Amino
acid
sequence
of
the penicillin-binding
protein/DD-peptidase
of Streptomyces
K15
Predicted
secondary
structuresof
the
low-M,
peniciliin-binding
proteins of class A Pilar PALOMEQUE-MESSIA,* Serge ENGLEBERT,* Melina LEYH-BOUILLE,*MartineNGUYEN-DISTECHE,* Colette DUEZ,* Simone HOUBA,* Otto DIDEBERG,*
Jozef VAN BEEUMENt and Jean-Marie GHUYSEN*t
*Centred'Ingenierie desProteines, Universitede Liege, Institut de Chimie, B6, B-4000 SartTilman (Liege 1), Belgium,
andtLaboratorium voorMicrobiologie enMicrobiele Genetica, Rijksuniversiteit-Gent, K.L. Ledeganckstraat 35, B-9000 Ghent, Belgium
Thelow-Mrpenicillin-binding protein (PBP)/DD-transpeptidase ofStreptomycesK15is synthesized in the formofa
291-aminoacid-residue precursor possessing acleavable 29-amino acid-residue signalpeptide. Sequence-similarity searches
and hydrophobic-cluster analysis show that the Streptomyces K15 enzyme, the Escherichia coli
PBPs/DD-carboxy-peptidases 5 and 6, the Bacillus subtilis PBP/DD-carboxypeptidase 5and the spollA product(aputativePBPinvolvedin
the sporulation ofB. subtilis) are structurally relatedand form a distinct class A of
low-M,
PBPs/DD-peptidases. The distribution of thehydrophobic clusters along theaminoacidsequencesalso shows that theStreptomyces K15 PBP, andby extension the other PBPs of class A, have similarityin the polypeptide folding, with the
,8-lactamases
of class A, with asreferencethe Streptomycesalbus G and Staphylococcusaureus,J-lactamasesof known three-dimensional structure.Thiscomparison allowsonetopredictmostof thesecondarystructures inthe PBPs and the amino acid motifs that definethe enzyme activesites.
INTRODUCTION
Thelow-Mrpenicillin-binding proteins (PBPs)/DD-peptidases catalyseruptureofthe C-terminal peptidebondofthebacterial wall peptidoglycan precursors and other R-D-alanyl-D-alanine-terminated analogues by sequential transfer ofthe R-D-alanyl moietytoanessentialserine residue(withreleaseofthe leaving
group D-alanine and formation of a serine ester-linked acyl-enzyme)and thentoanexogenousacceptor. Penicillin inactivates the DD-peptidase activity by immobilizing the essential serine residueinto a stable acyl-enzyme (Ghuysen, 1991).
Thelow-MrPBP/DD-peptidaseofStreptomycesK15 has been purifiedtoproteinhomogeneityinthepresenceof
cetyltrimethyl-ammonium bromide(Nguyen-Distecheetal.,1982).It ispeculiar
in at least two respects. (1) At variance with other low-Mr
PBPs/DD-peptidases that are anchored into the plasma
mem-brane bya non-cleaved C-terminal peptide segment (Spratt &
Cromie, 1988),theStreptomyces K15enzymecanbesolubilized
directly from the mycelium with the help of a high NaCl
concentration (M. Nguyen-Disteche & M. Leyh-Bouille,
un-published work). (2)Theefficacywithwhich thelow-Mr PBPs/DD-peptidases perform hydrolysis compared with transpeptidation
of thecarbonyldonordependsontheefficacywithwhichwater,
the leaving groupD-alanine andanexogenous peptide NH2-X
can attack the acyl (R-D-alanyl-)enzyme. Although all known
PBPs/DD-peptidases act mainly as DD-carboxypeptidases, the
relative acceptoractivityfor theacyl-(R-D-alanyl-)(Streptomyces
K15 enzyme)is water<< D-alanine <H2N-X. Hydrolysisofthe
carbonyl donor,inthe absenceofH2N-X, isnegligible because
attack of the acyl-enzyme by the leaving group D-alanine
regenerates theoriginal substrate. In thepresence ofa suitable
compound NH2-X, the carbonyl donor is quantitatively
con-verted intoatranspeptidated product, i.e. the Streptomyces K15
enzymefunctionsas astrictDD-transpeptidase(Nguyen-Disteche
etal., 1986).
Understanding thesepeculiar properties needsaprecise
knowl-edge of the structure of the Streptomyces K15
PBP/DD-pep-tidase.Genecloning andsequencing has yielded the amino acid
sequence of the protein. Pair-wise comparison with the other
low-M,
PBPs of known primary structure has led to the conclusion that(1) the Streptomyces K15 PBP, the Escherichia coli PBPs 5 and 6 (Broome-Smith etal., 1988), the Bacillus subtilisPBP5(Toddetal., 1986)and thespollA product,which isthought tobe aPBPspecificallyinvolved in sporulation (Wu&Piggot, 1991), belongto a sameclass Aof
low-M,
PBPs/DD-peptidases, and(2) these PBPsadopt apolypeptide scaffolding
comparablewith that ofthe class A fl-lactamases of
Staphylo-coccus aureus (Herzberg & Moult, 1987) and Streptomyces
albusG(Didebergetal., 1987; Lamotte-Brasseur etal., 1991).
MATERIALS AND METHODS
Bacterialstrains, plasmidsand media
Streptomyces K15wasfrom thislaboratory.PlasmidpBR322
(Bolivar etal., 1977) was used for cloning experiments.
Escherichia coli HB1O1 (Boyer&Roulland-Dussoix, 1969)served
as host of the recombinant plasmids. Streptomyces K15 was
grown at 28°C in tryptone/soya broth (Oxoid) with vigorous
orbitalshaking.E.coliwasgrownat37°CinLuriaBertani(LB)
orM9CA medium(Maniatisetal.,1982)withvigorous shaking.
TransformantswereselectedonLB/agar plates containing25,ug
ofampicillin/ml or 12.5,goftetracycline/ml.
Abbreviations used: PBP,penicillin-binding
protein;
HCA,hydrophobic-cluster analysis.
: Towhomcorrespondence should be addressed.
