interactions qui sont bien caractérisées. L’avancement constamment grandissant de la bio-informatique met à notre disposition des outils de plus en plus avancés, nombreux et précis. Nous pouvons parler ici d’outils d’enrichissement comme ALIGATOR [Holmans, Peter], Biofilter [Ritchie Lab], GSEA [BROAD Institute] et MAGENTA [BROAD Institute]. Nous pouvons aussi parler d’outils d’associations comme PLINK [Purcell] et JE [Cordell, Heather J, et Masao Ueki]. Il y a aussi des bases de données complexes comme DAVID [National Institute of Allergy and Infectious Diseases], GO [Open Biological and Biomedical Ontologies] et GEA [European Molecular Biology Laboratory - European Bioinformatics Institute]. Nous pouvons aussi inclure les logiciels de représentations graphiques comme Osprey [Tyers]. Il existe aussi plusieurs outils avancés de Bio-informatique. La communauté qui entoure cette discipline est aussi très active et dynamique. Nous pouvons parler ici par exemple de la communauté Bioconductor [Anon] qui développe des modules de bio-informatique pour le logiciel R [CRAN]. Comme une Maîtrise possède un cadre temporel limité, nous n‘utiliserons pas toutes ces ressources.
Nous allons travailler sur une population venant de l’Est du Québec, endroit reconnu pour avoir une population dérivée d’un effet fondateur entraînant une forte homogénéité génétique, et donc, étant un bon bassin pour la recherche de variante rare de gènes qui s’y retrouvent en plus grande fréquence que dans les autres bassins de populations selon [Moreau, 2007]. Nos données sont sous la forme d’étude cas-témoins telle que rapportée dans l’article de Ripke [Ripke et coll., 2013]. Il est cependant commun de voir que certaines études se font sur des
4
familles afin de minimiser l’impact négatif de la mauvaise compréhension de la maladie en minimisant l’hétérogénéité génétique puisque les atteints d’une même famille ont les même gènes [Maziade, 2005]. Il est aussi commun de voir des études sur des populations plus vastes [Jia, 2010]. Nous n’avons pas choisi cette approche en raison de la trop grande hétérogénéité de ce type de bassin génétique qui pourrait entraîner des faux négatifs. [Ritchie, 2011] adopte une démarche et une approche très similaire à la démarche de notre projet et des différentes approches que nous utiliserons. Elle démontre aussi l’importance de filtrer nos données afin de diminuer le nombre de calculs. De plus, ces nouvelles interactions que nous tenterons d’identifier pourront servir de biomarqueurs de la maladie.
5
1.2 Objectifs
La schizophrénie est une grave maladie multi génique complexe. Dans le cadre de mon projet, nous tenterons de trouver de nouvelles interactions géniques pouvant expliquer la maladie en complétant les étapes suivantes :
- Filtrer les SNPs du jeu de données pour assurer leur qualité et les gènes que l’on peut associer à ces SNPs pour garder le plus pertinent à la schizophrénie afin de diminuer le nombre de calculs.
- Comparer le nombre d’interactions trouvées avec la méthode JE sur un ensemble de SNPs enrichi par GSEA ou par Biofilter calibré par la maladie de Hirschsprung ou de Bardet-Biedl avec les résultats de l’ensemble de SNPs sélectionnés par association marginale.
- Trouver les interactions géniques déjà connues en utilisant Biofilter. - Identifier des voies qui peuvent être enrichi via GSEA.
- Chercher de nouvelles interactions avec une régression logistique et la méthode JE parmi les paires de gènes identifiés par GSEA ou Biofilter
-Tracer le graphe des interactions trouvées.
1.2.3 Aperçu des étapes
L’annexe A1 illustre très bien l’ensemble du projet. Nous partirons des résultats obtenus préalablement par l’équipe du Dr Bureau et qui nous serviront à classer/trier notre liste de gènes retenus pour, soit faire des tests d’interactions, soit faire de la prédiction de voies (pathways) pour ensuite faire des tests
6
d’interactions. Pour faire notre prédiction de voies (pathways), nous utiliserons la méthode d’enrichissement de jeux de gènes telle qu’implantée dans le logiciel GSEA que nous utiliserons avec une liste de voies (pathways) déjà connues en faisant 1000 permutations ou Biofilter qui nous donnera la liste des interactions déjà connues. Afin de limiter nos recherches au cerveau, nous utiliserons une liste de gènes s’exprimant dans le cerveau et provenant de GEA pour filtrer les prédictions d’interactions de Biofilter ainsi que nos tests d’interactions afin de diminuer le nombre de calculs. En parallèle, nous calibrerons Biofilter en nous basant sur les résultats obtenus en se référant à des maladies multigéniques complexes mieux comprises comme la maladie de Hirschsprung ou celle de Bardet-Biedl. Les interactions seront recherchées par des analyses d’association. Nous ferons aussi de nombreux tests sur nos différents logiciels afin de bien comprendre la portée de chaque paramètre. Le gène encore inconnu responsable de l’association du SNP rs1156026 à la schizophrénie trouvée par l’équipe du CRULRG (d’ailleurs, ce gène, sans nom actuellement, sera appelé ainsi pour mon projet) sera ajouté systématiquement à chaque analyse. Nous ferons nos analyses d’association sur les résultats de GSEA et Biofilter avec JE. Nous comparerons aux résultats d’association marginale directement soumis à JE. Bien sûr, nous résumerons et interpréterons nos résultats.
7
8
2.1 Matériel et méthode
Nous présenterons dans cette section les différentes méthodes et logiciels que nous avons utilisés dans le cadre de ce projet.
2.1.1 Population/données
Pour ce projet de maîtrise, nous avons à notre disposition les données génétiques de personnes de l’Est du Québec atteintes ou non de schizophrénie recrutées dans une étude dirigée par Dr M. Maziade [Bureau, 2013]. 247 personnes étaient atteintes et 250 étaient des contrôles. Un cas était défini comme étant une personne ayant reçu un diagnostic de schizophrénie. Pour établir ce diagnostic, une entrevue structurée pour le DSM-III-R ou le DSM-IV couplée aux informations parentales et aux dossiers médicaux complets a été faite. Par la suite, un panel de 4 psychiatres a fait un BED donnant le diagnostic [Maziade, 2005].
L’hybridation a été faite pour des SNPs sur une puce Illumina HumanHap300 personnalisée avec 57 000 SNPs additionnels. Nous avons donc une liste de SNPs. Un SNP est une variation d’une seule paire de bases qui peut se retrouver n’importe où dans le génome. Comme nous voulons travailler avec des gènes, nous devons trouver quels SNPs se retrouvent dans un gène ou proche d’un gène et quels SNPs se retrouvent dans une partie non codante de notre ADN. Nous avons donc rattaché nos SNPs à un ou des gènes. Nous disons « des », car il y a chevauchement de gènes pour certains SNPs. Pour rattacher un SNP à un gène, nous avons utilisé 2 approches. Premièrement, nous avons vérifié si le SNP était à l’intérieur de la partie codante du gène (exon) ou à moins de 50k bases de ce dernier (exon ou intron) afin de tenir compte du déséquilibre de liaison. Nous avons aussi refait les mêmes choses, mais pour une cartographie directe, c’est-à-dire que pour être associé à un gène, un SNP doit c’est-à-directement s’y retrouver (exon). L’équipe de notre centre de recherche a déjà fait quelques analyses sur
9
les données que nous avons notre disposition comme vérifier l’équilibre de Hardy-Weinberg par exemple ou encore vérifier les fréquences alléliques, ou bien vérifier le bon appariement des cas et témoins selon les composantes principales de variation génique, etc [Quackenbush, 2002]. Elle n’a cependant pas effectué d’analyse sur les chromosomes X et Y et l’ADN mitochondrial. Comme nous partons de ces analyses, nous nous retrouvons donc à avoir quelques gènes en moins. Afin de profiter au maximum des analyses précédentes, les SNPs restants ont ensuite été filtrés avec des critères définis préalablement par notre équipe, c’est-à-dire la moyenne de la fréquence de l’allèle mineur chez les cas et les témoins > 1 %, un taux cible (call rate) minimum de 98 % et une valeur-p au test de Hardy-Weinberg >= 2,5 x 10-5. Comme nous nous intéressons aux gènes exprimés dans le cerveau et que notre liste de SNP initiale couvrait le génome autosomal au complet, un autre filtre a été appliqué sur notre liste de gènes pour ne conserver que ceux étant exprimés dans le cerveau selon la GEA. Le but de ce dernier filtre est de minimiser les calculs. Pour nos 2 méthodes, nous avons ensuite sorti le minimum de SNPs référant à un gène, le maximum et la moyenne.
