Synthèse de dérivés stéroïdiens et évaluation de leur
capacité inhibitrice sur les enzymes de la
stéroïdogenèse pour
le traitement de la maladie
d’Alzheimer et
/ou du cancer de la prostate
Mémoire
Sophie Boutin
Maîtrise en médecine moléculaire - avec mémoire
Maître ès sciences (M. Sc.)
Québec, Canada
ii
Synthèse de dérivés stéroïdiens et évaluation de leur
capacité inhibitrice sur les enzymes de la
stéroïdogenèse pour le traitement de la maladie
d’Alzheimer et/ou du cancer de la prostate
Mémoire
Sophie Boutin
Sous la direction de :
iii
Résumé
Les stéroïdes et leurs dérivés sont connus pour leur importance et leur versatilité en biologie. Ce sont aussi des composés lipidiques ayant un rôle essentiel dans plusieurs processus physiologiques. Leur rôle dans certaines maladies et certains cancers hormono-dépendants n’est plus à prouver. Toutefois, sachant leur affinité pour les enzymes de la stéroïdogenèse, leur potentiel thérapeutique devient intéressant. Ainsi, des dérivés stéroïdiens synthétisés peuvent être utilisés comme inhibiteurs de diverses enzymes, empêchant leur activité oxydative, réductrice ou autres.
Dans le cadre de mes travaux, les composés stéroïdiens sont utilisés en tant que traitement thérapeutique potentiel de la maladie d’Alzheimer (MA) et du cancer de la prostate (CP). Dans la première section du mémoire, ils sont évalués en tant qu’inhibiteurs potentiels de la 17β-hydroxystéroïde déshydrogénase type 10 (17β-HSD10), une enzyme mitochondriale possédant des propriétés suggérant un rôle potentiel dans la MA. Les divers composés qui sont évalués, synthétisés par chimie classique en solution et/ou par chimie en parallèle sur support solide, dérivent du premier inhibiteur stéroïdien de la 17β-HSD10 qui a été identifié par notre laboratoire. Leur activité inhibitrice, leur sélectivité d’action et leur stabilité métabolique ont été évaluées au moyen de divers essais biologiques. De plus, un inhibiteur non stéroïdien de la 17β-HSD10 rapporté dans la littérature a été synthétisé afin de comparer son activité inhibitrice avec celle de notre inhibiteur stéroïdien. Des problèmes rencontrés durant sa préparation nous ont amené à effectuer une caractérisation complète de cet inhibiteur et, par la suite, à effectuer son évaluation biologique. Finalement, deux nouveaux essais biologiques utilisant comme substrats l’alloprégnanolone et l’estradiol, deux neurostéroïdes ayant un rôle dans la MA, ont été développés afin de mesurer l'activité inhibitrice des divers composés synthétisés contre la 17β-HSD10 purifiée.
Dans la seconde section du mémoire, les nouveaux analogues stéroïdiens ont été évalués pour un traitement potentiel du CP, en mesurant l’inhibition de la transformation du 3α-androstane-3α,17β-diol (3α-diol) en dihydrotestostérone (DHT). En effet, la 5α-DHT, étant le plus puissant androgène naturel, favorise la prolifération des cellules cancéreuses de la prostate. En empêchant sa production à partir du 3α-diol, les divers dérivés stéroïdiens peuvent alors agir comme traitement thérapeutique complémentaire contre le CP
iv
androgéno-dépendant. La synthèse d’un précurseur de la 5α-DHT marqué au carbone 14, le [4-14C]-3α-diol, ainsi que la mise au point et l’application de l’essai biologique évaluant la
capacité inhibitrice des mêmes composés synthétisés précédemment ont été effectuées, permettant d’identifier une première génération d’inhibiteurs efficaces de la transformation du 3α-diol en 5α-DHT.
v
Abstract
Steroids and their derivatives are known for their importance and versatility in biology. They are also lipid compounds possessing an essential role in several physiological processes. Moreover, their role in certain hormone-dependent diseases and cancers has already been proven. Knowing their affinity for the enzymes of steroidogenesis, their therapeutic potential becomes interesting to use. Thus, synthesized steroid derivatives can be used as inhibitors of various enzymes, preventing their oxidative, reductive or other activities.
In this study, steroid derivatives are used as a potential therapeutic treatment for Alzheimer's disease (AD) and prostate cancer (PC). In the first section of this thesis, they are assessed as potential inhibitors of 17β-hydroxysteroid dehydrogenase type 10 (17β-HSD10), a mitochondrial enzyme with properties suggesting a potential role in AD. The various compounds evaluated, synthesized by classical chemistry in solution and/or by solid-phase synthesis, derive from the first steroidal inhibitor of 17β-HSD10 previously identified in our laboratory. Their inhibitory potency, their selectivity of action and their metabolic stability have been evaluated by various bioassays. In addition, a non-steroidal inhibitor of 17β-HSD10 known from literature was synthesized to compare its inhibitory potency to our steroidal inhibitor. Problems encountered during its synthesis led us to carry out a complete characterization of this inhibitor and subsequently to carry out its biological evaluation. Finally, two new bioassays using the substrates allopregnanolone and estradiol, two neurosteroids having a role in AD, were developed to measure the inhibitory potency of the steroid derivatives against purified 17β-HSD10.
In the second section of this thesis, previously synthesized compounds were evaluated as a potential treatment for PC, by inhibiting the transformation of 3α-androstane-3α,17β-diol (3α-diol) into 5α-dihydrotestosterone (5α-DHT). Indeed, 5α-DHT, being the most potent natural androgen, promotes the proliferation of PC cells. By preventing its production from 3α-diol, then the various steroid derivatives can act as complementary therapeutic treatment against androgen-dependent PC. The synthesis of a carbon 14 labeled precursor of 5α-DHT, [4-14C]-3α-diol, as well as the development and application of the
biological test evaluating the inhibitory capacity of the compounds previously synthesized were carried out, making it possible to identify a first generation of effective inhibitors of the transformation of 3α-diol into 5α-DHT.
vi
Table des matières
Résumé ... iii
Abstract ... v
Table des matières ... vi
Listes des figures et schémas ... viii
Listes des tableaux ... xi
Liste des abréviations ... xii
Remerciement ... xvii Avant-propos ... xviii Introduction ... 1 1.1 La maladie d’Alzheimer ... 2 1.2 Le cancer... 6 1.3 Les stéroïdes ... 11 1.4 Les 17β-HSDs ... 19 1.5 Origine du projet ... 24
1.6 Synthèses chimiques et protocoles biologiques employés ... 26
1.7 Hypothèses et objectifs ... 29
Chapitre 1 ... 33
Synthèse d’inhibiteurs stéroïdiens de la 17β-hydroxystéroïde déshydrogénase type 10 (17β-HSD10): sélectivité, stabilité métabolique et inhibition ... 33
Avant-propos ... 34
Résumé ... 35
Synthesis of 17β-hydroxysteroid dehydrogenase type 10 steroidal inhibitors: selectivity, metabolic stability and enhanced potency ... 36
Abstract ... 37
Chapitre 2 ... 74
Confirmation de la structure chimique et évaluation biologique d’un inhibiteur non-stéroïdien de la 17β-hydroxystéroïde déshydrogénase type 10 ... 74
Avant-propos ... 75
Résumé ... 76