Thenucleotilesequence data
reported
willappearin theEMBL,GenBank and DDBK Nucleotide Sequence Databases under the accession number X59965.N-Terminal VTKPTI AAVG
YAMNNG1T
GT TLYTKAADTRRSTGSTT|KI M(T) AKVXLAO^SNLN aminoacidsequence
A T
5'G-GGA-T_CC-TAC-GCC-ATG-AAC-AAC-GGC3' 3TTC-TAG-TAC-TGG-CGG-TTC-CAC-TTA-AGG5'
BamHl EcoRI
Senseprimer Antisenseprimer
5'ATG-AAC-AAC-GG
C-AC G-GG C-AC C-ACG-CT C-T AC-AC C-AAG-GCC-GCG-GAC-ACC-AGG-PCRSall fragment
-CGG-TCG-ACC-GGC-TCC-ACC-ACC-AAG-ATC-ATG-ACC3
3 |GC-TGG-CCG-AGG-T
GG-TGG-TTC-TAG-T ACJ5Non-degenerated26-merprobe
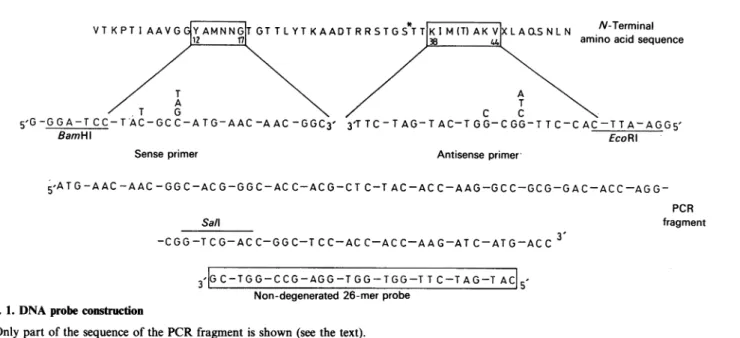
Fig.
1. DNAprobe
constructionOnlypartof thesequenceof thePCRfragment is shown(seethetext).
Enzymes, antibiotics, oligonucleotides and recombinant DNA techniques
Restriction endonucleases and T4 DNA ligase were from Boehringer (Mannheim, Germany) and Bethesda Research Laboratories (Gaithesburg, MD, U.S.A.); lysozyme and tetra-cyclinewerefrom Sigma ChemicalCo.(St.Louis,MO, U.S.A.). Sequenasewasfrom UnitedBiochemical Corp.(Cleveland, OH, U.S.A.), bacterial alkaline phosphatase was from Amersham International (Amersham, Bucks., U.K.) and ampicillin was fromBeecham (Brussels, Belgium). Oligonucleotides (see below) were synthesized by Eurogentec (Liege, Belgium). The
Strepto-myces chromosomal DNA was preparedas described by
Hop-wood et al. (1985). Preparation ofplasmid DNA, transformation ofE. coli,digestion ofDNAwithrestrictionenzymes, treatment withbacterial alkaline phosphatase, ligation,agarose-gel electro-phoresis of plasmids and digestedDNAsandelution ofseparated DNA fragments were performed essentially as described in Maniatisetal. (1982).
Radioactiveoligonucleotide probe andscreening of gene libraries
On the basis of the known amino acid sequence ofthe
N-terminal region ofthe mature Streptomyces K15 PBP
(Leyh-Bouilleetal., 1989) and the known Streptomyces codon usage, two nucleotide primers were synthesized (Fig. 1). The sense
oligonucleotide IhadaBamHI siteatthe5'-endand encodedthe
sequence Y12AMNNG17 of the protein and the antisense
oligonucleotide 2 had an EcoRI site at the 5'-end and was
complementary ofthenucleotidesequenceencodingthe sequence
K38IMTAKV44. Polymerase chain reaction (PCR) was
per-formed on 1001ul samples containing the Streptomyces K15 chromosomal DNA(1.5
jug),
theprimers (1 ,Meach),the dNTPs(200,UM
each),
the
Taq
DNApolymerase (2.5 units;
Perkin-Elmer-Cetus, Norwalk, CT, U.S.A.) andgelatin
(10,ug).
The buffer was 10mM-Tris/HCl
buffer, pH 8.3,
containing
50 mm-KCIand 1.5mM-MgCI2. Sampleswerecovered with mineral oil andsubmittedto 30amplification
cycles:
1mindenaturationat94°C, 1.5 min annealing at 55
°C
and 2minpolymerization
at72°C(Saikietal., 1988;Leeetal.,
1988).
The110bp-DNA
PCR product was partiallysequenced
and the sequence translated exactly into theexpected
peptide
segment(Fig. 1).
From this nucleotide sequence, anon-degenerated
26-merprobe (Fig. 1)
was
synthesized,
labelled with[y-32P]ATP
(Maxam
&Gilbert,
1980) and used to screen gene libraries by hybridization
[according to the procedure described in Focus (1984); BRL, Bethesda, MD, U.S.A.]. Southern blotting was performed as described in Maniatisetal. (1982).
Nucleotide sequence
DNA segments cloned into bacteriophages M13mpl8 and
M13mpl9 were sequenced by the dideoxynucleotide chain-termination method(Sanger et al., 1977, 1980). The sequencing reactions were conducted with the universal M13 sequencing primer or the two synthetic oligonucleotides shown in Fig. 2.
Zones of base compression due to high G+C content were
resolved by using dITP instead of dGTP (Sequenase kit). For eachpossible reading frame the codonusage wasanalysed with Staden's program (Staden & McLachlan, 1982; Staden, 1982), with the geneencoding the Streptomyces R61 PBP/DD-peptidase (Duezetal., 1987) asreference.
Amino
ac2'
analysisAmino acid analysis was performed on a Multichrom B
analyser(Beckman, PaloAlto, CA, U.S.A.). Before hydrolysis thesampleswereoxidizedeither inthegasphaseorintheliquid phaseand thenhydrolysed in vacuowith 6M-HCIat 106°Cfor 24h.
