DESIGN ET SYNTHÈSE DE MACROCYCLES
PSEUDOPEPTIDIQUES POUR LE
DÉVELOPPEMENT D’INHIBITEURS
D’INTERACTIONS PROTÉINE-PROTÉINE
Simon Vézina-Dawod
Mémoire
Maîtrise en sciences pharmaceutiques
Maître ès sciences (M. Sc.)
Québec, Canada
RÉSUMÉ
Pour que la vie puisse exister, des milliers de protéines doivent interagir ensemble pour permettre les divers processus biochimiques essentiels que l’on retrouve au niveau cellulaire. Le dérèglement d’une seule interaction protéine-protéine peut avoir des conséquences catastrophiques sur la qualité de vie d’une personne. C’est ainsi que de nombreux programmes de recherches sont basés sur l’exploration d’une ou de plusieurs cibles protéiques impliquées dans diverses maladies. Étant donné la localisation intracellulaire des nouvelles cibles thérapeutiques ainsi que l’absence de ligands connus, le développement d’agents thérapeutiques devient de plus en plus ardu. En effet, la chimie combinatoire classique et le design rationnel ont leurs limitations et il y a un besoin évident de développer de nouvelles approches pour faire l’étude et la modulation des interactions protéine-protéine. C’est dans cet esprit que s’imbrique les suivants travaux, qui portent sur les peptoïdes comme outil moléculaire peptidomimétique. Ces oligomères de glycine N-substitutée présentent notamment des propriétés pharmacologiques avantageuses et représentent une base moléculaire de choix pour élaborer de nouvelles méthodologies de criblage à haut débit. En passant par le développement de nouvelles approches de synthèse sur support solide, jusqu’à l’évaluation de la pénétration cellulaire de peptoïdes macrocycliques, le suivant mémoire devrait être lu comme un préambule à l’utilisation des peptoïdes en chimie combinatoire pour la découverte d’inhibiteurs ou de modulateurs d’interactions protéine-protéine.
ABSTRACT
In living organisms, thousands of proteins must interact together to enable essential biochemical processes found into cells. Misfunction of only one protein-protein interaction can have catastrophic consequences on one’s welfare. Many research programs are based on the exploration of target proteins involved into a disease. Because of the intracellular localization of new therapeutic targets and the lack of known ligands, development of drugs is harder than ever. Classical combinatorial chemistry and rational design have their limitations, showing the need of new tools for modulating protein-protein interactions. The following work is based on peptoids as a suitable peptidomimetic scaffold with great pharmacological properties. Those N-substituted glycine oligomers show promising attributes for high-throughput screening and protein-protein interaction inhibitors development. From new synthesis approaches improving chemical diversity to the evaluation of the cellular uptake of macrocyclic peptoids, the following memoire should be read as an effort for enhancing the use of peptoids in combinatorial chemistry for the discovery of protein-protein interaction modulators.
TABLE DES MATIÈRES
RÉSUMÉ ... III ABSTRACT ... V TABLE DES MATIÈRES ... VII LISTE DES TABLEAUX ... IX LISTE DES FIGURES ... XI ABRÉVIATIONS ... XV REMERCIEMENTS... XXI
CHAPITRE 1 ... 1
1.1 Les interactions protéine-protéine ... 3
1.2 Développement d’agents thérapeutiques ... 5
1.2.1 Développement d’inhibiteurs d’IPP : l’exemple du complexe p53-MDM2 ... 7
1.3 Les ligands peptidiques et le peptidomimétisme ... 10
1.3.1 Prototypes moléculaires mimant le squelette peptidique ... 11
1.3.2 Modifications du squelette peptidique ... 14
1.4 Les peptoïdes ... 15
1.4.1 Structure, synthèse et généralités ... 16
1.5 La pénétration cellulaire et les liaisons intramoléculaires ... 23
1.6 Les peptoïdes en chimie combinatoire ... 25