Structure confirmation and evaluation of a nonsteroidal inhibitor of 17β-hydroxysteroid dehydrogenase type 10 ... 77
Abstract ... 78
Chapitre 3 ... 95
Identification de dérivés stéroïdiens inhibant la transformation de l’alloprégnanolone et de l’estradiol par la 17β-hydroxystéroïde déshydrogénase type 10 ... 95
vii
Avant-propos ... 96
Résumé ... 97
Identification of steroidal derivatives inhibiting the transformations of allopregnanolone and estradiol by 17β-hydroxysteroid dehydrogenase type 10 ... 98
Abstract ... 99
Chapitre 4 ... 111
Formation de la 5α-dihydrotestostérone à partir du 5α-androstane-3α,17β-diol par des cellules LAPC-4 – identification d’inhibiteurs des voies non classiques de production du plus puissant androgène ... 111
Avant-propos ...112 Résumé ...113 Abstract ...115 Discussion et conclusion ... 129 1.1 Discussion générale ...130 1.2 Conclusion ...139 Références ... 141 Annexe 1 ... 151 Annexe 2 ... 201 Annexe 3 ... 226 Annexe 4 ... 237
viii
Listes des figures et schémas
Introduction
Figure 1. Les inhibiteurs de l’acétylcholinestérase, les composés reminyl, rivastigmine et donépézil,
et l’antagoniste du récepteur N-méthyl-D-aspartate (NMDA), le composé mémantine………5
Figure 2. Représentation simplifiée de la voie de synthèse des stéroïdes à partir du cholestérol et des
familles de stéroïdes………..………..12
Figure 3. Stéroïdogenèse simplifiée des progestagènes, androgènes et estrogènes………..……….15 Figure 4. Stéroïdogenèse simplifiée de la 5α-dihydrotestostérone (5α-DHT) via les voies classique,
alternative et détournée (backdoor).………..…..………17
Figure 5. Contribution de la 17β-hydroxystéroïde déshydrogénase type 3 (17β-HSD3) à la
biosynthèse des androgènes testostérone (T) et de la 5α-DHT à partir du 4-Androstène-3,17-dione (4-dione). Le RM-532-105 est un dérivé 3β-androstane qui inhibe l’enzyme stéroïdogénique 17β-HSD3……….…….20
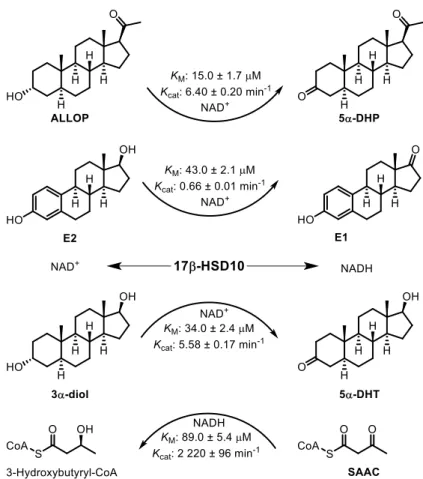
Figure 6. Transformation de trois stéroïdes et du S-acétoacétyl-CoA (SAAC) catalysées par la
17β-hydroxystéroïde déshydrogénase type 10 (17β-HSD10) en présence de nicotinamide adénine dinucléotide oxydée et réduite (NAD+ et NADH) comme cofacteur……….22 Figure 7. Exemple d’inhibiteurs non-stéroïdiens connus de la 17β-HSD10……….23 Figure 8. Le composé 1, un premier inhibiteur stéroïdien de la 17β-HSD10, l’epi-androstérone
(Epi-ADT) et la 5α-DHT………...25
Figure 9. Conditions pour l’addition du dérivé stéroïdien
3α-hydroxy-3β-piperazino-5α-androstane-17-one sur la résine polystyrène fonctionnalisée diol………27
Chapitre 1
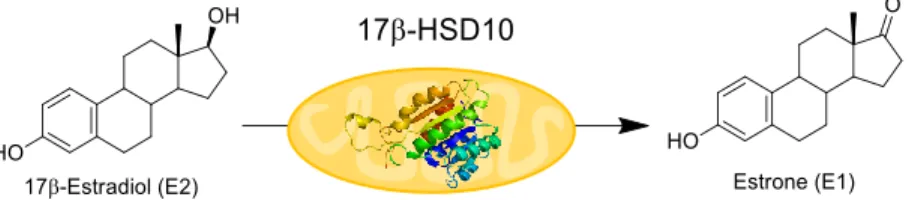
Figure 1. Oxidation of the 17β-OH of estradiol catalysed by 17β-HSD10 (Km = 43 μM) to form
estrone, a 17-ketosteroid………39
Figure 2. Derivatization of compound 1 to improve the metabolic stability and selectivity for
17β-HSD10 over 17β-HSD3 (D-ring derivatives 2-7, Schemes 1 and 2) and to optimize the inhibitory potency for 17β-HSD10 (libraries A-D, 130 compounds represented by 8-11, Scheme 3). Two hybrid inhibitors, compounds 25 and 26, were also synthesized (Scheme 4)………...40
Figure 3. Inhibition of E2 into E1 transformation by HEK-293[17β-HSD10] intact cells………...54 Scheme 1. Synthesis of D-ring steroid derivatives 2 (17β-OH), 3 (17β-OH/17α-CH3) and 4b
(17β-OH/17α-C≡CH)………..41
Scheme 2. Synthesis of D-ring steroid derivatives 5 (17α-OH), 6 (17-CF2) and 7
(17-C=O/16-C(CH3)2)……….42
Scheme 3. Parallel solid-phase synthesis of amide (A-1 and A-2), sulfonamide (B-1), urea and
thiourea (C-1) and amine (D-1, D-2 and D-3) libraries of steroid derivatives 8-11………...44
ix
Chapitre 2
Figure 1. Steroidal and nonsteroidal compounds 1 and 2, respectively, representative of two families
of 17β-HSD10 inhibitors……….80
Figure 2. Chemical synthesis of nonsteroidal 17β-HSD10 inhibitor 2………..81 Figure 3. 1H NMR spectra of compound 2 in DMSO-d
6 and full signal assignment………....82 Figure 4. ORTEP diagram resulting from X-ray analysis of compound 2. Methanol (CH3OH) is
present in the crystal in proportion 1:2 with compound 2 (represented as the R isomer). ………….82
Figure 5. Partial spectra of HSQC experiment showing key HC-correlations for both CH-1 and CH3O
signals……….84
Figure 6. Partial spectra of HMBC experiment showing four key correlations between CH-1 and
C-2, C-3, C-7 and C-12………..86
Figure 7. Inhibition by compounds 1 and 2 of the transformation of E2 into E1 by recombinant
17β-HSD10 (A) and HEK-293[17β-17β-HSD10] intact cells (B). Results of a triplicate expressed in % ± SEM………87
Chapitre 3
Figure 1. A. Transformation of two neurosteroids catalyzed by 17β-hydroxysteroid dehydrogenase
type 10 (17β-HSD10) in the presence of NAD+
as cofactor. Chemical structure of lead compound 1, a first steroidal inhibitor of 17β-HSD10 identified previously……….101
Figure 2. Transformation of ALLOP into 5α-DHP by purified 17β-HSD10 protein………..103 Figure 3. Inhibition of 17β-HSD10 protein by steroidal derivatives 1, 5, 6 and substrates ALLOP and
E2………..106
Figure 4. Inhibition of 17β-HSD10 protein by steroidal derivatives 1, 5 and 6, and substrate ALLOP
and E2………...107
Chapitre 4
Figure 1. Simplified steroidogenesis of potent androgen 5α-DHT, via classical, alternative and
backdoor pathways………...117
Figure 2. Chemical synthesis of 3α-diol and [4-14C]-3α-diol………..119 Figure 3. Transformation of [14C]-3α-diol/3β-diol (8:2) into [14C]-5α-DHT in LAPC-4 cells…...121
Figure 4. Inhibition curves and IC50 values for the formation of 5α-DHT from 3α-diol in LAPC-4
cells by unlabeled 3α-diol, A-2,16, C-1,12, D-3,6, D-3,8, D-3,7 and 25………125
Conclusion
Figure 1. Le composé 1, un premier inhibiteur stéroïdien de la 17β-HSD10………..130 Figure 2. Les composés D-3,7, 25 et 26………...132 Figure 3. Le dérivé benzothiazole phosphonate 2, un inhibiteur de la 17β-HSD10 non-stéroïdien, et
l’imine intermédiaire 5 nécessaire à la synthèse du composé 2 (voir chapitre 3)……….133
Figure 4. Les composés 4b, 5 et 6 (voir chapitre 1)……….136
Annexe 1
x
Figure S2. Chemical structures of library A-2 (amide derivatives)………...166
Figure S3. Chemical structures of library B-1 (sulfonamide derivatives)………...173
Figure S4. Chemical structures of library C-1 (urea and thiourea derivatives)………...179
Figure S5. Chemical structures of library D-1 (amine derivatives)……….184
Figure S6. Chemical structures of library D-2 (amine derivatives)……….189
Figure S7. Chemical structures of library D-3 (amine derivatives)……….195
Annexe 3
Figure S1. A. Concentration of compound 1 measured in the brains of mice after injection into the carotid artery by in situ perfusion; B. Vascular volumes assessed with 14C-sucrose brain perfusion in mouse groups, showing that infusion of compound 1 does not affect the integrity of the BBB in mice. C. Uptake coefficients (Kin) calculated for compound 1 (32.7 and 13.8 µLg-1s-1). D. Kin and CNS Score (ACD/Percepta software 14.0.0) for compound 1 and other molecules. ………..229Figure S2. Chemical synthesis of 5………..230
Fig. S3. IR spectra of compound 5………...231
Fig. S4. 1H NMR (CDCl 3) of compound 5………...232
Figure S5. 13C NMR (CDCl3) of compound 5……….233
Figure S6. HRMS spectra of compound 5………...234
Fig. S7. HPLC spectra of compound 5……….235
Annexe 4