Similaritysearches
TheGoad& Kanehisa (1982)procedure expressesthe extent
ofsimilarity betweenpairsof amino acid sequences bya score, the more negative the score, the better the similarity. The significance oftheextentofsimilarityfoundbetweentwoaligned amino acid sequences was estimated
by
theSEQDP
program. Scores with atleast five deviations above that expected from a run of20randomized pairs ofproteins having thesame aminoacid
compositions
as theproteins
under consideration indicatesignificant similarity. Pair-wise aminoacidsequences
alignment
wasalsooptimized usingthe localhomology
algorithm
of Smith & Waterman (1981)(Bestfit
program inthe GCGpackage;
gap weight, 5.0; lengthweight,
0.3).Prediction of secondarystructuresbythehydrophobic-cluster analysis
Hydrophobic-cluster analysis (HCA)
wasperformed
and theHCA
sequence-similarity
scores wereestimated as described inGaboriaud et al.(1987). HCA is a powerful method for analysing proteins that areweakly related in the primary structure. It rests upon a representation of the protein sequences on an
a-helical
two-dimensional pattern (in which the hydrophobic residues tend to form clusters that usually correspond to the secondary structure elements) and compares thedistributionof the clusters alongthesequences. Clusters of similar shapes, sizes and relative positions express similarity in the polypeptide folding of the proteins. HCA offers the following advantages: (1) the two-dimensional plots allow distant information to become visible morereadily than with methods based only on single amino acid property/identity; (2) deletions or insertions are easily introduced between the secondary structures (Henrissat et al., 1990). Proteins with very close polypeptide folding have HCA score > 75 %. Scores of 55-65 % indicate similarity inthe secondary-structure topology (Gaboriaud et al., 1987; Henrissat etal., 1990). RESULTSGenecloning and sequencing
The genomicDNAof Streptomyces K15 was partially cleaved withBamHI,withBglIIand with Bcll, and the DNA fragments
were cloned in pBR322 previously cut with BamHI and
de-(a)
km) 500 1000 13
,. 1 * 2
w 3 Il 4 *,
SphI N-Term. Sail
PCR A B C D 357bp SmaliC-Term Sphl E F G H
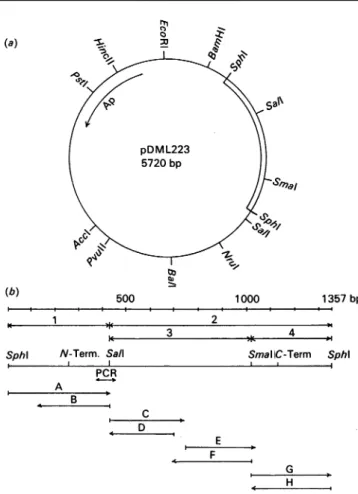
Fig.2. Restriction map of pDML223 (a) and strategy of nucleotide sequencing (b)
The bacteriophage vectors Ml3mpl8, M13mpl9, Ml3tgl3O and Ml3tgl31 were used to clone the various subfragments. The sub sequencesA, B, C, F,G andHwereobtainedby usingthe universal primer and the subsequences D and E by using the synthetic nucleotidescorrespondingtonucleotide residues 742 to723 of the sequence and nucleotide residues 710 to 729 (see Fig. 3). The positionscorrespondingtothe N-terminal and C-terminal residues of theDD-peptidaseprecursorareindicated.
phosphorylated. Of the 2990 ampicillin-resistant and tetracycline-sensitive E. coli transformants obtained, two clones from the BamHIlibrary and one clone from theBclI library gave a strong positive reaction with the radioactive probe after washing the filters at 75 °C
(Tm
-10°C). Two clones, one from each library, were analysed. PlasmidspDML220fromtheBamHIlibrary andpDML221 from the Bcll library had acquired respectively a
10.7 kb and 9.8 kb insert. They were digested with restriction endonucleases, andthe DNA fragments(separated by gel electro-phoresis and transferred to nitrocellulose) were submitted to hybridization with the radioactive probe. Among the smallest hybridizing fragments, the 1.3kb SphI DNA fragments origin-ating from both pDML220 and pDML221 were subcloned into pBR322, yieldingpDML223and pDML224 respectively. As derivedfrom the restriction maps (Fig.2a), these two plasmids containedthe sameinsert, but in opposite direction.
Establishment of the nucleotide sequence of 90% of each
strand of the 1357bpSphI DNAfragment(by using the strategy shown in Fig. 2b) revealed an 876-nucleotide-residue open reading frame (Fig. 3). This open reading frame starts with a GTGcodon,presents thebiasedpattern of codon usage typical toStreptomycesgenes(thepercentageof
G.
C being 49, 45 and 98 at the first, second and third positions of the triplets respectively), terminates with an amber stop TAG codon and is followed by a putative terminator sequence. The 5'-ATGAAGGG-3' segment at position -9 to -2 upstream of the translation start codon (GTG) complements five bases out of eight of the 3'-end of the 16 S rRNA of Streptomyces lividans (Bibb & Cohen, 1982) and resembles a ribosome-binding site (Hopwoodetal., 1986).Primary structure
The876-nucleotide-residueopenreading frametranslated into a 291-amino acid-residue protein precursor (Fig. 3). The 29-aminoacid-residueN-terminalregionhasthe features ofasignal peptide; it contains three arginine residues at positions -28, -26and -25, along hydrophobicstretch from Ala-25 to Pro-4 and the leader
peptidase-cleavage
site Ala--3-Thr-Ala--1. The aminoacidsequence of theproteinfromVal-1
toAsn-52 is exactly that ofthe N-terminal region ofthepurifiedmembrane-bound Streptomyces K15 enzyme (as determined by Edman
degradation; Leyh-Bouille etal., 1989) and the amino acid compositionofthepolypeptide chainfrom Val- 1toLeu-262 is in excellent agreement with thatofthemature protein (Table 1). Similaritysearches
Both the Goad & Kanehisa (1982)
procedure (Table
2) and Bestfit program (resultsnot shown) revealedhigh similarity,
in the primary structure, between the Streptomyces K15 enzyme, the E. coli PBPs 5 and 6(Broome-Smith
etal., 1988)
and theB. subtilis PBP5(Todd
etal., 1986) andspollA product (Wu
& Piggot, 1991). No ormarginal
similarity
was observedwith the Streptomyces R61PBP/DD-peptidase (Duez
etal.,
1987),
the serinefl-lactamases
ofclassA,
Cand D(Dehottay
etal.,
1987;
Ambler, 1980; Jaurin &
Griindstr6m,
1981; Dale etal., 1985)
and the
penicillin-binding
domains of the bifunctionalhigh-Mr
PBPs(Broome-Smith etal., 1985;Asohetal., 1986;
Nakamura etal., 1983).Hydrophobic clusteranalysisandamino acid
alignment
The upper part of Fig. 4 compares the distribution of the
hydrophobic
clustersalong
the amino acid sequences(see
theMaterials
and methodssection)
of theStreptomyces
K15PBP,
theE.coliPBP5
(and by
extensionPBP6;
notshown)
andthe B.subtilisPBP5
(and by
extension,
thespoIlA product;
notshown).
