Molecular dynamics simulation of CO2 hydrates: Prediction of three phase coexistence line
13
0
0
Texte intégral
Figure

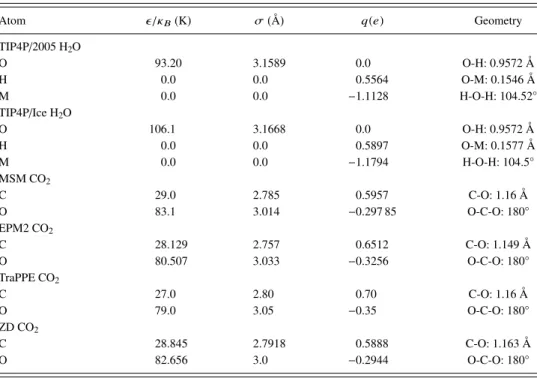
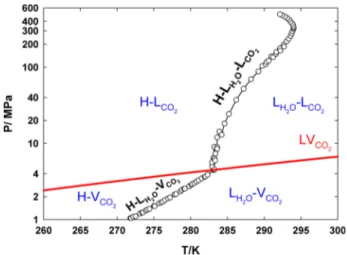
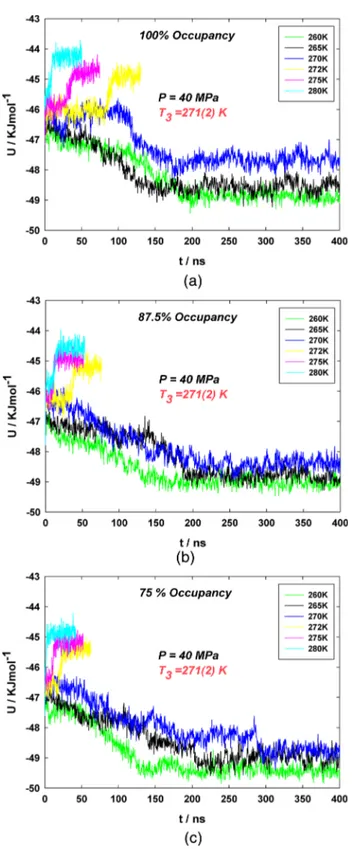
+4
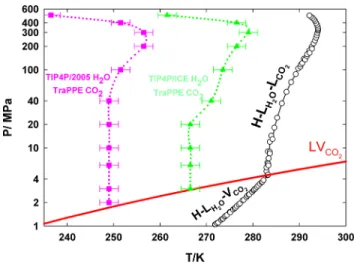
Documents relatifs
Crystalline phase transitions are usually simulated for small systems ( w 100 atoms) using metadynamics techniques [6] based on the constant pressure Parinello-Rahman Molecular
Molecular dynamics study of thermal diffusion in a binary mixture of alkanes trapped in a slit pore.. Jean Colombani, Guillaume Galliero, Bernard Duguay, Philippe Bopp,
In this work, we combine neutron scattering experiments in a specific mixture of H 2 O and D 2 O (the so-called null water) with ab initio molecular dynamics simulations in order
We conclude from the results that molecular dynamics simulation with explicit solvent is quite capable of treating the dynamics of interfacial water molecules, even though