Figure S1. Representative TLC elution plates of reference steroids (T, 5α-DHT, 3α-diol, 3β-diol and ADT) and non-radiolabeled synthetized 3α/3β-diol steroids……….240Figure S2. 1H NMR of 5α-DHT, synthetized from T using Birch reduction………241
Figure S3. 1H NMR of 3α-diol/3β-diol, synthetized from 5α-DHT using L-selectride………242
Figure S4. TLC elution plate of extracted labeled steroids after 24 h of incubation of LAPC-4 cells with [14C]-3α-diol/3β-diol (8:2) (non-radiolabeled/radiolabeled 20:1, 1 μM)……….243
Figure S5. 1H NMR of compound 25 in CDCl3………..245
Figure S6. 13C NMR of compound 25 in CDCl 3……….245
xi
Listes des tableaux
Chapitre 1
Table 1. Assessment of solid-phase chemical synthesis of libraries A1 to D3………..45 Table 2. Biological results for D-ring derivatives 2-7………48 Table 3. Biological results for a selection of compounds from libraries A-D………52
Chapitre 2
Table 1. Assignation of proton and carbon NMR signals related to compound 2………...83
Chapitre 3
Table 1. Inhibition of pure 17β-HSD10 protein by a series of steroidal derivatives (compounds 1 to 16)……….104
Chapitre 4
Table 1. Inhibition of the formation of 5α-DHT from 3α-diol in LAPC-4 cells by a first series of
steroid derivatives………...122
Table 2. Inhibition of the formation of 5α-DHT from 3α-diol in LAPC-4 cells by a second series of
steroid derivatives chose after the first screening………..123
Annexe 1
Table S1. Structure and screening results (inhibition of 17β-HSD10 and 17β-HSD3 at 0.3 μM and 3
xii
Liste des abréviations
Biologie3α-diol 5α-Androstane-3α,17β-diol
3α-HSD 3α-Hydroxystéroïde déshydrogénase/Δ5-Δ4-isomérase
3β-diol 5α-Androstane-3β,17β-diol
3β-HSD 3β-Hydroxystéroïde déshydrogénase/Δ5-Δ4-isomerase
4-dione 4-Androstène-3,17-dione 5α-DHT 5α-Dihydrotestostérone 5α-DHP 5α-Dihydroprogestérone 5-diol 5-Androstène-3β,17β-diol 17α-OH-PREG 17α-Hydroxyprégnénolone 17α-OH-PROG 17α-Hydroxyprogestérone 17β-HSD 17β-Hydroxystéroïde déshydrogénase
17β-HSD3 17β-Hydroxystéroïde déshydrogénase type 3 17β-HSD10 17β-Hydroxystéroïde déshydrogénase type 10
Aβ Amyloïde-β
ADN Acide désoxyribonucléique
ADT 5α-Androstan-3α-ol-17-one (androstérone)
ALLOP Alloprégnanolone
AR Récepteur des androgènes
ARN Acide ribonucléique
BBB Barrière hématoencéphalique (blood brain barrier)
CHOL Cholestérol
CNS Score de pénétration de la barrière hématoencéphalique
CP/PC Cancer de la prostate
DHEA Déhydroépiandrostérone
E1 Estrone
E2 Estradiol
Epi-ADT Epi-androstérone
ER Récepteur des estrogènes
FFMA Forme familiale de la MA
FSMA Forme sporadique de la MA
HBA Accepteur de liaison hydrogène (hydrogen bond acceptor) HBD Donneur de liaison hydrogène (hydrogen bond donor) IC50 Concentration de l’inhibiteur provoquant 50% d’inhibition
FSH Hormone folliculo-stimulante
Kcat Constante catalytique en enzymologie
Km Constante de Michaëlis
LBD Domaine de liaison du ligand
LH Hormone lutéinisante
LH-RH Hormone de libération des gonadotrophines hypophysaires (Luteinizing hormone-releasing hormone)
xiii
LogP Logarithme du coefficient de partage octanol-eau
MA/AD Maladie d’Alzheimer
mRNA ARN messager
NAD+ Nicotinamide adénine dinucléotide (forme oxydée)
NADH Nicotinamide adénine dinucléotide (forme réduite)
NADP+ Nicotinamide adénine dinucléotide phosphate (forme oxydée) NADPH Nicotinamide adénine dinucléotide phosphate (forme réduite)
NMDA Récepteur N-méthyl-D-aspartate
PREG Prégnénolone
PROG Progestérone
PSA Antigène prostatique spécifique
SAAC S-Acétoacétyl coenzyme A
T Testostérone
TPA Traitement de privation androgénique
TPSA Surface polaire topologique (topological polar surface area)
Méthodes d’analyse
APCI Ionisation chimique sous pression atmosphérique (Atmospheric pressure chemical ionization)
APT Test du proton attaché (Attached proton test)
COSY Spectroscopie de corrélation homonucléaire (Homonuclear correlation spectroscopy)
HMBC Corrélation de liaisons multiples hétéronucléaires (Heteronuclear multiple bond correlation)
HPLC Chromatographie liquide haute performance (High performance liquid chromatography)
HPLC-MS Chromatographie liquide haute performance couplée à un spectromètre de mase
HRMS Spectrométrie de masse à haute résolution (High-resolution mass spectrometry)
HSQC Expérience de corrélation quantique hétéronucléaire (Heteronuclear single quantum correlation experiment)
IR Spectroscopie infrarouge (Infrared spectroscopy)
LRMS Spectrométrie de masse à basse résolution (Low-resolution mass spectrometry)
MS Spectrométrie de masse (Mass spectrometry)
NMR (RMN) Résonance magnétique nucléaire (Nuclear magnetic resonance)
NMR 1H Résonance magnétique nucléaire du proton
NMR 13C Résonance magnétique nucléaire du carbone
TLC (CCM) Chromatographique sur couche mince (Thin-layer chromatography)
xiv
Produits chimiques et solvants
(CH3)3SOI Iodure de triméthylsulfoxonium
AcOH Acide acétique
CDCl3 Chloroforme deutéré
CH3Li Méthyllithium
CH3MgI Iodure de méthylmagnésium
CH(OCH3)3 Orthoformiate de triméthyle
DCM Dichlorométhane
DEAD Azodicarboxylate de diéthyle
DIPEA N,N’-Diisopropyléthylamine
DMF Diméthylformamide
DMSO Diméthylsulfoxyde
Et2O Éther diéthylique
EtOAC Acétate d’éthyle
EtOH Éthanol
FMOC Fluorénylméthoxycarbonyle
H2O Eau
HBTU Hexafluorophosphate d’o-(benzotriazol-1-yl)-N,N,N’,N’-tétraméthyluronium
HCl Acide chlorhydrique
HPO(OCH3)2 Diméthylphosphite
K2CO3 Carbonate de potassium
KOH Hydroxyde de potassium
MeOH Méthanol
Mg(ClO4)2 Perchlorate de magnésium
MgSO4 Sulfate de magnésium
Na2SO4 Sulfate de sodium
NaBH3CN Cyanoborohydrure de sodium
NaBH4 Borohydrure de sodium
NaH Hydrure de sodium
NaHCO3 Bicarbonate de sodium
NH4Cl Chlorure d’ammonium
NMO N-Oxyde de N-méthylmorpholine
PPh3 Triphénylphosphine
p-TSA Acide paratoluènesulfonique
TEA Triéthylamine
THF Tétrahydrofurane
TPAP Perruthénate de tétrapropylammonium
NH3 Ammoniac
H2O2 Peroxyde d’hydrogène
Li Lithium métallique
Symboles RMN et IR
xv
δ Déplacement chimique (ppm)
ν Fréquence (cm-1)
broad s or m Singulet/multiplet large
d Doublet
dd Doublet de doublet
dt Doublet de triplet
J Constante de couplage en Hertz
m Multiplet q Quadruplet s Singulet t Triplet Unités °C Degré Celcius Ci Curie cm Centimètre cm-1 Réciproque du centimètre h Heure Hz Hertz M Molaire
m/z Masse sur charge
mCi Millicurie MHz Mégahertz min Minute mL Millilitre mmol Millimole mol Mole nm Nanomètre
ppm Partie par million
rpm Rotation par minutes
μCi Microcurie μL Microlitre μM Micromolaire Autres eq/equiv Équivalent g Gaz lq Liquide
rf Facteur de rétention (Retention factor)
rt Température ambiante: 22°C (Room temperature)
s Solide
xvi
« Oui, le passé est douloureux; mais à mon sens, on peut soit le fuir, soit tout en apprendre. »
xvii
Remerciement
Tout d’abord, je désire transmettre mes plus grands remerciements à mon directeur de recherche, le Dr Donald Poirier. Merci d’avoir cru en moi, mes capacités ainsi que pour toutes les connaissances que ma maîtrise m’a permis d’acquérir. La combinaison entre la synthèse chimique ainsi que l’évaluation biologique m’a permis d’apprendre énormément sur la chimie médicinale et les éléments nécessaires à l’obtention d’un traitement efficace contre une maladie donnée. Je suis reconnaissante d’avoir ainsi pu obtenir une formation unique, auprès d’un expert passionné, disponible et encourageant.