2.1.2 Association marginale
L’association marginale se trouve à être l’association entre le phénotype et fréquences génotypiques dans les marges du tableau 1. Elle est un moyen plus rapide que les modèles multivariés de voir si un gène semble lié à un phénotype d’intérêt. Une bonne partie du projet de recherche repose sur les associations marginales qui ont été précédemment calculées. Si la schizophrénie était une maladie basée sur un seul gène, nous pourrions en rester là, car la solution serait déjà trouvée avec le signal que notre équipe a obtenu sur le chromosome 13 [Bureau, 2013], mais la maladie est multigénique. Nous devons donc poursuivre notre investigation en regardant les interactions gènes-gènes. Il existe deux maladies multigéniques complexes, la maladie de Bardet-Biedl et la maladie de Hirschsprung, qui sont de bons exemples de maladies impliquant des interactions
10
gènes-gènes. Ces deux maladies ont l’avantage d’être très bien connues, c’est-à-dire que nous connaissons bien chacun des gènes impliqués dans la maladie et que les différentes interactions qu’ils ont entre eux sont connues et bien documentées [Cantrell, 2004] [Owens, 2005] [De Pontual, 2009] [Tam, 2009] [Badano, 2005]. Elles nous servirons donc de modèles. Les valeurs-p obtenues suite à l’association marginale servent donc entre autres à ordonner notre liste de gènes retenus pour GSEA par exemple ou servent de critère de sélection comme défini dans la section sur Biofilter (2.1.5.5).
Locus G \ Locus H H1H1 H1H2 H2H2
G1G1 q22 q21 q20
G1G2 q12 q11 q10
G2G2 q02 q01 q00
Tableau 1 : Tableau de fréquence de génotype à 2 locus (G et H)
q00 = nombre de cas avec aucun allèle mineur pour le 1er SNP et aucun allèle mineur pour le 2e SNP. q01 = nombre de cas avec aucun allèle mineur pour le 1er SNP et 1 allèle mineur pour le 2e SNP. q02 = nombre de cas avec aucun allèle mineur pour le 1er SNP et 2 allèles mineurs pour le 2e SNP. q10 = nombre de cas avec 1 allèle mineur pour le 1er SNP et aucun allèle mineur pour le 2e SNP. q11 = nombre de cas avec 1 allèle mineur pour le 1er SNP et 1 allèle mineur pour le 2e SNP. q12 = nombre de cas avec 1 allèle mineur pour le 1er SNP et 2 allèles mineurs pour le 2e SNP. q20 = nombre de cas avec 2 allèles mineurs pour le 1er SNP et aucun allèle mineur pour le 2e SNP. q21 = nombre de cas avec 2 allèles mineurs pour le 1er SNP et 1 allèle mineur pour le 2e SNP. q22 = nombre de cas avec 2 allèles mineurs pour le 1er SNP et 2 allèles mineurs pour le 2e SNP.
2.1.3 Interaction gène-gène/Épistasie
L’épistasie est définie comme l’interaction entre 2 gènes quand le phénotype dépend de cette interaction et peut être masqué par l’un des 2 gènes ou les 2 dans certains cas « symétriques » [Cordell, 2002]. L’étude des interactions gènes-gènes constitue donc la principale tâche de mon projet. Plusieurs logiciels et méthodes permettent d’analyser l’épistasie [Cordell, 2009]. Nous ferons une
11
régression logistique sur nos données afin de voir les possibilités d’épistasie ainsi qu’une analyse selon la méthode JE. [Kooperberg, 2008] définit bien ce que devrait être une telle régression. Il nous démontre que le test de l’effet d’interaction sous un modèle logistique est approximativement indépendant du test de l’effet marginal. Nous pouvons donc grâce à cette indépendance choisir des gènes en fonction de leur valeur-p d’un test marginal sans craindre d’avoir de biais de sélection sur le paramètre d’interaction et donc, nous pourrons réduire le nombre de corrections pour contrôler les erreurs de type1. En effet, plutôt que de corriger pour les paires entre tous nos SNPs, nous n’aurons qu’à corriger pour les paires entre les SNPs testés. Pour faire la régression logistique, nous utiliserons la formule log(/1- ) = B0 + B1X1 + B2X2 + B3X1X2 où est le risque
théorique, B0 est notre ordonnée à l’origine, B1X1 et B2X2 sont nos variables
indépendantes (codage allélique) avec leur coefficient (pente) pour nos 2 gènes et B3X1X2 est notre terme d’interaction entre nos 2 gènes. [Cordell, Ueki, 2011] nous
allume cependant un feu rouge lors de sa présentation sur certaines des méthodes existant pour tester l’épistasie, nous démontrant leurs failles comme par exemple la possibilité de considérer comme résultat positif la présence d’un effet principal sur un seul locus et non une interaction et proposant des corrections. Elle nous propose aussi sa propre méthode, la méthode JE. Un élément clé de ce projet étant la comparaison de méthodes, nous essayerons aussi la méthode JE que nous suggère le Dr Cordell. Un résumé de notre réflexion se trouve à l’Annexe A4
2.1.4 Méthodes de filtrage basées sur les connaissances
biologiques
Depuis longtemps, diverses données biologiques sont collectées et assemblées dans de grosses bases de données. Nous allons voir dans cette section comment tirer parti de ce travail colossal.
12
[Bush, 2009] recommande d’utiliser au préalable le savoir biologique déjà disponible pour enrichir nos données. L’enrichissement pourrait se définir comme étant l’utilisation d’information des bases de données pour cibler plus spécifiquement de l’information comme des gènes ou des paires de gènes. Il nous parle aussi du logiciel Biofilter [Ritchie Lab] qui permet de faire cet enrichissement à priori. Ce logiciel a été retenu pour mon projet. Nous en parlerons donc plus dans la sous-section Biofilter (2.1.5.5). L’un des avantages de cette méthode est l’utilisation des catégories GO [Open Biological and Biomedical Ontologies] afin de diminuer le nombre de calculs. [Chasman, 2008] utilise aussi GO comme base de données principale dans son approche. Nous examinerons la base de données GO plus tard, dans la sous-section GO (2.1.5.1). [Köhler, 2008] utilise une approche similaire à celle de Bush en ce sens qu’il utilise des bases de données pour avoir le savoir biologique comme point de départ, mais son approche repose sur les réseaux d’interactions. [Wang, 2007] nous présentent une autre approche en utilisant le logiciel GSEA (BROAD Institute), un logiciel conçu à la base pour de l’enrichissement d'expression génique, mais qui selon leur article peut être utilisé aussi pour l’enrichissement de voies (pathways) pour des SNPs par de simples modifications. Cette solution étant aussi retenue pour mon projet, elle sera, elle aussi, discutée plus en profondeur dans la section sous-section GSEA (2.1.5.6). Une autre solution d’enrichissement proposée par la BROAD Institute est le logiciel MAGENTA qui se spécialise dans l’enrichissement lorsque le génotype n’est pas connu [Segrè, 2010]. [Holmans, 2009] et [Manning, 2009] utilisent eux aussi le principe d’enrichissement, mais avec une autre méthode. Cette revue s’arrête cependant en 2011 par manque de temps.