Alignment of theconservedclusters
requires
veryfew deletions.Ih) k
GCA
TGCGCACAGCATAGATACAACGCGTACGCATAGCAGTTCCCGTCCACCATCCGGACICCACGGAGGCCGCCGGACCCCA 82 CGCGCCCCTCCTCCCTCACCTTTCAAAGGGACCCCGCCCACGAACTGCACAACCATTCGTGCTACTTACGTGTCAGAGG
AGAAGCGGCATCCCCATGTGGCCGCACTCTTGGGGCATTTAACGTATTCGGGGTATTCGACTTGATAACTGGCATGAAG 240
Val Arq Leu Ar Arq Ala Ala Ala Thr Val Ile Thr Thr Gl Ala Leu Leu Ala Ala
GGT GTG CGC CTC CGT AGA GCT GCC GCC ACC GTC ATC ACC ACC GGC GCG CTG CTC GCG GCC 300
G1 Thr Leu G1 Ala Thr Pro Ala Thr Ala
lThr
Lys Pro Thr Ile Ala Ala Val G yGGX
ACC CTC GGC GCG ACT CCC GCG ACC GCC ACC MG CCC ACC ATC GCC GCT GTG GGC360
20 30
Gliqr Ala Met Asn Asn Gl Thr Gl Thr ThrLeuTrThr Lus AloA aAsA hr Ar
GGCM
Tc WC TG AAC MC GGC ACO GGC ACC ACG CTC TIC ACC AAG GCC GCGGAt
ACC AGG 420*40 s50
Arq Ser Thr Gl Se Thr Thr L s lie Met Thr Ala
Lws
Vol Vol Leu Ala Gln Ser AsnCG TCG ACC GO TCC ACC ACC AAG ATC ATG ACC GCC AAG GIG GTC CTG GCC CAG TCG MC
480
Leu Asn Leu Asp Ala Lqs Val Thr
CTG MC CTC
GAT
GCC MG GTC ACGL4s Pro Ser Gln Ala H's Leu Ile
GG CCC TCG CAG GCG CAC CTG ATC
60
Ile Gln Lus Ala
Tr
SerATC CAG AAG GCC TCAGC
80
Val Gi A Ls Val Thr
GTC GGC
GAE
AXG
GTC ACC70
Asp Tjr Val Val Ala Asn
GAC TXC GTG GTC GCC AAC 540
90
Val Arq Gln Leu Leu T
GTC CGC CAG CTC CTG TXC 600
100 110
GlU Leu Met Leu Pro Ser GI Cs A¶ Ala Ala
Tr
Ala Leu Ala Am L sTr
Gl SerGGG CTG ATG CTG CCG TCC
GM
TI GAC GCC GCG T GCG CTG GCCGA GTCG ICG 660120 130
Gi Ser Gln Ala Ala Ala Ar Val Ls Ser Phe Ile Gl Ls Met Asn Thr Ala Ala Thr
GGC AGC CAG GCT GCC GCG CG GTG AMG TCG TTC ATC GGC
AXG
AIG AAC ACC GCC GCG ACC 720140
Asn Leu Gi1 Lou His Asn Thr Hts Phe Asp Ser Pho Asp
MC CTC GGT CTG CAC MC ACG CAC TTC GAt TCG TIC GAt
150
Glu Ile Glu A Gly Ala Asn
GG ATC GGC MC GGC GCC MC 780
160 170
Tr Ser Thr Pro Arg His Leu Thr Lgs Ile Ala Ser Ser Ala Met L AsneSr Thr Phe
TAC TCG ACG CCG CGG CAC CTG ACG AAG ATC GCC AGC AGC GCG ATG AXG MC TCG ACG TTC 840
Ar Thr Val Val
Lws
Thr LysCGC ACG GTC GTC AMGACC
AXG
180 190
Ala Tr Thr Ala Ls Thr Val Thr Ls Thr Gl Ser Ile
GCGTXC ACG GCG A G ACG GTC ACC AG ACC
GGM
AGC ATC 900200 210
ArlThr Met As Thr Tr LsAsn Thr Asn Glu Leu Leu Ser Ser
Tr
Ser Gl Ala IleCGCACC AG GA ACGTIGG
AXG
AAC ACC AAC GGQ CTG CTC AGC AGC TIC AGC GGY GCG ATC 960Gig Val Ls Thr Gl Ser Gl Pro
GGC GTG MG ACC GGC TCC GGC CCC
220
Glu Ala
Lws
rGAG GCC AAG TAC
230
Cus Leu Val Phe Ala Ala Th Arq
TGC CTC GTC TTC GCC GCC ACC CGG1020
240 250
Gli GG Ls Thr Val Ile Glu Tr Val Leu Ala Scr Thr Scr Ile Pro Ala
Ar2
Glu SerGGC GGO
AXG
ACG GTC ATC GGC ACC GTC CTC GCC TCC ACG TCC ATC CCG GCC CGC GAG TCG1080Asp Ala Thr Lus Ile Met
GA GOCC ACC
AXG
ATC ATG260 A
Asn Tr Gi1 Phe AIa
AAC T C GGC TTC GCC | TAG CCCACGCACACGCAGAAGGGGCCGACC 1146
GCTTGTCGCGGTCG
CGCCCTTCTGCCGTTCGTGCGGCCCCTGGCGCCGTCTCAGGCCGGGTTGCCGGTCGAGACGAAGAG
CTGGATGTCCGGCATGTTCTTCGGS-TCCTCATCGGIITCCGCCGATTGGOCAGTTGGGCCATGACCGTGCCCTCGCCGTA1304
CGTGTICTCGTCCTGGTGCTTGATCGTCCACGACCAGCCCGCCGCCCGCATGC
Fig.3.Nucleotide sequence of the geneencodingtheStreptomycesK15PBP/DD-peptidaseprecursor and deducedamino acid sequence of theprotein
TheN-terminal valine residue and C-terminal leucine residue ofthematureenzymeareboxed.Theputative ribosome-bindingsite is underlined.