Je veux aussi dire un grand merci aux Dr René Maltais et Dr Jenny Roy, pour tous les conseils et connaissances transmises et le soutien démontré au cours de ces deux ans. J’ai appris énormément à leur contact et ils ont su me guider dans la réalisation de mes divers projets. Leur savoir-faire ainsi que leur professionnalisme m’ont beaucoup appris.
Ensuite, je souhaiterais remercier les différents organismes subventionnaires qui, par leur soutien financier, ont permis la réalisation de mon projet de maîtrise: soit la Fondation CHU de Québec (Axe Endocrinologie et Néphrologie), Mitacs Inc. (Vancouver, BC, Canada) ainsi que la faculté de médecine de l’Université Laval.
De plus, je remercie mes collègues, le Dr Francisco Cortés-Benítez, Raphaël Dutour, Maxime Lespérance, Dr Jean-Yves Sancéau et Dr Martin Perreault. Les discussions que nous avons eues sur la chimie et la biologie m’ont tellement apporté en termes de connaissances et de croissance personnelle. Je remercie également tous les gens que j’ai côtoyé au Centre de recherche du CHU de Québec-Université Laval.
Finalement, je remercie du fond du cœur mes parents, mes sœurs et mon conjoint, Alain, Lucie, Valérie, Josée, Émilie et Mathieu, sans qui je ne serai pas qui je suis aujourd’hui. Leur soutien indéfectible est ce qui m’a permis d’avancer aussi loin et de viser aussi haut dans la vie.
xviii
Avant-propos
Ce mémoire est rédigé sous le format de thèse par articles et contient l’introduction, quatre chapitres, puis la conclusion.
Le chapitre 1 est un manuscrit qui a été soumis au journal European Journal of
Medicinal Chemistry par mon directeur de recherche le 23 juin 2020. Étant première auteure
de l’article, il a été inclus sous sa forme actuelle dans ce mémoire. Je suis l’auteure principale et je l’ai composé avec mon directeur de maîtrise, Donald Poirier. Jenny Roy et René Maltais ont contribué à l’amélioration du manuscrit.
Le chapitre 2 est un article publié dans le journal Magnetochemistry le 23 juillet 2018. Étant première auteure de l’article, il a été inclus dans ce mémoire et la mise en page a été adaptée aux critères de présentation du document. Je suis l’auteure principale de l’article, ayant composé les sections 2.1, 2.3, 2.4, 3.1, 3.2 et 4 de l’article. Donald Poirier a grandement aidé dans la rédaction de l’article, en écrivant la section 1 et en améliorant les autres sections.
Le chapitre 3 est un article publié dans le journal Bioorganic & Medicinal Chemistry
Letters le 28 septembre 2018. Je suis première auteure et la mise en page a été adaptée aux
critères de présentation du document. Je suis l’auteure principale du manuscrit, l’ayant rédigé dans sa totalité. Les coauteurs sont Jenny Roy, René Maltais, Wael Alata, Frédéric Calon et Donald Poirier.
Le chapitre 4 est un manuscrit publié dans le journal Bioorganic & Medicinal
Chemistry Letters le 15 janvier 2020. Étant première auteure de l’article, la mise en page a
été adaptée aux critères de présentation du mémoire. J’ai rédigé le manuscrit. Les coauteurs sont Jenny Roy, René Maltais et Donald Poirier.
Un avant-propos incluant la contribution de chaque coauteur est inclus avant chaque chapitre correspondant.
1
2
1.1 La maladie d’Alzheimer
1.1.1 Généralité
La démence, fléau du 21e siècle, est définie comme un trouble altérant progressivement la mémoire et les capacités cognitives au-delà de ce qui est provoqué normalement par le vieillissement.1,2 Parmi les différentes formes de démence, la plus
fréquente est la maladie d’Alzheimer (MA), un trouble neurodégénératif et chronique.1,3
564 000 canadiens en seraient affectés, dont 65 % sont des femmes. Il est estimé que 25 000 nouveaux cas seront diagnostiqués cette année et que d’ici 2031, le nombre de personnes aux prises avec la maladie atteindra 937 000.4 Même si la prévalence de la MA augmente dès l’âge de 65 ans et elle atteint majoritairement les gens entre 75 et 84 ans,5 il se peut tout de
même qu’une forme précoce de la maladie se développe à un âge moyen de 45 ans.1,3
L’espérance de vie après le diagnostic est de 8 à 10 ans, mais cette phase caractérisée par des symptômes cliniques observables succède aux stades précliniques et prodromal, ce dernier aussi appelé pré-démentiel. Ces deux premières phases peuvent durer jusqu’à 20 ans avant que des symptômes cliniques permettent de porter un diagnostic de MA.
Cliniquement, la MA provoque un déclinement de la mémoire et d’autres fonctions cognitives tels que le langage, l’orientation visuospatiale, le raisonnement abstrait et l’apprentissage.1,2 Neuropathologiquement, la MA se définie par l’accumulation de plaques
de peptides amyloïdes-β (Aβ), dites séniles, par les lésions tau neurofibrillaires intraneuronales et par l’atrophie du cerveau due à la mort progressive des cellules neuronales.6,7 La maladie affecte grandement l’hippocampe et le cortex cérébral en altérant
les lobes frontaux et temporaux et progresse ensuite dans d’autres zones du cortex.3
Il existe deux formes de MA: les formes familiale et sporadique. Elles se différencient par l’âge d’apparition de la maladie et la prédisposition génétique de la personne atteinte. La forme familiale de la MA (FFMA) est plutôt rare, ne représentant qu’environ 5% des cas de MA. Elle se manifeste autour de l’âge de 50 à 60 ans et est due à des facteurs génétiques.8-10
En effet, les mutations de trois gènes ont été identifiées comme responsable de la FFMA: sur les gènes de la préséniline 1 et de la préséniline 2, ainsi que sur le gène du précurseur de la protéine Aβ. Plus de 270 mutations de ces trois gènes ont été identifiées à ce jour.10 La forme sporadique (FSMA) atteint les gens de plus de 65 ans et représente plus de 95% des cas de
3
MA.8 Cette forme de MA est causée par une combinaison de facteurs autant environnementaux que génétiques. Toutefois, peu de ces risques génétiques ont été identifiés. Le gène le plus prédominant pouvant causer la MA identifié à ce jour est celui de l’apolipoprotéine E, dont l’allèle epsilon 4 est associé à une augmentation du risque de développer la MA.9
Plusieurs facteurs de risques de cette maladie ont été identifiés à ce jour.Le fait de vieillir, un niveau de scolarité peu élevé et la présence sur l’apolipoprotéine E de l’allèle epsilon 4 ont été associés à un risque accru de développer la MA au moyen d’une étude pancanadienne.11 Dans cette même étude, l’utilisation d’anti-inflammatoires non-stéroïdiens,
la consommation modérée de vin et de café, les activités de stimulation cognitive ainsi que l’activité physique régulière ont été liées à un risque moins élevé de développer la MA et sembleraient retarder son apparition et sa progression.11,12 Ils existent aussi des facteurs de risques dits modifiables, qui pourraient être contrés si une modification du mode de vie était effectuée.13 Par exemple, les maladies cardiovasculaires (hypertension, accident vasculaire cérébral et autres), le diabète, l’épilepsie, la dépression et un historique de trauma crânien sont associés à un risque accru de MA.13
Une personne atteinte de la MA passera par trois stades durant le développement de la maladie.14,15 Un patient se trouve dans le stade léger (ou précoce) de la maladie lorsque les symptômes se limitent à des déficits légers, tels que les pertes de mémoire, les difficultés à communiquer ainsi que les changements d’humeurs et de comportements. Cette période dure généralement de 2 à 4 ans.15 Le stade modéré est associé à un déclin plus rapide de la mémoire ainsi que des fonctions cognitives et fonctionnelles.14 Le besoin d’aide dans la réalisation des
tâches quotidiennes commence à se manifester à ce stade qui peut durer de 2 à 10 ans.15
Finalement, le dernier stade dénommé avancé (ou grave) est marqué par l’incapacité de la personne atteinte à communiquer verbalement ou à prendre soin d’elle-même.14 Une aide
constante est alors nécessaire pour assurer la meilleure qualité de vie possible dans ce stade qui peut durer en moyenne de 1 à 3 ans.15
4
1.1.2 Marqueurs biologiques