2.1.5 Logiciel et base de données
Cette section est dédiée à la partie la plus importante du projet : Les différents logiciels et bases de données que nous avons utilisés.
13 2.1.5.1 GO
GO est une grosse base de données ontologique qui permet de classifier les différents gènes selon leurs fonctions. Dans un premier temps, les gènes sont répartis en 3 catégories. Les gènes servant de composante cellulaire, ceux servant dans des processus biologiques et ceux ayant une fonction moléculaire. Chacune de ces catégories est ensuite divisée en sous catégories plus spécifiques qui sont elles-mêmes divisées en sous-catégories, etc. Cette structure hiérarchisée permet de trouver plus facilement des liens entre différents gènes, car il est reconnu que des gènes avec des fonctions similaires oeuvrent souvent sur les mêmes ligands ou dans les mêmes voies (pathways). En termes d’informaticien, les gènes qui se retrouvent dans chaque feuille de l’arbre sont réputés pour interagir entre eux ou avoir des fonctions très similaires et donc si un gène d’intérêt se retrouve dans une feuille, il serait bien d’examiner les autres gènes de cette même feuille pour voir s’ils pouvaient faire partie de nouvelles interactions.
2.1.5.2 GEA
GEA est une grosse base de données sémantiquement enrichie de méta-analyse provenant de statistiques obtenues d’archives de puce à expression. Elle contient donc des informations sur les expressions des gènes. Les expériences sur chaque gène y sont référencées et identifient une expression à la hausse ou à la baisse dans le cadre de leur étude en se basant sur les valeurs des tests statistiques effectués lors de l’expérience. Nous avons donc utilisé cette base de données pour y en extraire la liste des gènes qui sont exprimés dans le cerveau afin de pouvoir ne garder que ces gènes lors de nos analyses et ainsi diminuer le nombre de calculs. Pour agir ainsi, nous supposons que les gènes impliqués dans la schizophrénie sont tous exprimés dans le cerveau et donc qu’il n’y a pas de
14
gènes uniquement exprimés dans le cœur ou les poumons qui sont impliqués dans la maladie par exemple.
2.1.5.3 MSigDB
La MSigDB est une base de données contenant une collection de voies (pathways) annotées pour différents organismes et dans un format utilisable par GSEA. Lors de nos analyses, nous nous intéresserons aux voies (pathways) qui sont présentes dans notre cerveau. Nous avons donc interrogé cette base de données pour en extraire une sous-base de données de voies (pathways) retrouvées chez l’homme avec le mot clé Homo Sapiens et nous avons ensuite extrait une sous-sous-base de données ne contenant que les voies (pathways) d’intérêt en nous servant de la liste de mots clés : « schizophrenia depressive bipolar brain neur* cortex spinal cogni* glial astrocytes synap* dendrite axon* nerv* acetylcholine actin anion cation channel volt* sodium calcium potassium gaba tgf glutamate vesicle». Un résumé plus complet se retrouve à l’Annexe A5
2.1.5.4 DAVID
Le logiciel DAVID (National Institute of Allergy and Infectious Diseases.) a été utilisé pour annoter notre liste de SNPs après les avoir cartographiés (mappés) sur un gène. En gros, il prend une liste de symboles et cherche la description du gène auquel il réfère lorsque cette dernière est connue. Il retourne ensuite le résultat dans un format texte que l’on peut greffer facilement avec Perl. Cette annotation constitue un artifice facilitant la compréhension des résultats en permettant de rapidement voir ce que fait un gène et ainsi mieux comprendre son lien avec un autre gène avec lequel il interagirait.
15 2.1.5.5 BIOFILTER
Biofilter est un logiciel développé par le laboratoire du Dr Ritchie. Il prend une liste de SNPs et cherche dans plusieurs bases de données (GO, KEGG, Net Path, pfam, Reactome, DIP et BioCarta. Voir Annexe A3) les interactions biologiques qui sont déjà connues [Turner, 2011]. Nous nous sommes basés sur les résultats d’association marginale (valeur p marginale < 0.2), ainsi que sur les résultats de cartographie (mapping) positif pour définir notre liste de SNPs. Pour avoir une interaction déjà connue, GO par exemple, comme expliqué dans la section sur GO (2.1.5.1), va donner l’information à Biofilter qu’il y a interaction entre les gènes qui se retrouvent dans la même feuille. En utilisant sa propre base de données, Biofilter associe chaque SNP à un gène puis il vérifie pour chaque paire de gènes la présence d’interaction connue dans les bases de données pour l’une des sources. Chaque fois qu’une interaction est trouvée dans une base de données (source), un indice est incrémenté de 1. La recherche se poursuit ainsi pour chaque source. Un autre indice est incrémenté de 1 pour chaque source différente où une interaction a été trouvée. Nous avons donc un indice final au format source-interaction (ex. : 6-78). Nous pouvons aussi fournir nos propres informations reliées à la maladie qui nous intéresse. La liste ainsi obtenue est un meilleur point de départ pour nos analyses d’interactions, car elle diminue le nombre de calculs. En effet, nous utilisons la prémisse que si une paire de gènes est reconnue pour avoir une interaction, les chances pour que ces gènes de façon individuelle aient d’autres interactions avec d’autres gènes sont plus élevées que pour une sélection au hasard et ces interactions pourraient contribuer à causer une maladie. Afin de paramétrer convenablement Biofilter, nous avons fait les différents tests présentés à l’Annexe A6. Il est à noter que nous avons demandé à Biofilter dans ses paramètres de configuration de se limiter aux 50 000 meilleures paires de gènes car nous avons déterminé dans nos tests que ce serait suffisant pour englober notre point de coupure. Ce dernier sera fixé en se basant sur les résultats qu’obtiendra Biofilter sur nos maladies connues de Hirschsprung et de
16
Bardet-Biedl. La version 0.5 de Biofilter fut utilisée pour effectuer ces tests, mais celle utilisée pour les résultats du projet fut la version 2.0 [Pendergrass, 2013].