The invertedrepeatsof theputative transcriptionterminationsignalareshownbyhorizontalarrows.Ser* represents the active-site serine residue.
Theconserved clusterscoverapprox.30%of theproteins.Of the total 14 alignedclusters, ten for the
pair
Streptomyces Ki5-E. coli (pair I)andeightfor thepair
Streptomyces K15-B. subtilis (pairII)have HCAscoreshigherthan 65%.Within theconserved area, the overall HCA scores are800%
forpair
I and71% for pair II. Many charged and additional hydrophobic residues are also conserved around the clusters showing considerable sequence similarity. The lower part ofFig.4 shows the aminoacid sequence alignment as derived from the
hydrophobic-cluster distribution. The cost of this comparison, expressed in percentage of residues of the
original
sequences that are noteffectively aligned,is 8%for bothpairs.Thealignmentgenerates
26% strict identities and 34% similarities for pair I, and 21 % strict identities and 29 % similarities for pair II. Note that, at variancewithStreptomyces K15 PBP, theE.coli andB. subtilis PBPs have a long C-terminal extension. These extensions are excluded from the present analysis.
As shown above, the Goad & Kanehisa (1982) and Bestfit procedures didnothighlight similarity,intheprimarystructure, between the three PBPs and the class A
,J-lactamases.
HCA,
however, revealedsimilarity, inthepolypeptide folding,
between the PBPs and the ,-lactamases ofStaph.
aureus(Herzberg
& Moult, 1987)andStrep.albus G(Dideberg
etal., 1987;
Lamotte-Brasseur etal., 1991).Thuspair-wise comparison
(upper
partofTable 1. Amino acid composition oftheStreptomyces K15DD-peptidase
The amino acid composition is given as number of residues per
proteinmolecule asderived from chemical analysis (A) and from the
nucleotidesequence of the encoding gene (B).
Amino acid composition (residue/molecule) Amino acid A B Lys His Arg Cys Asp Asn f Met Thr Ser Glu Gln Pro Gly Ala Val Ile Leu Tyr Phe Trp Total 20 22 4 4 10 9 2 2 28
110
14 8 7 34 34 21 22 8 2 7 6 28 25 34 33 17 18 12 12 19 18 10 12 6 6 1 268 267Fig. 5) of the Streptomyces K15 PBP with the Strep. albus G /-lactamase(pair III)and with theStaph.aureus/1-lactamase(pair IV)shows that the conserved areasrepresent 25 %(pairIII) and 30% (pair IV)of the proteins. Within the conserved areas, the overall HCA scores are 70% (pair III)and 73% (pair IV). The HCA scores for the individual clusters vary from 50% to89% (pair III) and from 22% to 100% (pair IV). The cost of the amino acid alignment (lower part of Fig. 5) does not exceed 12%. The sequences effectively aligned contain 15.6% strict identities and 23 % similarities for pair III, and 11.5% strict identities and 19% similarities forpair IV. The
significance
ofthe similarity is expressed by a low but significant standard deviation of 9units (pair III) and 6 units (pair IV).
DISCUSSION
The Strep. albus G and Staph. aureus
,-lactamases
of class A areofknownthree-dimensionalstructure. These proteins consist ofan all-a domain (helices a2-a9) and ana//,
domain (helicesal,alIO,al1andstrands
/11-/15).
Their active sites are defined by four conserved amino acid motifs. Using the ABL numberingscheme, the S70VFK motif in the Streptomyces enzyme or
S70TSKinthe staphylococcal enzyme is central to the enzyme cavity with the essential S70 at theN-terminus of helixa2.The S13ODN motif (inboth enzymes), on a loop connecting helices
a4 and a5, occurs on one side of the cavity. The E166PELN
motif in the Streptomyces enzyme or E166IELN motif in the
staphylococcal enzyme is on a loop at the entrance of the cavity. Finally, the K234TG motif in the Streptomyces enzyme or
K234SG motif in the staphylococcal enzyme occurs on the
innermost
/13
strandofthefl-sheet
on the other side ofthe cavity. Onthe basisof the principlethat similarity in the hydrophobic-clusterdistribution expresses similarity in the polypeptide folding of the proteins, the present study leads to the following con-clusions.(1)TheStreptomyces K15 PBP, the E. coli PBP5 (and 6) and the B. subtilis PBP5(andspoIIAproduct)arestructurally related (Fig. 4).Itisproposed that they belongto a same class Aof low-MrPBPs/DD-peptidases.