Les deux marqueurs biologiques associés à la MA sont les plaques Aβ et les lésions neurofibrillaires. Tout d’abord, la formation extracellulaire des plaques amyloïdes serait due à un mécanisme de progression de la maladie nommé hypothèse de la cascade amyloïde. Elle implique l’oligomérisation d’un peptide neurotoxique Aβ de 42 acides aminés, ci-après nommé Aβ-42, qui mènerait à l’accumulation des plaques extracellulaires.16,17 L’Aβ-42 est
un produit de la dégradation de la protéine précurseur de l’Aβ (APP), obtenu à la suite de plusieurs réactions enzymatiques subséquentes sur l’APP. Les fonctions de l’APP sont de promouvoir la croissance cellulaire, la motilité, la croissance des neurites et la survie des cellules.18 Dans la voie non-amyloïdogène de la dégradation de l’APP, les α-sécrétases fragmentent l’APP en α-APP, des protéines solubles moins dommageables que l’Aβ-42 pour les neurones.6 Dans la voie amyloïdogène, l’APP subit l’action d’abord des β-sécrétases, qui produisent alors les peptides fragmentaires β-APP (soluble) et C99. Le peptide C99 est par la suite clivé par des γ-sécrétases, menant à la formation de peptides Aβ de 40 ou 42 acides aminés.6,19 Normalement, les fragments d’APP sont expulsés dans la cellule et seront alors détruits.19,20 Toutefois, leur sécrétion sous forme d’Aβ-42, non-soluble, est toxique pour les neurones.21 Par son action, l’Aβ-42 réduit la quantité d’acétylcholine libérée et la durée d’action de ce composé sur les récepteurs du neurone postsynaptique. Ceci provoque alors une diminution de la communication neuronale, ce qui cause alors problème au bon fonctionnement du système nerveux.22,23
En ce qui concerne les lésions neurofibrillaires, elles sont le produit de l’agglutination à l’intérieur des cellules neuronales de la protéine tau hyperphosphorylée. À l’état normal, cette phosphoprotéine est essentielle à l’assemblement et à la stabilisation des microtubules ainsi qu’à la stabilité du cytosquelette des neurones.24,25 Dans un cerveau normal,
l’expression des divers isoformes et leur degré de phosphorylation sont régulés par le développement du système nerveux. Toutefois, dans un cerveau atteint de la MA, les différentes isoformes se retrouvent sous une forme hyperphosphorylée. Ceci change la structure de la protéine tau, provoquant son agrégation qui empêche alors son rôle normal et entraîne un malfonctionnement du transport axonal.2,24,25
5
1.1.3 Traitements
Malgré l’ampleur des travaux dans la recherche sur la MA, les plus grands défis demeurent l’absence de biomarqueurs fiables permettant un diagnostic précoce de la maladie ainsi que le manque de stratégies et de traitements préventifs.5,26,27 De plus, il n’existe aucun traitement curatif de la MA.28 Les traitements présentement disponibles n’agissent que sur les symptômes, et donc ne retardent pas la progression de la maladie et ne la guérissent
pas.21,26,27 Les deux types de traitements disponibles sont les inhibiteurs
d'acétylcholinestérase et les antagonistes du récepteur N-méthyl-D-aspartate (NMDA), un des récepteurs post-synaptiques du glutamate.29 Le premier est administré généralement aux
patients étant aux stades léger à modéré, alors que le second est administré aux patients aux stades modéré à avancé.29
L’acétylcholinestérase est une enzyme qui dégrade l’acétylcholine, un neurotransmetteur jouant un rôle dans l’apprentissage et la mémoire.30 Chez un patient atteint
de la MA, l’Aβ-42 agit de façon à diminuer l’action synaptique de l’acétylcholine en réduisant sa durée d’action ainsi que sa concentration.22,23 L’inhibition de
l’acétylcholinestérase permet une dégradation plus lente de l’acétylcholine. Elle peut donc effectuer son action sur une plus longue durée, permettant d’aider les terminaisons nerveuses détériorées à quand même transmettre les influx nerveux.31,32 Les composés galantamine, rivastigmine et donépézil (Figure 1) sont disponibles au Canada et sont des inhibiteurs de l’acétylcholinestérase.3,34
Figure 1. Les inhibiteurs de l’acétylcholinestérase, les composés galantamine, rivastigmine et donépézil, et
l’antagoniste du récepteur N-méthyl-D-aspartate (NMDA), le composé mémantine.
Les antagonistes du NMDA agissent, quant à eux, pour empêcher la liaison du glutamate à son récepteur. Le glutamate est un neurotransmetteur médiateur majeur du
6
système nerveux central impliqué dans plusieurs fonctions du cerveau, dont la cognition, la mémoire et l’apprentissage.33 Chez un patient atteint de la MA, une surproduction de
glutamate provoque l’excitoxicité, un processus pathologique qui altère et détruit les cellules neuronales.34 Seul le composé mémantine, qui est disponible au Canada, est un antagoniste du NMDA.3,30,32 À ce jour, il n’y a que ces 4 médicaments qui sont disponibles pour atténuer les symptômes de la MA.
1.2 Le cancer
1.2.1 Généralité
Le terme cancer est le nom donné à un ensemble de maladies apparentées différentes les unes des autres possédant toutefois des caractéristiques communes.35,36 Il s’agit d’une pathologie caractérisée par une division cellulaire anarchique et inarrêtable de cellules anormales.37,38 Ces cellules, ne répondant plus aux mécanismes normaux de différenciation et de régulation, peuvent alors se propager dans un tissu, muscle ou organe environnant et l’endommager jusqu’à ce qu’il ne soit plus fonctionnel. À force de multiplication cellulaire, les cellules peuvent former une masse dénommée tumeur maligne.35-38 Cependant, il est possible qu’aucune tumeur solide ne se forme, comme dans le cas d’une leucémie qui est un cancer du sang.36 De plus, par la circulation sanguine et le système lymphatique, les cellules cancéreuses peuvent se propager à un nouveau tissu à partir du foyer tumoral d’origine, produisant de nouvelles tumeurs dénommées métastases.39,40 Pour être nommée cancer, une
tumeur doit posséder l’ensemble des propriétés énumérées précédemment.41
Une tumeur peut être décrite soit comme maligne ou bénigne. Une tumeur bénigne est une croissance anormale d’un tissu, sans gravité directe, qui ne se propage pas et qui n’envahie pas les tissus environnants, contrairement à une tumeur maligne.42 Une tumeur
maligne est ce qui est communément nommé un cancer. La tumeur bénigne se développe généralement plus lentement qu’une tumeur maligne, mais peut tout de même atteindre un grand volume.42 Elle devient dangereuse lorsqu’elle compresse des tissus et cause notamment
des dommages nerveux. Ceci mène alors à une réduction du flux sanguin et donc diminue l’apport en oxygène, ce qui peut provoquer la nécrose de tissus.43
7
Les cellules cancéreuses acquièrent progressivement six altérations majeures de leur physiologie cellulaire, ce qui leur font acquérir des caractéristiques communes à plusieurs types de cancers.44 Elles sont auto-suffisantes en facteur de croissance; sont insensibles aux
signaux et mécanismes antiprolifératifs; évitent le phénomène d’apoptose (autodestruction d’une cellule due à un signal donné); possèdent une capacité illimitée à se multiplier; provoquent le phénomène d’angiogenèses (création de nouveaux vaisseaux sanguins à partir de ceux existants); envahissent les tissus environnants; et forment des métastases.44
Il existe une grande quantité de types de cancers. Ils sont différenciés par les mutations génétiques qui y sont associées, par la diversité des causes ayant mené à la cancérogenèse et par la variation du profil biologique des cellules cancéreuses due à leur localisation dans le corps humain. Par conséquent, il n’existe pas de traitement universel pour le cancer.44,45 De façon générale, un cancer est traité en suivant un plan de traitement personnalisé basé sur les caractéristiques spécifiques du cancer. Il s’agit souvent d’une combinaison d’options thérapeutiques, améliorant les chances de guérison. Autant les options de traitement peuvent être utilisées pour traiter directement le cancer, autant elles peuvent être utilisées pour diminuer le risque de récidive. Il existe plusieurs types de traitements thérapeutiques, dont les principaux sont : la chirurgie, la radiothérapie, la chimiothérapie, l’hormonothérapie, la thérapie ciblée et l’immunothérapie.