2.1.5.6 GSEA
GSEA est un logiciel qui à la base permet de faire de l’enrichissement d’expression de gènes. Cet enrichissement d’expression de gènes est fait par une méthode de calcul qui détermine si un groupe de gènes défini à priori montre une différence d’expression statistiquement significative entre les différents états biologiques (phénotypes). Il est cependant possible de lui donner notre propre liste de gènes ordonnée selon notre test statistique préféré. Nous pouvons donc lui donner une liste avec la valeur-p allélique minimum pour chacun des gènes de notre liste de gènes retenus [Holden, 2008]. Le gène avec la plus petite valeur se retrouvant ainsi au début de la liste et celui avec la plus grande à la fin. Pour calculer la statistique d’enrichissement, GSEA fait en gros une marche dans notre liste de gènes ordonnée en augmentant un indice quand un gène est présent (hit) dans une voie (pathway) et en le diminuant lorsqu’il ne l’est pas (miss). Le score d’enrichissement est calculé à partir de la valeur la plus éloignée de 0 de cette variable que GSEA a rencontrée lors de cette marche. Lors du calcul de la valeur d’enrichissement, il est possible d’accorder un poids au calcul de l’indice. La valeur de 0 est la valeur « classique » et permet de faire un test statistique standard de Kolmogorov–Smirnov. Nous pouvons aussi la changer pour 1, 1,5 et 2. Rappelons rapidement que le test de Kolmogorov–Smirnov permet de tester si un échantillon suit une loi donné ou si deux échantillons suivent la même loi. La formule pour calculer l’enrichissement est :
17
[Subramanian, 2005] Figure 1 : Formule pour le calcul de l’enrichissement de GSEA
Formule pour le calcul de l’enrichissement de GSEA tel que défini par [Subramanian, 2005]
La valeur d’enrichissement est la déviation maximum de 0 de Phit - Pmiss
S est notre voie (pathway) qu’on analyse actuellement, Nh est le nombre de gènes
dans S, N est le nombre de gènes de notre liste ordonnée, i est la position du gène dans notre liste ordonnée, gi est le gène à la position i dans notre liste ordonnée et
P est le poids. Le paramètre rj dépendrait du phénotype si nous utilisions cette
possibilité de GSEA. Cependant, dans notre situation, sa valeur est simplement la valeur numérique du rang donné en entrée quand on fait une analyse avec une liste pré ordonnée. Si S est distribué aléatoirement, alors le score d’enrichissement est petit.
GSEA utilise une base de données contenant différentes voies (pathways) connues (MSigDB). Il vérifie ensuite parmi notre liste de gènes retenus ceux qui sont dans ces voies (pathways) puis fait son test d’enrichissement. Normalement, il procède à des permutations des gènes, mais cela ne convient pas à des données de SNP car sa méthode considère que nous sommes dans une situation d’enrichissement d’expression de gènes. Comme il est important de faire des permutations pour contrer la dépendance créée lors de tests multiples, nous les ferons manuellement comme nous le suggère [Holmans, 2009]. Nous allons pour chaque SNP tirer une valeur-p aléatoire d’une loi uniforme. Nous prenons ensuite pour chaque gène le SNP avec la valeur-p minimum. Cette valeur p est ensuite transformée en –log base 10 pour avoir la plus grande valeur possible pour la plus petite valeur-p. GSEA a besoin de cette transformation pour ordonner convenablement notre liste. Pour finir, nous resoumettons nos données à GSEA.
18
Nous referons le tirage ainsi que les étapes qui le suivent un total de 1000 fois pour avoir nos 1000 permutations. Bien entendu, il faut aussi en plus des 1000 permutations soumettre notre liste originale. La méthode de Holmans est cependant différente de la nôtre. Nous, nous regardons la valeur d’enrichissement maximale. Lui il la regarde à différents points de coupure (cutoff). Il est aussi possible de soumettre à GSEA notre propre liste de voies (pathways) alors nous pourrons rester proches de la littérature récente et donc comparer notre approche avec celle de [Jia, 2010] ou aller chercher des informations dans d’autres bases de données. Afin de paramétrer convenablement GSEA, nous avons fait les différents tests présentés à l’Annexe A7.
2.1.5.7 JE
JE est une méthode inventée par les Dr Cordell et Ueki [Ueki, 2012]. Comme cette méthode était nouvelle lors de ma Maîtrise, nous l’avons programmée dans R. Cependant, elle fut implantée depuis peu dans la suite logicielle CASSI [Cordell, et Ueki, 2013]. À cause du phénomène de bruit dans les échantillons qui tire son origine dans la trop grande hétérogénéité génétique de la schizophrénie, du côté multigénique complexe de la maladie, de l’effet très faible sur le phénotype que peut avoir une interaction génique, des erreurs de manipulation de la biopuce qui nuisent à la qualité de l’échantillon, etc., les méthodes statistiques traditionnelles pour détecter les possibles interactions ne donnent pas toujours de bons résultats. En effet, il arrive que la simple présence d’un effet principal pour l’un des 2 locus amène les tests traditionnels à détecter une interaction alors que dans les faits il n’en existe aucune, donnant un faux positif. Une très faible interaction peut aussi se perdre dans le bruit et passer inaperçue en donnant un faux négatif. La méthode JE vient pallier ces problèmes. Cette méthode n’est sensible qu’aux véritables effets d’interactions, c’est-à-dire aux effets qui sont détectés sur les 2 locus et qui sont statistiquement significatifs pour décrire une interaction génique, ou, lorsque la maladie n’est pas suffisamment rare, à un effet principal aux 2 locus.
19
Le fonctionnement et l’implantation du JE ne sont pas si complexes. Nos données sont sous la forme d’un tableau (tableau 2).
SUBID groupe rs1642 rs7323 rs1816 rs2884 … 17 000 1 2 1 0 1 … 17 001 1 0 1 1 2 … 17 002 1 1 1 0 0 … 18 000 0 2 0 0 0 … 18 001 0 1 0 0 2 … … … … …
Tableau 2 : Exemple de notre fichier de données
SUGID est le numéro unique attribué à chaque sujet et permettant d’assurer la confidentialité. Groupe permet d’identifier nos cas et nos témoins. Cas = 1 et témoins = 0. Viens ensuite nos différents SNPs et leurs valeurs. 0 = aucun allèle mineur. 1 = présence de l’allèle mineur sur l’un des deux chromosomes. 2 = présence de l’allèle mineur sur chaque chromosome.
Par la suite, nous prenons une paire de SNPs qui ne sont pas situés dans le même gène. Pour les cas, nous faisons les sommes pour chaque situation possible afin d’obtenir les qij tels que définis au tableau 1.
Nous définissons ensuite les rapports marginaux suivants :
i
22= q
22q
00/ q
20q
02i
21= q
21q
00/ q
20q
01i
12= q
12q
00/ q
10q
02i
11= q
11q
00/ q
10q
01Posons Y(Ø) qui est défini comme étant le log du rapport de cotes des allèles tel qu’estimé par la méthode de [Wu, 2010].
20
Y(Ø) = log(P
11P
22/P
12P
21)
Où Pjk est la probabilité de l’haplotype Gj -- Hk
Nous pouvons maintenant définir l’estimateur de Y(Ø) suivant :
Y
cas= w
22(log i
22) / 2 + w
21log i
21+ w
12log i
12+ w
11log(2 * i
11− 1)
Où les poids wij somment à 1 et sont choisis pour minimiser la variance Ycas.
En suivant les mêmes étapes pour les témoins, nous pouvons calculer Ytémoins.
JE = [Y
cas- Y
témoins]
2/ (V
cas+ V
témoins)
Où V représente la variance estimée de Y
2.1.5.8 R
R est un environnement de programmation en langage S développé par le Projet R (CRAN). Fonctionnant sous le principe des modules, il est virtuellement possible de lui faire faire n’importe quel type de tâche en lui fournissant les librairies appropriées. Très utilisé par les Statisticiens, les Actuaires et les Physiciens, il l’est aussi par les bio-informaticiens. L’essentiel de ce projet repose sur cette architecture. La cartographie de nos SNPs, les différents filtres que nous avons appliqués aux données, l’adaptation aux SNPs et son implantation, l’utilisation de la régression logistique, les appels à Biofilter, l’implantation de JE et la production des graphiques ont tous été faits dans R.