(2) The secondary structures in the Streptomyces K15 PBP (and the other PBPs of class A) have a spatial disposition comparable with that foundin the
,1-lactamases
of classA(Fig. 5).Identificationinthe PBPs of strands /11,
/82,
/13, /14
and/5
and of helices a2, a4,a5,a6,ac8 andacl 1 is not a matterofcontroversy. Identificationin the PBPsofclustersequivalent to helicesa3, 7, a9andalO in the/1-lactamases
is hypothetical. Note, however, the presence of one glycine residue in helix a7 and of two asparagine residues on the N-terminal side of helix alO in all PBPs.(3)Threeofthe four motifs that define the active site of the /-lactamases (see above) have homologues in the Streptomyces
K15, E. coli and B. subtilis PBPs. Thus the STTK, SLTK or
SMTK tetrad of the PBPs(astheyarelisted)arehomologues of
the S70XXK motifofthe
,-lactamases,
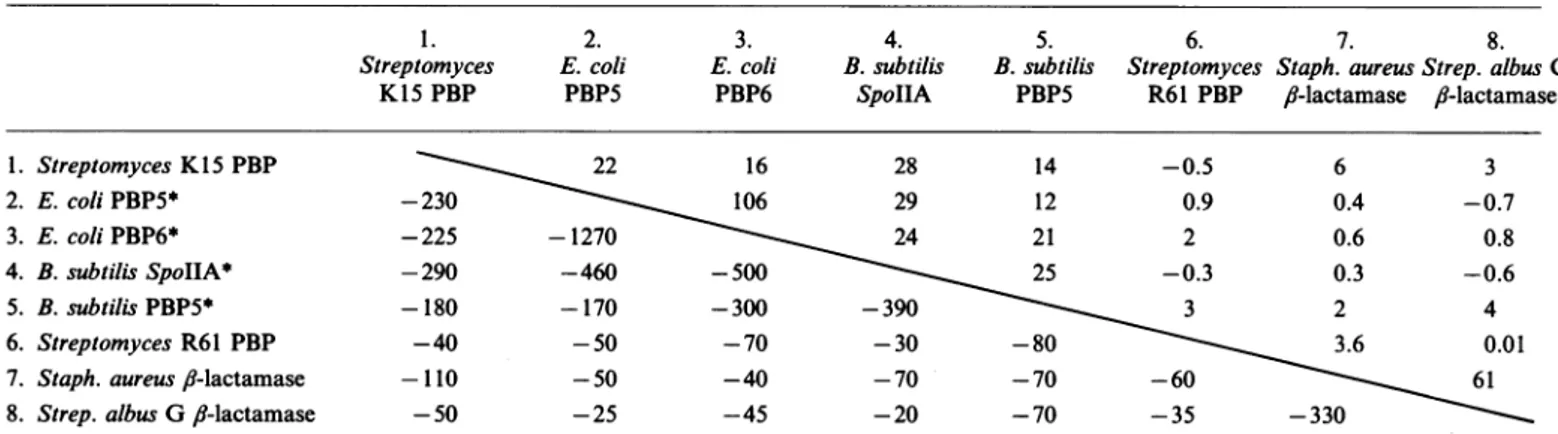
theSGC, SGN or SANTable2.Search forsequencesimilaritybetweenthe aminoacidsequencesof severallow-Mr PBPs andII-lactamasesbyusingtheGoad & Kanehisa
(1982)
method
Comparisonscores (belowthediagonal line)andsignificancein SDU(abovethediagonal line) areshown.
2. E.coli PBP5 3. E.coli PBP6 4. B.subtilis SpoIIA 5. B.subtilis PBP5 6. 7. 8.
Streptomyces Staph. aureusStrep. albus G
R61 PBP 8-lactamase /1-lactamase 1. StreptomycesK15PBP 2. E. coliPBP5* 3. E. coliPBP6* 4. B.subtilisSpoIIA* 5. B.subtilis PBP5* 6. StreptomycesR61 PBP 7. Staph.aureus,b-lactamase 8. Strep.albus G /-lactamase
~~~22
16 28 14 -0.5 6 3-230 ~~~~106
29 12 0.9 0.4 -0.7 -225 -1270 24 21 2 0.6 0.8 -290 -460 -500 25 -0.3 0.3 -0.6 -180 -170 -300 -390 3 2 4 -40 -50 -70 -30 -80 3.6 0.01 -110 -50 -40 -70 -70 -60 61 -50 -25 -45 -20 -70 -35 -330* TheC-terminal extensionsof the E. coliPBP5(fromP267 toG374),PBP6(fromT262toS373),the B. subtilisPBP5(fromE283 toW412)and
SpollA (fromP284 to K389)have been eliminated for thecomparison. 1.
Streptomyces
_ - s -rnL= , S: c ct * ~ ><QUIH>CHOEq X>24 X I0 * HH>4 ¢ k/) Hx'C > CCH 0iU3@1. EA < 4 1-4 1G>4 EnVD U)a CHN4 OP XH HI: .p E>Z* @ h: ~~~xmE. 0 W4 C S-4HH. H Hfh74H *@'C0 CHd >o uH >0 *COX mHHC' CCH~~*>~~>4 0 'DcOH-4 0 Z 0 >H4In * Ce H Oa'Clp -,* 'oiH HHH HIi'000'*eo2 9>@6
-,i kor OHH*EqE fiM *
*F
oP
a;W;z:;
:< >< *HHHVo . U
*H:
LO-o
s E* P;U)0 >-A H,ixK.Qz
7 fH:> Hf\vU:> uz C,)i~-l is Z0¢clfC .000.~~~~~~0 HC-)ES I SFju~_n V E 0a_ :H *>4CC. (D. :z' c)zcf H Z n xHt.- t-
x
ff <.
LD H OXn LC a rT ,x>4 040 0 cnE a4 L,i*D o *>412% 0 0 L 'C'CqXZ t E >cHO X 01- 00 0X : OH4H H L 2~~~*4 0'7 Co i 3 *a,>C>EPH4E,XX F, f X_ x* f uHZ 004~ HH > 4> Oe0Ct'CH 3g3 -EH4 Zr'' Z C 2 2pHOv)U 00XH >CX 0p[ZOH ¢ > <>HH L^iO H3H 00a , o CO> O H V H'CH >4C 0 U V HO Z V * <H >1* H .00 * E-;( ( * E S>q H x N(:) * 2* + -:HF1 > :> (D 1- E- <: E-4tn C PnI< lHCx g;Z,XU) Ui0< ] X :> o~~~~~>~~~~~az :>>U)x4 >
t P4 H * - a~~~ 4 HX > apF I ~ ~~>>SZ~~~ a X [
=
H Icl
>
<~~0
01E
1U
1- Enll x * @ NN4 1-o1 k S4 Eq > * o ; >4 Ef C: M b M @ X g D~~~~~C' cCflcCfl d-cl.-I 4z ~~~- <OLan.