Souvent utilisée en première ligne de traitement, la chirurgie permet de retirer physiquement la tumeur cancéreuse ou même complètement l’organe ou tissu atteint.46
Lorsque tous les tissus cancéreux sont retirés par chirurgie, la maladie est alors éradiquée.47 La chirurgie est souvent combinée à d’autres options de traitements due à la difficulté de physiquement retirer toutes les cellules cancéreuses.47 De plus, son utilisation est déterminée
par le type de cancer et la localisation des tumeurs. En effet, certains cancers ne forment pas de tumeurs physiques pouvant être retirées; la chirurgie n’est donc pas appropriée.36 Parfois,
la localisation des tumeurs, tel qu’au cerveau ou sur la moelle épinière, en font des chirurgies à haut risque de complication.47
La radiothérapie est l’usage de rayons ou de particules de haute énergie pour provoquer la mort des cellules cancéreuses en endommageant l’acide désoxyribonucléique (ADN). Les rayons sont souvent dirigés localement, sur la tumeur, le tissu ou l’organe ciblé.48
8
La chimiothérapie est l’utilisation de médicaments anticancéreux qui endommagent les cellules cancéreuses et, ultimement, mènent à la mort cellulaire. Elle regroupe une grande quantité de molécules ayant des cibles et des effets biologiques variés.49 Toutefois, ces
traitements peuvent aussi attaquer des cellules saines et sont donc souvent accompagnés d’effets secondaires tels que nausées, vomissements, perte des cheveux, affaiblissement du système immunitaire, etc.50 L’hormonothérapie permet de créer un environnement non
propice à la prolifération cellulaire, plutôt que de viser la mort de la cellule cancéreuse.51
Utilisée dans les cas de cancers hormono-dépendants (cancers dont la prolifération des tissus cancéreux est stimulée par la présence d’hormones stéroïdiennes agissant sur un récepteur spécifique),52 l’hormonothérapie vise à réguler des taux d’hormones en venant agir sur le système endocrinien en administrant soit des hormones stéroïdiennes, soit des composés agissant sur la biosynthèse ou sur le récepteur des hormones.53 La thérapie ciblée est l’utilisation d’un agent chimiothérapeutique qui agit spécifiquement sur un récepteur ou une cible biologique de la cellule cancéreuse et donc de la tumeur. Elle permet donc moins d’effets secondaires que la chimiothérapie classique, puisqu’elle atteint moins les cellules saines.54
Finalement, l’immunothérapie est la stimulation du système immunitaire pour provoquer une destruction des cellules cancéreuses. Dans ce type de thérapie se retrouve l’utilisation d’anticorps monoclonaux, l’utilisation de cytokines et l’immunothérapie cellulaire.55,56
1.2.2 Le cancer de la prostate
La prostate est une glande du système reproducteur masculin de la grosseur d’une noix située sous la vessie et en avant du rectum. La fonction de cette glande est de produire, de sécréter et d’entreposer le liquide prostatique, l’un des constituants du liquide séminal. Ce liquide est laiteux et alkalin et contient des protéines, des enzymes et des minéraux.57,58 Le
fonctionnement de la prostate nécessite des hormones sexuelles, les androgènes, qui sont produites par les testicules et les glandes surrénales. De plus, les tissus prostatiques contiennent l’enzyme réductase qui produit le plus puissant des androgènes, la 5α-dihydrotestostérone (5α-DHT) à partir de la testostérone (T).59
9
Le cancer de la prostate (CP) est l’un des plus prévalents chez les hommes avec plus de 23 300 nouveaux cas diagnostiqués au Canada en 2020.60 Il s’agit d’un cancer dont la
vitesse de progression varie grandement d’un patient à l’autre. Sa prévalence est d’autant plus grande chez les hommes âgés de plus de 65 ans, puisqu’ils représentent plus de 85% des cas de CP.61 Bien que les causes spécifiques ne soient pas connues, certains facteurs de risques ont été identifiés. L’âge, l’ethnicité, l’historique familiale, la génétique, les infections et inflammations, les androgènes, la diète et l’exercice ainsi que le mode de vie (tabac, alcool, activité sexuel) sont tous des facteurs de risques connus.61,62 Par ailleurs, plus de 80% des CP
sont androgéno-dépendants, c’est-à-dire que le développement, la croissance et le fonctionnement de ces CP nécessitent la présence de stéroïdes sexuels spécifiques, les androgènes. En effet, leur liaison au récepteur des androgènes (AR) favorise la prolifération de cellules saines ou cancéreuses de la prostate. De plus, dans les débuts du CP, aucun symptôme n’est apparent. Au fil de son développement, divers symptômes apparaissent tels qu’un fréquent besoin d’uriner, une difficulté à uriner, une difficulté à obtenir une érection et de la douleur pendant l’éjaculation.63
La détection du CP se fait de plusieurs façons. La première est le toucher rectal, permettant de détecter s’il y a une anomalie quant à la grosseur de la prostate. La seconde est le dosage sanguin de l’antigène spécifique de la prostate (PSA). Le PSA est sécrété par la prostate, sert à conserver le sperme à l’état liquide et se retrouve à l’état normal en faible quantité dans le sang (sous 4 ng/mL).64 Lors d’un CP, la désorganisation des cellules du tissu prostatique augmente la sécrétion de PSA dans le sang. Toutefois, divers autres facteurs tels que l’inflammation, l’éjaculation ou une intervention sur la prostate peuvent également augmenter la sécrétion de PSA.65 Ainsi, une concentration élevée en PSA ne signifie pas la
présence d’un CP. Toutefois, cela augmente le risque d’en avoir un. Un dosage de PSA élevé peut alors être suivi d’une biopsie pour confirmer le diagnostic. Les cellules de la prostate présentant une anomalie sont prélevées durant la biopsie. Par l’analyse microscopique et la recherche de biomarqueurs spécifiques du CP, il est possible de déterminer si les cellules sont bel et bien cancéreuses.66
Une fois le diagnostic du CP établi positivement, les cliniciens catégorisent le risque du CP par la combinaison de trois paramètres cliniques et pathologiques afin de déterminer
10
le traitement approprié. Le premier est le score de Gleason, qui est utilisé pour établir le niveau d’agressivité d’un CP et son grade.67 Le second est le système TNM, qui définit les
stades du CP. Dans le stade T (pour extension de la tumeur primitive), les différents stades sont : la non-détection de la tumeur (T0), la confirmation d’une tumeur maligne par dosage sanguin du PSA et une biopsie (T1), la palpation du CP par toucher rectal et une localisation sur la glande (T2), le déploiement du CP en dehors de la prostate et aux vésicules séminales (T3) et l’envahissement par le CP des organes et tissus adjacents à la prostate (T4). Le stade N (pour invasion lymphatique) se définit par le score N0, soit l’absence d’envahissement lymphatique, ou N1, soit la présence de métastases aux ganglions lymphatiques. Finalement, le stade M (pour métastases à distance) se décrit par soit l’absence de métastases (M0) ou par leur présence (M1), soit sur des ganglions non régionaux, sur des os ou d’autres sites.67 Finalement, le dernier paramètre clinique est le dosage du PSA. La combinaison de ces trois paramètres permet d’évaluer le risque du CP et établir le traitement approprié (faible risque, risque intermédiaire ou élevé).67,68
1.2.3 Traitement du CP
Plusieurs voies de traitements sont possibles dans le cas du CP. La surveillance active est la première option envisagée pour les cancers à faible risque.67 Il s’agit de surveiller sporadiquement la progression du cancer au moyen d’une analyse du niveau sanguin de PSA et de biopsie. La radiothérapie (par irradiation externe ou par curiethérapie) est une autre option thérapeutique envisagée. L’irradiation externe implique l’utilisation de rayonnement à haute énergie pour détruire les cellules cancéreuses. Elle permet de traiter des CP localisés ou qui ont atteint les tissus voisins. La chirurgie, soit la prostatectomie radicale, est l’une des options thérapeutiques les plus pratiquées lorsque le cancer est localisé.67,69
De plus, l’utilisation d’anti-androgènes donnés en combinaison avec la radiothérapie est utile dans le cas de CP localisés et avancés. En bloquant la liaison des androgènes au domaine de liaison du ligand (LBD) du AR, les anti-androgènes empêchent la prolifération cellulaire qui serait autrement induite.70 Deux classes d’anti-androgènes existent : les stéroïdiens et les non-stéroïdiens.
11
Le traitement par privation androgénique (TPA) s’effectue lorsque le CP est détecté à un stade avancé, par la castration chirurgicale ou chimique. La castration chirurgicale est obtenue en enlevant physiquement les testicules, tandis que la castration chimique est atteinte par l’administration d’un agoniste du LH-RH.71,72 Ces traitements peuvent être jumelés ou non à de la chimiothérapie. Lors de la castration chimique, les agonistes du LH-RH viennent stimuler les cellules hypophysaires qui augmentent alors la sécrétion de LH-RH, qui induisent alors une production de testostérone (T). Lorsque les agonistes sont administrés à forte dose, une désensibilisation des récepteurs hypophysaires se produit, éliminant ainsi la production de T testiculaire.71,72 Il s’agit du traitement de prédilection des CP
androgéno-dépendants. Ces techniques entraînent une diminution de 80 à 90% des niveaux de T circulants, mais diminuent seulement de 50 à 70% les concentrations en 5α-DHT intraprostatiques.73,74 Il est important de noter que la 5α-DHT peut être produit dans le tissu prostatique (intracrine) ou dans les tumeurs du CP (de novo) à partir de précurseurs stéroïdiens en circulation, tels que le 4-androstène-3,17-dione (4-dione) et le 5α-androstane-3α,17β-diol (3α-diol).75-78 Combiner la TPA à un anti-androgène ou à des inhibiteurs des
enzymes de la stéroïdogenèse est donc une voie à favoriser.