2.1.5.9 Perl
Perl (Perl) est un langage spécialisé dans la manipulation de chaîne de caractères que nous avons utilisé afin de jumeler les annotations de DAVID à la cartographie que nous avons faite des SNPs à notre disposition.
21 2.1.5.10 Osprey
Osprey (Tyers) est un logiciel spécialisé permettant de créer des représentations graphiques des réseaux d’interactions protéine-protéine. Nous avons utilisé ce logiciel pour représenter nos nouvelles interactions et tenté de déceler un réseau d’interaction qui pourrait suggérer la présence d’une nouvelle voie (pathway) que nous ne connaissons pas encore impliquée dans la schizophrénie. Il nous permet entre autre de faire une sélection en éliminant automatiquement toutes les paires de gènes qui ne possèdent aucune autre interaction et donc qui ne semblent rattachés à aucun réseau. Par la suite, nous pouvons éliminer facilement manuellement tous les réseaux qui sont composés de 3 gènes ou plus et qui ne sont pas rattachés au réseau principal, soit le plus grand réseau d’interaction que nous pouvons trouver.
23
24
3.1 Résultats
Nous présenterons dans cette section divers résultats dont nous discuterons plus tard dans la section Discussion (4.1).
3.1.1 Résultats de cartographie et filtrage
Nous sommes partis d’une liste de 348 411 SNPs répartis partout dans le génome, à l’exception des chromosomes X et Y et de l’ADN mitochondrial qui n’ont pas été considérés. Certains sont dans des régions codantes, d’autres non. Comme nous nous intéressons aux gènes, une première étape fut d’éliminer tous les SNPs n’étant pas directement dans un gène ou à proximité d’un gène tel que défini dans la section méthode (2.1.1). Comme certains SNPs ont eu des problèmes d’hybridation et pour s’assurer de la qualité générale des données, un filtre a été appliqué tel que défini dans la section Population/Donnée. Notre liste de SNPs avant cartographie est donc passée à 327 729 SNPs. Dans la cartographie Directe, nous avons réduit notre liste de SNPs à 153 110 localisés dans 15 559 gènes différents. Pour la cartographie Étendue, notre liste de SNPs fut réduite à 232 380 localisés dans ou proche de 21 397 gènes différents. Nous avons déjà établi que nous nous intéressions aux gènes exprimés dans le cerveau. Afin de tenir compte de cette réalité, un autre filtre a été appliqué à partir de la liste des SNP exprimés au cerveau de GEA. La cartographie a ensuite de nouveau été appliquée pour nous donner une cartographie Directe de 132 657 SNPs directement localisés dans 13 472 gènes différents ayant une expression au cerveau. La cartographie Étendue a donné 205 162 SNPs directement localisés ou à proximité de 16 856 gènes différents ayant une expression au cerveau. Certains gènes sont très petits et ne possèdent qu’un seul SNP. D’autres sont énormes et vont contenir jusqu’à 751 SNP. Nous avons donc une moyenne de
25
9.92028 SNPs par gène avec une cartographie Directe et de 19.58003 avec une cartographie Étendue. Le tableau 3 résume ces différents résultats.
Méthode de cartographie (mapping) Directe Étendue (50 kb de chaque côté)
Nombre de SNPs au départ 348 411 348 411
Nombre de SNPs après cartographie (mapping) 153 110 232 380
Nombre de gènes pointé par un ou des SNPs 15 559 21 397
Nombre de SNPs total après filtre pour contrôle de qualité
327 729 327 729
Nombre de gènes après filtre pour contrôle de qualité
14 960 20 460
Nombre de SNPs après filtre pour garder gènes exprimés au cerveau
132 657 205 162
Nombre de gènes après filtre pour garder gènes exprimés au cerveau
13 472 16 856
Nombre de SNPs minimum par gène 1 1
Nombre de SNPs maximum par gène 721 751
Nombre de SNPs moyen par gène 9.92 19.58
Tableau 3 : Résumé cartographie et filtre
Tableau résumé des résultats pour une cartographie directe (Le SNP se retrouve directement dans le gène) et une cartographie étendue (Le SNP se retrouve directement dans le gène ou jusqu’à une distance de 50 000 paires de bases de ce dernier). Il contient aussi les résultats du filtrage.
26
3.1.2 Résultats GSEA
L’enrichissement de notre jeu de données par la méthode GSEA a permis de faire ressortir différentes voies (pathways) dont les gènes semblaient intéressants pour la suite, c’est-à-dire pour nos tests pour trouver de nouvelles interactions.
Cartogr aphie
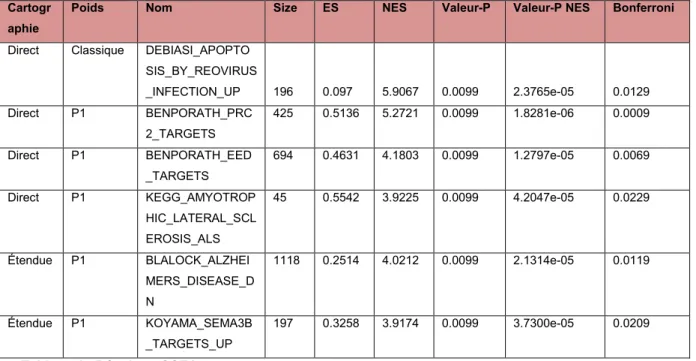
Poids Nom Size ES NES Valeur-P Valeur-P NES Bonferroni
Direct Classique DEBIASI_APOPTO SIS_BY_REOVIRUS _INFECTION_UP 196 0.097 5.9067 0.0099 2.3765e-05 0.0129 Direct P1 BENPORATH_PRC 2_TARGETS 425 0.5136 5.2721 0.0099 1.8281e-06 0.0009 Direct P1 BENPORATH_EED _TARGETS 694 0.4631 4.1803 0.0099 1.2797e-05 0.0069 Direct P1 KEGG_AMYOTROP HIC_LATERAL_SCL EROSIS_ALS 45 0.5542 3.9225 0.0099 4.2047e-05 0.0229 Étendue P1 BLALOCK_ALZHEI MERS_DISEASE_D N 1118 0.2514 4.0212 0.0099 2.1314e-05 0.0119 Étendue P1 KOYAMA_SEMA3B _TARGETS_UP 197 0.3258 3.9174 0.0099 3.7300e-05 0.0209
Tableau 4 : Résultats GSEA
Tableau des résultats concluant de GSEA. Une voie (pathway) a été retenue lorsqu’elle présentait un résultat au test de Bonferroni inférieur à 0.05. Nous y retrouvons la cartographie pour identifier nos gènes, le paramètre de poids utilisé dans GSEA, le nom de la voie (pathway) identifiée, le nombre de gènes qui la composent, la valeur obtenue pour le score d’enrichissement (ES), la valeur normalisée du score d’enrichissement (NES), la valeur-p du ES, la valeur-p du NES et la valeur corrigée selon Bonferroni du NES qui a servi à discriminer nos résultats.
La voie DEBIASI_APOPTOSIS_BY_REOVIRUS_INFECTION_UP contient la liste des gènes liés à l’apoptose dont l’expression est modifiée suite à une infection par rétrovirus. La voie BENPORATH_PRC2_TARGETS contient la liste des gènes qui possèdent la marque de tri-méthylation H3K27 dans leur région promotrice et qui sont capable de se lier au gène PRC2. La voie BENPORATH_EED_TARGETS contient la liste des gènes capable de se lier au gène EED. La voie KEGG_AMYOTROPHIC_LATERAL_SCLEROSIS_ALS contient la liste des gènes
27
impliqués dans la maladie de la Sclérose latérale amyotrophique. La voie BLALOCK_ALZHEIMERS_DISEASE_DN contient la liste des gènes présentant une baisse d’expression dans la maladie d’Alzheimer. Pour finir, la voie KOYAMA_SEMA3B_TARGETS_UP contient la liste des gènes dont l’expression est augmentée lorsque le gène SEMA3B est exprimé.