CHAPITRE 2 ... 27 2.1 Hypothèses de travail ... 29 2.2 Objectifs ... 31 CHAPITRE 3 ... 33 Avant-propos ... 35 Résumé ... 37
N-Substituted Arylsulfonamide Building Blocks as Alternative Submonomers for Peptoid Synthesis ... 39
- Supporting Information (SI) - ... 53
CHAPITRE 4 ... 75
Résumé ... 78
Toward the development of optimized macrocycle-based peptidomimetic scaffolds: probing the structural requirements to improve cellular uptake ... 79
- Supporting Information (SI) - ... 91
CHAPITRE 5 ... 113
5.1 Discussion générale ... 115
5.2 Conclusion ... 117
LISTE DES TABLEAUX
CHAPITRE 3
Table 1. Bromine displacement by N-(2-methoxyethyl)-o-nitrobenzenesulfonamide 2a under various conditions ... 44
Table 2. Removal of the o-NBS group from 6a under various conditions in DMF ... 47
Table 3. Crude purities and isolated yields for oligomers (8b-i) containing amino
alcohols 1b-h ... 48
Table S1. Sequence, characterization data and yields for peptoid oligomers 8b-8i ... 60
CHAPITRE 4
Table 1. Sequence of the synthesized peptide and peptoid analogs for the cellular
uptake study ... 82
LISTE DES FIGURES
CHAPITRE 1
Figure 1. Schématisation de certaines interactions protéine-protéine impliquées lors
de l’apoptose. ... 3
Figure 2. Représentation de l’interaction entre deux protéines (A et B) et de la très
grande surface d’interaction. ... 4
Figure 3. Distribution de ligands peptidiques connus en fonction de leur taille
(gauche) et de leur surface d’interaction avec leur protéine partenaire (droite). ... 5
Figure 4. Exemple d’une chimiothèque pour le développement d’inhibiteur d’IPP par
la stratégie de la cartographie épitopique. ... 6
Figure 5. Schéma montrant le rôle biologique de la protéine pro-apoptotique p53. ... 8 Figure 6. Site d’interaction entre la protéine MDM2 et un fragment peptidique de
p53. ... 8
Figure 7. Exemples de molécules inhibitrices du complexe p53-MDM2. ... 9 Figure 8. Le peptidomimétisme pour le développement d’inhibiteurs d’IPP. ... 10 Figure 9. Comparaison des structures de la morphine (5), phénazocine (6) et
met-encéphaline. ... 11
Figure 10. Exemples de structures moléculaires mimant une conformation en
hélice-. ... 12
Figure 11. Exemples de structures moléculaires mimant une conformation en
feuillet-β. ... 13
Figure 12. Exemples d’approches pour mimer les structures en coude β. ... 14 Figure 13. Exemples d’isostères du lien peptidique. ... 15
Figure 14. Évolution des articles, des revues et des brevets portant sur les
peptoïdes. ... 16
Figure 15. Comparaison entre les structures générales d’un peptide et d’un
peptoïde. ... 17
Figure 16. Différentes approches pour la synthèse des peptoïdes sur support
solide. ... 17
Figure 17. Exemple d’amines utilisées (protégés s’il y a lieu) dans l’approche par
submonomère de Zuckermann et al.. Entre parenthèse la nomenclature du monomère correspondant au sein d’un oligomère. ... 18
Figure 18. Comparaison entre le protocole à température ambiante et celui sous
irradiations micro-ondes. ... 19
Figure 19. Modification post-oligomérisation par chimie click. ... 20 Figure 20. Tendances et isomérisation des liens amides secondaires et tertiaires
retrouvés chez les peptides et les peptoïdes. ... 21
Figure 21. Isomérisation d’une liaison N-arylamide. a) Nomenclature des atomes de
carbone et angles dihèdres pour un résidu peptoïde N-aryle i; b) Répulsion entre la densité électronique de l’oxygène du carbonyle et celle de l’anneau aromatique; c) Énergie d’isomérisation calculée pour les dérivés N-méthylacétamides. ... 22
Figure 22. Modèle de (Nphg)6 présentant une conformation d’énergie minimale
ressemblant celle d’une hélice polyproline type II. ... 22
Figure 23. Structure cristallographique d’un macrocycle possédant 2 résidus Nphg. .... 23 Figure 24. Le repliement conformationnel d’une molécule grâce aux LHI encourage