-Rbwd
O 'C ucfl CA 0) 0) c Q 0. Ct4 ;e 0 o c-i O -' *; g 00 C. o4 '4-04 I. O 04 _ Q C , 0O4 >' m o 0 0 .- 0 H ~ Ci ,-, 0. t 0. U '10 112, 1 -4 r-I c c K Cc 1991m
u *@ 0c AAo
u) -X4 >~~~~~~0 0~~~~ CY~~~~~ t) *.~~ a* o x00> * x 'U C H >U) *;|> *h-> X*x{ X S B B H<>404 t Kc r4;<z~ ~ z0 el,¢:<MM00s)K : .5 0>4 e. . 0>>H XL SOH 0V Z5 ZO t i: ) O 4< VH EA0 >40<oH (HO s0 E-4p U) *EH C H X @nH V)a 040~
01 LO E-4H <E z co >% z E4Er-1 OH<g Z S:H Xa > pn 4 a4z > z- X > .Z . 0 *-4 H04H
n4 w a4a4ZkD 4 > >X4*ZZZZ *>>XEqsazLI'I' I)(
a;>G b XL0 s GU% XL Z Wz1 a;>c>>4 0 < 14 :> a)M t V XH H U.X = X< Nzrv x>< wg u0>-el) i) 0 > E- p > XQ>4z u) < L1FQ > rz5 >4 H E-14 HIE-4; 4 O£<: im In X>, >a ° >H>- nV~~~L <>u < 4 ur_cn~~IV ' Z Z *o w| :gn *Esx> 13 u 0- Q *na ..~~ : C PHP >COZH~~~~0 C X;>>4 0E404o4 0 0>>4X H£Xw O P~~~~ ~<Eq~~;P Xxe X4> XLO LI) E- X :L4H-< O<y Z CY -.> soon. *HO4Z* E--I E-4 >4 I H HVU -CE CX 0 H0>:X el > m ~~~~~4
pe
04S~~~~~~~
f
Ee
t>
Cox 7. Z UZ E- z_, SZ .v 600015N
xS
tv
L00H4O> * H uXg H> <>0 4 h* 000 OH>4 pH ~ c;z 5 H 0EX-*U' < - ° UF-~~x zco 00H ~~wa000 0ZH SH0x co U)~~ ~ ~ V (A 0.-I~~~~~~~~~ ._.00
rJ~~~~~~~~~~i
triad arehomologuesof theS13ODNmotif,and the KTG motif is the homologue of the K234T(S)G motif. In addition, the
DSFD, DAD or ENKD groupings of the PBPs might be
homologues of the E166XELN motif of the
fl-lactamases.
The conserved motifsplay essential roles in thecatalytic mechanism of thefl-lactamases.
Theirhomologues may fulfil similar func-tions inthe PBPs.(4) At variance with the ,-lactamases and the Streptomyces K1 5 PBPwhosepolypeptidechainterminates about60residues downstream from theKT(S)G motif, the E. coli and B. subtilis PBPs have long additional C-terminal extensions themselves each terminated byanamphiphilic a-helical membrane anchor (Ferreira et al., 1988). With reference to the
fl-lactamases,
the Streptomyces K15 PBP appears to have much shorter al anda 11 helices andtherebyitsf-sheetispartiallyuncovered on one face,thusexposinga zoneofhydrophobicity potential.This zone might be the site of interaction between the protein and the plasma membrane.
(5)TheStreptomyces K15 PBP(Leyh-Bouille et al., 1987) and E. coli PBP5 (Curtis &Strominger, 1978) are susceptible to thiol-specific reagents. In the Streptomyces K15 PBP, one cysteine residue(the SGCmotif)occurs within the active site on the loop connecting helices a4 and a5 and another occurs ten residues downstream of thelysine residue of the KTGmotif,i.e. onstrand
fl4.
Which ofthese two cysteine residues is the site ofreaction withp-chloromercuribenzoatecan bedefinitely established only by site-directed mutagenesis. The E. coli PBP5 possesses one singlecysteine residue at the third position immediately down-streamfrom the SGNmotif, i.e. in the immediate vicinity of the active site. Finally, one single cysteine residue, ten residues downstream from the KTG motif(i.e.onstrand,/4)
isalsofound inthe B.subtilisPBP5 and in the SpoIIAprotein. Whether or not these two proteins are susceptible to thio-specific reagents is unknown.The work inLiegewas supported in partby the Fonds de la Recherche
Scientifique Medicale (Contractn°3.4537.88),anAction Concertee with
the Belgian Government (Convention 86/91-90), the Fonds de Recherche de laFacult6deMedecineULg and a ConventionTripartite
between the Region Wallonne, SmithKline Beecham, U.K., and the
University ofLiege. Thistext presents researchresults of the Belgian
programme on Interuniversity Poles of Attraction initiated by the
Belgian State,Prime Minister'sOffice, SciencePolicy Programming.The
scientificresponsibility is assumed by its authors.P. P.-M. was afellow of
theEuropeanCommunities(TrainingContract n°010197).Someof the
work described in thispaperformspartofadissertation presentedby
P.P.-M. as partial fulfilment fora Ph.D. degree at the University of
Granada (Spain). The work in Ghentwas supported by the National
IncentiveProgramof Fundamental ResearchinLife Sciences initiatedby
theBelgian SciencePolicy Programming Department (Bio22).Wethank
D.Vaira (University of Liege) for assistance in PCR and D.Allaer
(Eurogentec, Liege) for helpful biochemical discussion.
REFERENCES
Ambler, R. P. (1980) Philos. Trans. R. Soc.LondonB 289, 321-331 Asoh, S., Matsuzawa, H., Ishino, F., Strominger, J. L., Matsuhashi, M.