3.1.3 Résultats Biofilter
La méthode Biofilter a permis de faire ressortir différentes paires de gènes reconnus pour avoir des interactions et donc qui ont un fort potentiel pour avoir d’autres interactions que nous ne connaissons pas encore. Ces paires de gènes sont donc intéressantes pour la suite, c’est-à-dire pour nos tests pour trouver de nouvelles interactions. Le logiciel a été paramétré pour identifier les 50 000 meilleures paires de gènes parmi celles qu’il a cartographiées avec notre liste de SNPs afin de s’assurer de trouver toutes les paires avec un indice égal ou supérieur à notre seuil. Il est à noter que les résultats pour la cartographie Directe et Étendue furent identiques (tableau 5). Ces résultats s’expliquent par le fait que Biofilter fait lui-même sa propre cartographie et considère le déséquilibre de liaison avec son paramètre de population qui permet de faire une cartographie étendue. Sa cartographie de nos deux listes de SNPs fut donc pratiquement identique. Il y a eu cependant quelques petites différences mais elles ne sont pas ressorties dans les résultats. Ces différences mineures viennent du fait que nous avons appliqué une distance de 50 KB de chaque côté d’un SNP pour notre cartographie étendue alors que dans les faits, le déséquilibre de liaison n’est pas toujours identique d’une région chromosomique à une autre. Biofilter est donc beaucoup plus rigoureux que nous à ce sujet, même si cela n’a pas présenté de différence sur les résultats.
28 Cartographie Nombre de SNPs soumis Nombre de paires de gènes identifiés avant coupure Point de coupure Nombre de paires de gènes identifiés après coupure Nombre de gènes différents Directe 25 799 50 000 4-156 6105 373 Étendue 64 511 50 000 4-156 6105 373
Tableau 5 : Résultats Biofilter
Tableau résumé des résultats concluants de Biofilter. Nous y retrouvons le type de cartographie pour déterminer les SNPs soumis, le nombre de SNP que nous avons soumis, le nombre de paires de gènes que Biofilter a identifiées comme étant reconnues pour avoir des interactions tel que nous l’avons défini dans le fichier de configuration, le seuil qui représente l’index minimal que pouvait avoir une paire de gènes pour être retenue, le nombre de paires de gènes retenues après coupure et le nombre total de gènes différents qui se retrouvent dans les paires retenues.
3.1.4 Résultats Régression logistique
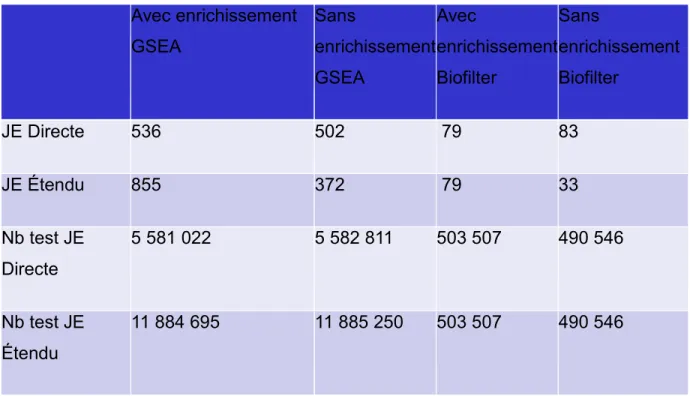
Aucun résultat statistiquement significatif n’a été identifié par les tests d’interactions, que nous utilisions ou non un enrichissement par GSEA. Au vu de ces résultats, il a été décidé de ne pas faire ce test avec l’enrichissement par Biofilter afin de minimiser les temps de calculs.
3.1.5 Résultats JE
Nous avons voulu comparer les résultats des tests avec enrichissement à ceux faits avec l’association marginale. Pour ce faire, nous avons fixé un point de coupure comparatif permettant de choisir les Y meilleurs SNPs de l’association marginale afin d’avoir un nombre de tests à peu près égal à ceux faits lors des tests avec enrichissement. Le point de coupure comparatif sans enrichissement a été défini comme étant
29
Y = ceiling ( (-1/2) + sqrt( (1/4) + (2 * nbTest) ) )
où ceiling : ceiling (plafond) prend un argument numérique simple x et retourne un vecteur numérique contenant le plus petit entier qui n’est pas inférieur à l’élément correspondant x
sqrt : Fonction racine carré
nbTest : Nombre de tests total faits pour lequel on veut trouver le nombre de SNPs nécessaire afin de faire un nombre de test similaire.
Avec enrichissement GSEA Sans enrichissement GSEA Avec enrichissement Biofilter Sans enrichissement Biofilter JE Directe 536 502 79 83 JE Étendu 855 372 79 33 Nb test JE Directe 5 581 022 5 582 811 503 507 490 546 Nb test JE Étendu 11 884 695 11 885 250 503 507 490 546
Tableau 6 : Résumé des résultats de JE
Tableau qui résume nos résultats pour JE avec enrichissement par GSEA, sans enrichissement pour un nombre de test équivalent à GSEA, avec enrichissement par Biofilter et sans enrichissement pour un nombre de test équivalent à Biofilter. Nous y retrouvons le nombre de résultats statistiquement significatif de tests d’interactions suite à une correction de Bonferroni pour chacune de nos 2 cartographies ainsi que le nombre de tests totaux effectués.
30
3.1.6 Résultats retenus
Après avoir passé une batterie de contrôles de qualité, de critères de sélections et de validations de concepts (section 2.1.1, 2.1.2, 2.1.3, 2.1.4, 2.1.5.5, 2.1.5.6, 2.1.5.7 et 2.1.5.10) depuis le début de notre projet, une petite liste de paires de gènes a su sortir du lot. Voici les différentes paires de marqueurs que nous avons retenues. Chacun des graphiques 2 à 14 est un graphique des rapports de cotes pour les paires de marqueurs dont les interactions ont une valeur p sous un seuil de Bonferroni de 0.05 pour le test de JE et qui visuellement parlant démontrent bien un effet d’interaction. Les graphiques 2 à 14 comportent aussi la particularité d’être sous un seuil de 0.006 pour le test de la régression logistique. Chacun de ces graphiques est accompagné d’une courte interprétation de l’interaction qu’on y retrouve. À l’annexe A2 se trouve une table avec quelques informations supplémentaires sur les différents gènes présentés dont les annotations. Chacune de ces paires de gènes semble présenter une interaction statistiquement significative non connue jusqu’à présent. Certaines de ces interactions donnent une susceptibilité pour développer la maladie. D’autres donnent une résistance. Dans certains cas, la présence de l’allèle mineur pour un seul des deux gènes entraîne une plus forte susceptibilité pour développer la maladie, mais la présence de l’allèle mineur de l’autre gène crée une interaction qui corrige le phénotype introduit par l’allèle du premier gène pour ramener le risque de la maladie à celui du génotype de référence. Nous qualifions ce phénomène de retour à la normale. Le tableau 7 résume les interactions selon cette classification.
Légende pour les figures 2 à 14 :
Y : Rapport de cotes par rapport au génotype homozygote pour l’allèle majeur aux deux marqueurs.