1.3 Les stéroïdes
1.3.1 Généralité
Les stéroïdes et leurs dérivés sont reconnus pour leur importance ainsi que leur versatilité en biologie. Ils représentent une grande famille de molécules lipidiques naturelles, nécessaires à l’équilibre ainsi qu’à l’homéostasie de systèmes biologiques et de processus physiologiques. La structure de base d’un stéroïde est le cyclopentanophénanthrène, formée de 17 carbones se retrouvant dans quatre cycles (Figure 2).79,80 Les cycles A, B et C sont composés de six carbones, alors que le cycle D est formé de cinq. Le terme hormone stéroïdienne désigne la classe de stéroïdes qui agissent comme molécules de signalisation dans grands nombres de processus biologiques.81 La production de stéroïdes chez l’être humain se produit lors de la stéroïdogenèse à partir du précurseur cholestérol (Figure 2). Ce composé à 27 carbones est essentiel pour la fluidité des membranes cellulaires, mais également pour la production des différents stéroïdes.81,82 L’apport en cholestérol provient majoritairement de l’alimentation, bien qu’il puisse être produit de façon endogène à partir
12
du lanostérol, le produit final de la voie du mévalonate. Cette voie de synthèse permet la production des précurseurs des terpènes, des terpénoïdes et des stéroïdes.83,84
Figure 2. Représentation simplifiée des classes de stéroïdes et de la voie de synthèse des stéroïdes à partir du
cholestérol. La numérotation des carbones du noyau stéroïdien est indiquée en rouge sur le cholestérol. L'identification des cycles A/B/C/D du noyau stéroïdien est représentée en bleu sur le noyau de la prégnénolone (la stéréochimie des protons de la prégnénolone n’est pas indiquée pour permettre une meilleure lisibilité).
Les hormones stéroïdiennes sont divisées en deux classes principales dues à leur fonction : les corticostéroïdes et les stéroïdes sexuelles. Les corticostéroïdes sont produits dans le cortex surrénal et sont sous-divisés en deux familles selon la structure des stéroïdes : les minéralocorticoïdes et les glucocorticoïdes (Figure 2). Ces types d’hormones stéroïdiennes sont impliqués dans la réponse immunitaire, la régulation inflammatoire, le catabolisme des protéines, le métabolisme des glucides, la diffusion des électrolytes et le comportement.85,86 Les représentants actifs de ces sous-classes sont respectivement l’aldostérone et le cortisol. Les hormones sexuelles quant à elles sont produites au niveau des gonades et de la glande surrénale. Cette classe est sous-divisée en progestagènes, androgènes et estrogènes.85,86 Les progestagènes, dont le seul représentant naturel est la progestérone, sont impliquées dans le système reproducteur masculin et féminin. Plus précisément, elles affectent la spermiogenèse, la capacitation des spermatozoïdes, la production de la T, la régulation du cycle menstruel, le maintien de la grossesse et la lactation.87,88 Les androgènes,
13
sont impliqués dans le développement et le maintien des caractères mâles, ainsi que la libido et le désir sexuel.89,90 Les estrogènes affectent le développement et la régulation de l’appareil
reproducteur féminin, ainsi que les caractéristiques sexuelles secondaires. Les trois principaux estrogènes sont l’estrone (E1), l’estradiol (E2) et l’estriol (E3).
Les stéroïdes et leurs dérivés sont également une classe de molécules intéressantes en ce qui concerne leur potentiel thérapeutique. En effet, ils peuvent être utilisés pour le traitement des cancers hormono-dépendants, mais aussi comme inhibiteurs d’enzymes de la stéroïdogenèse. Certaines de ces enzymes jouent des rôles importants dans plusieurs pathologies, et leur inhibition serait une avenue intéressante dans le but de traiter celles-ci. Cette stratégie thérapeutique vise soit à empêcher la production d’une hormone stéroïdienne ayant un effet néfaste dans la maladie, soit à empêcher la transformation d’une hormone stéroïdienne ayant un effet bénéfique contre la maladie.De plus, la stéroïdogenèse débute par une cascade de réactions enzymatiques linéaires. Cibler une enzyme qui agit dans ces premières étapes mènerait à l’arrêt complet de la stéroïdogenèse et la synthèse de tous les stéroïdes, ce qui aurait des effets néfastes sur beaucoup de processus physiologiques.
Plusieurs exemples sont disponibles dans la littérature montrant le potentiel thérapeutique des dérivés stéroïdiens. Lelaboratoire du Dr Poirier a d’ailleurs développé une large expertise pour plusieurs enzymes de la stéroïdogenèse. Les dérivés stéroïdiens peuvent être employés comme inhibiteurs des 17β-hydroxystéroïdes déshydrogénases (17β-HSDs), de l’aromatase, de la P450c17, de la 5α-réductase et de la stéroïde sulfatase.52,91-98
1.3.2 Stéroïdogenèse
La stéroïdogenèse est le processus biologique par lequel les hormones stéroïdiennes sont générées à partir du cholestérol (Figure 3).98 Tout d’abord, les progestagènes sont les
précurseurs de tous les stéroïdes humains. En effet, le cholestérol est converti en prégnénolone par l’action du cytochrome de clivage de la chaîne latérale du cholestérol (P450scc), une enzyme mitochondriale. Celle-ci hydroxyle la chaîne latérale du cholestérol en position C20 et C22, puis la clive.99 La prégnénolone est majoritairement produite dans les glandes surrénales, les gonades et le cerveau.100 Afin de produire les androgènes, la prégnénolone subit l’action subséquente du cytochrome P450c17. Ce cytochrome hydroxyle
14
le stéroïde en position 17, puis clive la chaîne latérale produisant la déhydroépiandrostérone (DHEA) à partir de la prégnénolone, ou le 4-dione à partir de la progestérone.101 Les
différentes 17β-HSDs permettent d’obtenir la T par leur action oxydante ou réductrice en position 17 du noyau stéroïdien. Le plus puissant des androgènes, la 5α-DHT, est par la suite produite à partir de la T par l’action de la 5α-réductase.102 L’E2 est produit en premier lieu en suivant la séquence DHEA – dione – E1, et en second lieu à partir de la T. Ainsi, le 4-dione et la T sont transformés en E1 et E2 par l’action de l’aromatase, qui tel que son nom l’indique aromatise le cycle A du noyau stéroïdien.102 Finalement, la synthèse de
l’alloprégnanolone (ALLOP) commence par la conversion de la progestérone en 5α-dihydroprogestérone (5α-DHP) par l’action de la 5α-réductase. Puis, des 3α-hydroxystéroïdes déshydrogénases (3α-HSD) ou la 17β-HSD type 10 (17β-HSD10) réduisent la cétone en position 3 du noyau stéroïdien en alcool.103
15
Figure 3. Stéroïdogenèse simplifiée des progestagènes, androgènes et estrogènes.CHOL: cholestérol; PREG: prégnénolone; PROG: progestérone; 17α-OH-PREG: hydroxyprégnénolone; 17α-OH-PROG: 17-hydroxyprogestérone; DHEA: déhydroépiandrostérone; 5-diol: 5-androstène-3β,17β-diol; dione: 4-androstène-3,17-dione; ; T: testostérone; 5α-DHT: 5α-dihydrotestostérone; ALLOP: alloprégnanolone; DHP: 5α-dihydroprogestérone; E2: estradiol; E1: estrone; 17β-HSDs: 17β-hydroxystéroïde déshydrogénase; 3β-HSD: 3β-hydroxystéroïde déshydrogénase/Δ5-Δ4-isomerase; 3α-HSD: 3α-hydroxystéroïde déshydrogénase/Δ5
16
1.3.3 Le 3α-diol, la 5α-DHT et la T dans le CP
La 5α-DHT, tel qu’énoncé précédemment, est le plus puissant des androgènes. Sa liaison avec le AR est la plus forte, et par son action la 5α-DHT peut promouvoir la prolifération des cellules saines de la prostate, mais aussi des cellules cancéreuses.59 La T se lie aussi au AR, mais moins fortement que la 5α-DHT. De façon classique, la 5α-DHT est produite à partir de la T par l’action de la 5α-réductase dans la prostate (Figure 2). Quant à elle, la T est produite par la réduction du 4-dione grâce à l’action de la 17β-HSD3, dont les caractéristiques seront discutées à la section 1.4.2.52,104 Dans le cas de cancer androgéno-dépendant, les traitements préférentiels sont la castration physique ou chimique. Toutefois, il arrive que le cancer continue sa progression malgré la déprivation en T. Le cancer est alors déclaré résistant à la castration, puisque les cellules cancéreuses continuent à proliférer. Ceci signifie, entre autres, que le AR est toujours activé par des androgènes produites par les enzymes de la stéroïdogenèse. En effet, la 5α-DHT peut être directement produite dans les tissus prostatiques, mais aussi dans les tumeurs cancéreuses à partir de divers précurseurs stéroïdiens en circulation dans l’organisme, tel que le 4-dione et le 3α-diol.75-78 Ainsi,
développer des inhibiteurs empêchant la transformation de ces deux précurseurs seraient une stratégie intéressante et complémentaire à la castration.