& Ohta, T. (1986) Eur. J. Biochem. 160, 231-238
Bibb, M. J. & Cohen, S. N. (1982) Mol. Gen. Genet. 187, 265-277
Bolivar, F.,Rodriguez, R. L., Greene, P. J., Betlach, M. C., Heyneker,
H.L., Boyer, H.W., Crosa, H. J. & Falkow, S. (1977) Gene 2, 95-113
Boyer,H. W.&Roulland-Dussoix, D. (1969) J. Mol. Biol. 41, 459-472
Broome-Smith, J. K., Edelman,A., Youssif, S. &Spratt, B. G. (1985)
Eur.J.Biochem. 147, 437-446
Broome-Smith,J.K.,,Ioannidis, I.,Edelman,A.&Spratt,B.G.(1988)
Nucleic Acids Res. 16, 1617
Curtis, S. J. &Strominger,J. L.(1978)J.Biol.Chem. 253. 2584-2588
Dale, J. W., Godwin, D.,Mossakouska, D.,Stephenson, P. &Wall,S.
(1985) FEBS Lett. 191, 39-42
Dehottay, P., Dusart, J., De Meester, F.,Joris, B., Van Beeumen, J.,
Erpicum, T., Frere,J.-M.&Ghuysen,J.-M.(1987)Eur.J. Biochem.
166,345-350
Dideberg, O., Charlier, P., Wery, J. P.,Dehottay,P., Dusart,J.,Erpicum,
T., Frere,J.-M.&Ghuysen,J.-M. (1987) Biochem. J. 245, 911-913
Duez, C.,Piron-Fraipont, C., Joris, B., Dusart, J.,Urdea, M. S., Martial,
J. A., Frere, J.-M. & Ghuysen, J.-M. (1987) Eur. J. Biochem. 162,
509-518
Ferreira, L. C. S., Schwarz, U., Keck, W.,Charlier,P.,Dideberg,0. &
Ghuysen,J.-M.(1988)Eur.J.Biochem. 171, 11-16
Gaboriaud,C.,Bissery,V.,Bencheritt,T. &Mornon, J.P.(1987)FEBS
Lett. 224, 149-155
Ghuysen, J. M. (1991) Annu. Rev. Microbiol. 45, 37-67
Goad, W.B.&Kanehisa, M.T.(1982) Nucleic Acids Res. 10, 247-263
Henrissat, B.,Saloheimo, M., Lavaitte, S. & Knowles, J.K.C.(1990)
Proteins Struct.Funct. Genet.8, 251-257
Herzberg, 0. &Moult,J.(1987) Science236,694-701
Hopwood,D.A., Bibb, M.J.,Chater, K.F.,Kieser, T., Bruton, C. J.,
Kieser,H.M., Lydiate,D.J.,Smith, C. P.,Ward,J. M.&Schrempf,
H. (1985) Genetic Manipulation of Streptomyces: A Laboratory
Manual, The John InnesFoundation, Norwich
Hopwood,D.A.,Bibb, M. J., Chater, K. F., Janssen, G. R.,Malpartida,
F.&Smith, C.P.(1986) inRegulation of GeneExpression: 25 Years
On (Brooth, E. R. & Higgins,C.F., eds.), pp. 251-276, Cambridge
University Press, Cambridge
Jaurin, B. &Gruindstr6m, T. (1981) Proc. Natl. Acad. Sci. U.S.A. 78,
4897-4901
Lamotte-Brasseur, J., Dive, G., Dideberg,O., Charlier, P., Frere, J.-M &
Ghuysen,J.-M.(1991) Biochem. J. 279, 213-221
Lee, C. C., Wu, X., Gibbs, R. A., Cooks, R. G., Muzny, D. N. &
Caskey, C.T. (1988) Science 239, 1288-1291
Leyh-Bouille, M.,Nguyen-Dist6che, M., Bellefroid-Bourguignon, C. &
Ghuysen,J.-M.(1987) Biochem.J.241, 893-897
Leyh-Bouille, M., Van Beeumen, J., Renier-Pirlot, S.,Joris,B.,
Nguyen-Disteche, M.&Ghuysen,J.-M.(1989) Biochem. J. 260,601-604
Maniatis, T., Fritsch,E.F. & Sambrook, J. (1982)Molecular Cloning:
ALaboratory Manual, Cold Spring Harbor Laboratory, Cold Spring
Harbor
Maxam, A.M.&Gilbert,W. (1980)Methods Enzymol. 65, 499-560
Nakamura, M., Maruyama, I.N., Soma, M., Kato, J., Suzuki, H. &
Hirota, Y.(1983) Mol. Gen. Genet. 191, 1-9
Nguyen-Disteche, M., Leyh-Bouille, M. & Ghuysen, J.-M. (1982)
Biochem.J.207, 109-115
Nguyen-Disteche, M., Leyh-Bouille, M., Pirlot, S., Frere, J.-M. & Ghuysen,J.-M.(1986) Biochem.J.235, 167-176
Saiki,R.K.,Gelfand,D.H.,Stoffel, S., Scharf, S. J., Higuchi, R.,Horn, G.T.,Mullis,K. B. &Erlich,H.A.(1988) Science 239,487-491
Sanger, F.,Nicklen, S. & Coulson, A.R.(1977) Proc.Natl. Acad. Sci.
U.S.A.74, 5436-5467
Sanger, F., Coulson, A. R., Barrell, B. G., Smith, A. J. H. & Roc, B. A.
(1980) J. Mol. Biol. 143,161-178
Smith,J.F.&Waterman,M.S.(1981)Adv.Appl. Math 2, 482-489 Spratt,B.G.&Cromie, K.D.(1988) Rev.Infect. Dis. 10, 699-711 Staden, R.(1982) Nucleic Acids Res. 10, 2951-2961
Staden, R.&McLachlan,A. D.(1982)Nucleic Acids Res. 10, 141-156
Todd,J.A., Roberts,A.N., Johnstone, K., Piggot, P. J., Winter, G. &
Ellar,D.(1986)J. Bacteriol. 167,257-264
Wu,J. J.&Piggot,P.J.(1991)inGenetics andBiotechnologyofBacilli,
vol. 3(Zukowski, M.M., Ganesan, A.T. &Hoch, J.A., eds.), pp.
321-327,AcademicPress,NewYork
Received4March 1991/24 April 1991; accepted 8 May 1991