X : qij
Où i = nombre d’allèles mineurs pour le SNP1 Où j = nombre d’allèles mineurs pour le SNP2
31
Figure 2 : KCNQ1 VS NAV2
Voie BENPORATH_EED_TARGETS, cartographie Directe avec poids configuré à 1 dans GSEA. Ici, nous voyons que quand les sujets ont au moins un allèle mineur dans les deux gènes, il y a une susceptibilité pour développer la schizophrénie. Il faut cependant noter que les homozygotes pour l’allèle mineur du gène NAV2 sont très rares.
32
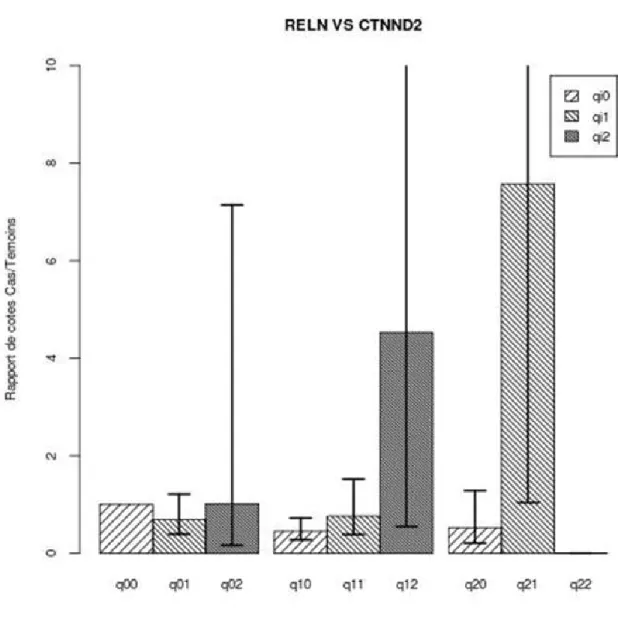
Figure 3 : RELN VS CTNND2
Voie BENPORATH_EED_TARGETS, cartographie Directe avec poids configuré à 1 dans GSEA. Ici, nous voyons que quand les sujets ont au moins un allèle mineur dans les deux gènes, il y a une susceptibilité pour développer la schizophrénie. Il faut cependant noter que les homozygotes pour l’allèle mineur du gène CTNND2 sont rares.
33
Figure 4 : GRM3 VS GRM7
Voie BENPORATH_EED_TARGETS, cartographie Directe avec poids configuré à 1 dans GSEA. Ici, nous voyons que quand les sujets ont au moins un allèle mineur dans les deux gènes, il y a une résistance pour développer la schizophrénie. Cependant, l’absence de l’allèle mineur pour un des 2 gènes en présence d’un homozygote de l’allèle mineur de l’autre gène entraine une susceptibilité pour développer la schizophrénie. Il faut aussi noter que les homozygotes pour l’allèle mineur du gène GRM3 sont rares.
34
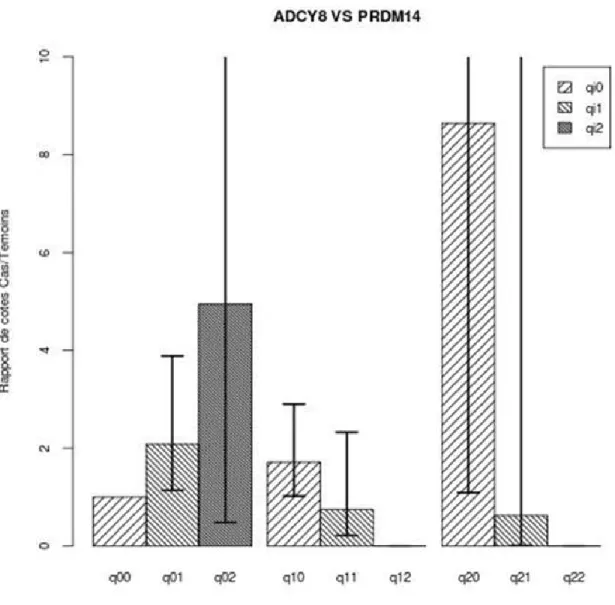
Figure 5 : ADCY8 VS PRDM14
Voie BENPORATH_EED_TARGETS, cartographie Directe avec poids configuré à 1 dans GSEA. Ici, nous voyons que quand les sujets ont au moins un allèle mineur dans les deux gènes, il y a une résistance pour développer la schizophrénie. Cependant, l’absence de l’allèle mineur pour un des 2 gènes en présence d’un homozygote de l’allèle mineur de l’autre gène entraine une susceptibilité pour développer la schizophrénie. Il faut aussi noter que les homozygotes pour l’allèle mineur du gène PRDM14 sont rares.
35
Figure 6 : ROBO1 VS NRXN1
Voie BLALOCK_ALZHEIMERS_DISEASE_DN, cartographie Étendue avec poids configuré à 1 dans GSEA. Ici, nous voyons que quand les sujets ont au moins un allèle mineur dans les deux gènes, il y a une forte susceptibilité pour développer la schizophrénie. Il faut cependant noter que les homozygotes pour l’allèle mineur du gène ROBO1 sont rares.
36
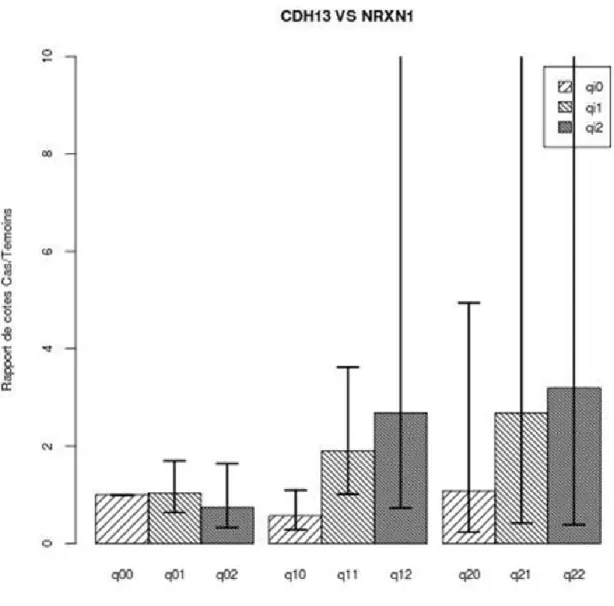
Figure 7 : CDH13 VS NRXN1
Voie BLALOCK_ALZHEIMERS_DISEASE_DN, cartographie Étendue avec poids configuré à 1 dans GSEA. Ici, nous voyons que quand les sujets ont au moins un allèle mineur dans les deux gènes, il y a une forte susceptibilité pour développer la schizophrénie. Il faut cependant noter que les homozygotes pour l’allèle mineur du gène CDH13 sont rares.
37
Figure 8 : TLK1 VS PDIA6
Voie BLALOCK_ALZHEIMERS_DISEASE_DN, cartographie Étendue avec poids configuré à 1 dans GSEA. Ici, nous voyons que quand les sujets ont au moins un allèle mineur dans les deux gènes, il y a une très forte susceptibilité pour développer la schizophrénie. Il faut cependant noter que les homozygotes pour l’allèle mineur du gène TLK1 sont rares. Notons que le rapport de cotes de q12 sort du cadre du graphique, car sa valeur est 15
38
Figure 9 : PLCB1 VS PLCL2
Voie BLALOCK_ALZHEIMERS_DISEASE_DN, cartographie Étendue avec poids configuré à 1 dans GSEA. Ici, nous voyons que quand les sujets ont au moins un allèle mineur dans les deux gènes, il y a une forte susceptibilité pour développer la schizophrénie. Il faut cependant noter que les homozygotes pour l’allèle mineur du gène PLCL2 sont rares.