Dans le cas présent, un intérêt est porté sur la voie alternative de production du DHT à partir du 3α-diol (Figure 4). Dans cette voie, le 4-dione est transformé par la 5α-réductase en 5α-androstane-3,17-dione (5α-dione). Puis, ce stéroïde est transformé en androstérone (ADT) par l’action d’une 3α-hydroxystéroïde déshydrogénase. Finalement, le 3α-diol est obtenu par l’action d’enzymes qui réduisent la cétone en position 3 de l’ADT en α-hydroxyle.78 Par la suite, la 5α-DHT serait obtenue directement du 3α-diol par l’action
d’une 17β-HSD (6 ou 10), de la rétinol déshydrogénase 5 ou d’autres enzymes oxydatives inconnues.77,91 La production d’ADT serait aussi possible à partir de l’ALLOP dans la voie dite détourné (« backdoor »). En inhibant les enzymes catalysant la transformation du 3α-diol en 5α-DHT, l’activation du AR, et par le fait même la prolifération des cellules cancéreuses, serait empêchée.
17
Figure 4. Stéroïdogenèse simplifiée de la 5α-dihydrotestostérone (5α-DHT), via les voies classique, alternative
et détourné (backdoor ).CHOL: Cholestérol; PREG: Prégnénolone; DHEA: Déhydroépiandrostérone; 5-diol: 5-Androstène-3β,17β-diol; 4-dione: 4-Androstène-3,17-dione; T: Testostérone; 5α-dione: Androstane-3,17-dione; DHT: Dihydrotestostérone; ALLOP: Alloprégnanolone; ADT: Androstérone; 3α-diol: 5α-Androstane-3α,17β-diol; CYP11A1: Cytochrome P450scc; 17β-HSDs: 17β-Hydroxystéroïde déshydrogénase; 3β-HSD: 3β-Hydroxystéroïde déshydrogénase/Δ5-Δ4-isomérase; 5α-R: 5α-Réductase; HSD:
3α-Hydroxystéroïde déhydrogénase/Δ5-Δ4-isomérase; CYP17A1: Cytochrome P45017A1; AR: Récepteur des
androgènes. Figure publiée dans S. Boutin et al., 2020, (voir chapitre 4).
1.3.4 L’E2 dans la MA
L’E2 est une hormone sexuelle féminine, et elle agit sur la régulation du système reproducteur féminin (Figure 3).105 Au niveau du cerveau, E2 agit de façon neuroprotectrice de plusieurs façons. D’une part, la forte liaison de E2 sur les récepteurs des estrogènes (ER) α et β agit comme un facteur de transcription, ce qui augmente l’expression de certaines protéines anti-apoptoses, en plus de diminuer l’expression de facteurs pro-apoptose. Ainsi, E2 prévient la phase d’initialisation de la mort cellulaire programmée par la mitochondrie.106,107 De plus, E2 protège les cellules du processus d’apoptose en activant un
18
espèces oxygénées réactives.108 Finalement, il a été rapporté que les estrogènes réduisent les niveaux de protéines Aβ et régulent leur production.109
1.3.5 L’ALLOP dans la MA
L’ALLOP est une progestagène agissant sur le système nerveux central de diverses façons. Ces effets sont très variés : dépresseur, anxiolytique, sédatif, analgésique, anti-convulsivant, neuroprotecteur, etc.110-112 L’ALLOP est un modulateur positif allostérique du
récepteur GABAAet a un grand impact sur la plasticité de ce récepteur.113-115 Par son
interaction avec le GABAA, l’ALLOP agit sur l’ouverture des canaux d’ions chlorures (Cl-)
dans les neurones à proximité du GABAA. À faible concentration, l’ALLOP se lie au GABAA
à des sites de liaison différents de ceux liés aux ligands conventionnels (benzodiazépines, éthanol et barbituriques) et peut agir comme un modulateur positif ou négatif des fonctions du récepteur.116-118 Le mécanisme d'action menant à l'activation de l’expression génique du cycle cellulaire par l’ALLOP dans les cellules souches neurales est médié par sa liaison à GABAA. Cette liaisonprovoque un efflux d'ions Cl- et un afflux simultané d'ions calcium
(Ca2+). Cette dynamique d'échange d'ions induit des signalisations cellulaires qui conduisent à la transcription des gènes mitotiques et à une régulation négative des gènes antimitotiques.118 Une augmentation rapide de la concentration intracellulaire de Ca2+ et une activation ultérieure du cycle cellulaire déclencheraient la neurogenèse.119 De plus, l’ALLOP régule aussi l’homéostasie du cholestérol à cause de mécanismes qui augmentent l’expression du récepteur nucléaire des oxystérols. Ce récepteur provoque la dégradation du cholestérol et prévient la sur-activation de la γ-sécrétase, ce qui vient prévenir la formation du peptide Aβ-42.120,121 Finalement, l’ALLOP diminue significativement la production des
peptides Aβ dans l'hippocampe, le cortex et l'amygdale en plus d’induire une diminution de l’expression de la 17β-HSD10, l’enzyme la métabolisant.122 Cette diminution de l'expression
de la 17β-HSD10 permet de limiter le métabolisme de l'ALLOP et d’augmenter sa concentration ainsi que tous les effets positifs associés.
19
1.4 Les 17β-HSDs
1.4.1 Généralités
Parmi les enzymes impliquées dans la stéroïdogenèse se retrouvent les 17β-HSDs, une famille d’enzymes contenant 15 membres qui catalysent stéréosélectivement l’oxydation de 17β-hydroxystéroïdes ou la réduction de 17-cétostéroïdes, en utilisant comme cofacteur le nicotinamide adénine dinucléotide oxydé (phosphaté ou non) (NAD(P)+) ou le nicotinamide adénine dinucléotide réduit (phosphaté ou non) (NAD(P)H), respectivement.91,123,124 Certaines de ces enzymes catalysent aussi des réactions d’oxydation et de réduction à la position 3 du squelette stéroïdien. Leurs actions permettent d’interconvertir les formes actives et inactives des hormones stéroïdiennes dans les dernières étapes de leur biosynthèse.125 Ces enzymes régulent par leurs actions les niveaux d’estrogènes et d’androgènes, mais aussi ceux de certains neurostéroïdes.126,127 Elles peuvent
aussi agir sur d’autres substrats que les stéroïdes, tel que les acides gras et les acides biliaires.125 De plus, la réversibilité des réactions qu’elles catalysent est une particularité propre à cette famille d’hydroxystéroïdes déshydrogénases. Toutefois, bien que l’activité enzymatique puisse être oxydative ou réductrice dans une préparation d’enzyme pure ou un homogénat de cellules selon le cofacteur utilisé, il semble qu’en cellules intactes leur activité est unidirectionnelle.128,129 Les activités enzymatiques de ces isoformes se retrouvent donc
dans un grand répertoire de tissus stéroïdogéniques, tel que les testicules, les ovaires et le placenta, ainsi que dans des tissus périphériques. Chaque isoforme des 17β-HSDs possède d’ailleurs une distribution tissulaire spécifique et a une affinité sélective pour son substrat. 130-132 En ciblant sélectivement ces enzymes, il est donc possible de régulariser les
concentrations d’androgènes et d’estrogènes, une avenue intéressante comme approche thérapeutique dans les maladies hormono-dépendantes, tel que les cancers du sein et de la prostate, l’ostéoporose, l’endométriose, mais aussi d’autres maladies tel que la MA.133-135
1.4.2 La 17β-HSD3
La 17β-HSD3 est presque exclusivement exprimée dans les testicules et est connue pour son rôle dans le cancer de la prostate. En effet, cette enzyme est impliquée dans la production de la T dans les testicules à partir du 4-dione dans la voie classique de la formation du 5α-DHT.59,128,136 Par la suite, la T peut être transformée en DHT par l’action de la