39
Figure 10 : GPC5 VS PKNOX2
Voie BLALOCK_ALZHEIMERS_DISEASE_DN, cartographie Étendue avec poids configuré à 1 dans GSEA. Ici, nous voyons que quand les sujets ont au moins un allèle mineur dans les deux gènes, il y a une très forte susceptibilité pour développer la schizophrénie.
40
Figure 11 : FAIM2 VS SHANK2
Voie BLALOCK_ALZHEIMERS_DISEASE_DN, cartographie Étendue avec poids configuré à 1 dans GSEA. Ici, nous voyons que quand les sujets ont au moins un allèle mineur dans les deux gènes, il y a une résistance à développer la schizophrénie. Il faut cependant noter que les homozygotes pour l’allèle mineur du gène FAIM2 sont rares.
41
Figure 12 : CDH13 VS CYCS
Voie BLALOCK_ALZHEIMERS_DISEASE_DN, cartographie Étendue avec poids configuré à 1 dans GSEA. Ici, nous voyons que quand les sujets ont au moins un allèle mineur dans les deux gènes, il y a une résistance à développer la schizophrénie. Il faut aussi noter que les homozygotes pour l’allèle mineur du gène CYCS sont rares.
42
Figure 13 : DLG2 VS RGS7
Voie BLALOCK_ALZHEIMERS_DISEASE_DN, cartographie Étendue avec poids configuré à 1 dans GSEA. Ici, nous voyons que quand les sujets ont au moins un allèle mineur dans les deux gènes, il y a une résistance à développer la schizophrénie.
43
Figure 14 : CAMK2D VS KCNQ5
Enrichissement fait par Biofilter. CAMK2D provient de la paire CAMK2B VS CAMK2D et possède un indice de 5-56. KCNQ5 provient de la paire KCNQ3 VS KCNQ5 et possède un indice de 5-12. Ici, nous voyons que quand les sujets ont au moins un allèle mineur dans les deux gènes, il y a un retour à la normale du phénotype qui a tendance autrement en présence d’allèle mineur pour un seul des deux gènes à entraîner une susceptibilité pour développer la schizophrénie.
Catégorie Paires de gènes
Susceptibilité KCNQ1-NAV2, RELN-CTNND2, NRXN1-ROBO1,NRXN1- CDH13, TLK1-PDIA6, PLCB1-PLCL2, GPC5-PKNOX2
Résistance GRM3-GRM7, FAIM2-SHANK2, CDH13-CYCS, DLG2-RGS7 Normale ADCY8-PRDM14, CAMK2D-KCNQ5
Tableau 7 : Résumé interaction
Tableau résumé qui liste les paires de gènes selon leur catégorie d’interaction. Une définition plus précise des gènes est disponible au tableau 11.
44
Dans la première catégorie, nous avons retenu plusieurs paires de gènes dont la présence de l’allèle mineure dans les 2 gènes pourrait créer une susceptibilité pour développer la schizophrénie. Le gène KCNQ1 code pour une protéine de sous unité de canal potassium contrôlé par le voltage. L’allèle mineur du SNP de ce gène semblerait avoir une interaction causant une susceptibilité pour développer la schizophrénie avec l’allèle mineur du SNP du gène NAV2, un gène qui code pour une protéine qui est impliquée dans la croissance et migration des neurones (figure 2). Le gène RELN code pour une protéine de matrice extracellulaire qui contrôle les interactions cellule-cellule critiques du positionnement et de la migration neuronale. L’allèle mineur du SNP de ce gène semblerait avoir une interaction causant une susceptibilité pour développer la schizophrénie avec l’allèle mineur du SNP du gène CTNND2, un gène qui code pour une protéine de jonction adhésive impliquée dans le développement du cerveau (figure 3). Le gène NRXN1 code pour une protéine d’adhésion cellulaire du système nerveux. L’allèle mineur du SNP de ce gène semblerait avoir une interaction causant une susceptibilité pour développer la schizophrénie avec l’allèle mineur du SNP du gène ROBO1, un gène qui code pour une protéine qui est impliquée dans la guidance des axones (figure 6), et une autre avec l’allèle mineur du SNP du gène CDH13, un gène qui code pour une protéine qui sert de régulateur négatif de la croissance de l’axone pendant la différenciation neurale et de protection contre l’apoptose dû à un stress oxydatif (figure 7). Le gène TLK1 code pour une protéine de régulation de l’assemblage de la chromatine. L’allèle mineur du SNP de ce gène semblerait avoir une interaction causant une susceptibilité pour développer la schizophrénie avec l’allèle mineur du SNP du gène PDIA6, un gène qui code pour une protéine qui catalyse la formation, la réduction et l’isomérisation de ponts disulfure (figure 8). Le gène PLCB1 code pour une protéine de Phospholipase C. L’allèle mineur du SNP de ce gène semblerait avoir une interaction causant une susceptibilité pour développer la schizophrénie avec l’allèle mineur du SNP du gène PLCL2, un gène qui code pour une protéine de Phospholipase C (figure 9). Le gène GPC5 code pour une protéine qui joue un rôle important dans la division cellulaire. L’allèle mineur du SNP de ce gène semblerait avoir une interaction causant une
45
susceptibilité pour développer la schizophrénie avec l’allèle mineur du SNP du gène PKNOX2, un gène qui code pour une protéine qui joue un rôle important dans la mort cellulaire (figure 10).
Dans la seconde catégorie, nous avons retenu quelques paires de gènes dont la présence de l’allèle mineure dans les 2 gènes pourrait créer une résistance à développer la schizophrénie. Le gène GRM3 code pour une protéine de récepteur de glutamate métabotropique. L’allèle mineur du SNP de ce gène semblerait avoir une interaction causant une résistance à développer la schizophrénie avec l’allèle mineur du SNP du gène GRM7, un gène qui code pour une protéine de récepteur de glutamate métabotropique (figure 4). Le gène FAIM2 code pour une protéine qui a un rôle d’inhibiteur de l’apoptose. L’allèle mineur du SNP de ce gène semblerait avoir une interaction causant une résistance à développer la schizophrénie avec l’allèle mineur du SNP du gène SHANK2, un gène qui code pour une protéine qui fait partie de l’échafaudage de la densité post synaptique et attache les mGluRs aux récepteurs NMDA lors de la synaptogénèse (figure 11). Le gène CDH13 code pour une protéine qui sert de régulateur négatif de la croissance de l’axone pendant la différenciation neurale et de protection contre l’apoptose dû à un stress oxydatif. L’allèle mineur du SNP de ce gène semblerait avoir une interaction causant une résistance à développer la schizophrénie avec l’allèle mineur du SNP du gène CYCS, un gène qui code pour une protéine qui sert à l’initiation de l’apoptose (figure 12). Le gène DLG2 code pour une protéine qui est impliquée dans la clustérisation des récepteurs et canaux ioniques postsynaptiques. L’allèle mineur du SNP de ce gène semblerait avoir une interaction causant une résistance à développer la schizophrénie avec l’allèle mineur du SNP du gène RGS7, un gène qui code pour une protéine qui a le rôle de régulateur des protéines-G postsynaptiques (figure 13).
Dans la troisième catégorie, nous avons retenu quelques paires de gènes dont la présence de l’allèle mineure dans les 2 gènes pourrait ramener à la normale le phénotype de développer la schizophrénie alors que la présence de l’allèle mineur