le passage en milieu hydrophobe. ... 24
Figure 25. Structure cristallographique de la CsA à partir de l’eau (gauche) ou à
CHAPITRE 2
Figure 1. Approche proposée pour le développement d’un prototype macrocyclique
rigide à pénétration cellulaire améliorée qui sera récupéré en chimie combinatoire. ... 30
CHAPITRE 3
Scheme 1. a) Peptoid synthesis by the submonomer method and b) alternative approach using N-substituted arylsulfonamide building blocks and their compatibility with the submonomer method. ... 42Scheme 2. Protection of the amino alcohols. ... 43
Scheme 3. Synthesis of peptoid oligomers 8a-h. ... 46
Figure S1. HPLC traces (λ = 220 nm) of submonomers 2a and 3b-h. ... 61
Figure S2. 1H NMR and 13C NMR spectra of submonomers (2a, 3b-h). ... 63
Figure S3. LC-MS characterization for peptoid oligomers (8b-i). ... 71
CHAPITRE 4
Scheme 1. Synthesis of cyclic peptide analogs 1-6. ... 83Scheme 2. Synthesis of the labeled peptoid monomer o-NBS-NLys(Dns)-OH 17. ... 84
Scheme 3. Synthesis of cyclic peptoid analogs 10-14. ... 85
Figure 1. Comparative cellular uptake of compounds 1-16 on HeLa cells as measured by flow cytometry. ... 86
Figure S1. LC-MS characterization for macrocycles 1-17. ... 98
ABRÉVIATIONS
Produits chimiques et solvants ACN acétonitrileAcOH acide acétique
AcOEt acétate d’éthyle
BTC bis(trichlorométhyl) carbonate CCl4 tétrachlorure de méthane CHCl3 chloroforme DBU 1,8-diazabicyclo[5.4.0]undéc-7-ène DIC diisopropylcarbodiimide DCM dichlorométhane DIPEA N,N-diisopropyléthylamine DMSO diméthylsulfoxyde DMF diméthylformamide Fmoc 9-fluorénylméthyloxycarbonyl
HATU O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tétraméthyluronium hexafluorophosphate
HCl acide chlorhydrique
HCTU O-(6-chlorobenzotriazol-1-yl)-N,N,N′,N′-tétraméthyluronium hexafluorophosphate
HFIP 1,1,1,3,3,3-hexafluoroisopropanol
HOAt 1-hydroxy-7-azabenzotriazole
iPrOH isopropanol
MeOH méthanol
MgSO4 sulfate de magnésium
NaCl chlorure de sodium
NaHCO3 hydrogénocarbonate de sodium
NMM N-méthylmorpoline
NMP N-méthyle-2-pyrrolidone
Pip pipéridine
PyBOP benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate
TEA triéthylamine
TFA acide trifluoroacétique
TIS triisopropylsilane
THF tétrahydrofurane
Unités
°C degré Celsius
g gramme h heure Hz hertz M molaire mg milligramme MHz mégahertz min minute mL millilitre μL microlitre mmol millimole mol mole
m/z masse sur charge
W watt
s/sec seconde
nm nanomètre
ppm parties par million
cm centimètre
μm micromètre
mm millimètre
Å Amstrong
équiv. équivalents
m/v masse sur volume
v/v volume sur volume
cat catalytique
mAu unité d’absorbance x 10-3
rpm rotation per minute
Méthodes d’analyse
APCI «Atmospheric pressure chemical ionization»
APT «Attached proton test»
ESI electrospray ionization
RP-HPLC chromatographie liquide à haute performance en phase inversée
RMN résonance magnétique nucléaire
2D RMN résonance magnétique nucléaire à 2 dimensions 1H RMN résonance magnétique nucléaire du proton 13C RMN résonance magnétique nucléaire du carbone
CCM/TLC chromatographie sur couche mince
MS spectromètre de masse
LCMS chromatographie liquide couplée à un spectromètre de masse
MO/MW micro-ondes
Résonnance Magnétique Nucléaire
1H un proton δ déplacement chimique d doublet dd doublet de doublet s singulet br s singulet large m multiplet q quadruplet t triplet J constante de couplage Autres λ longueur d’onde fréquence RT/rt room temperature
IPP/PPI Interaction protéine-protéine
ONG oligomère de glycine N-substituée
LHI/IHB liaison hydrogène intramoléculaire
SPPS solid-phase peptide synthesis
tBu tert-butyl
Boc tert-butyloxycarbonyl
Calcd calculé
tR temps de retention
PBS phosphate buffer saline
FBS fetal bovine serum
DMEM Dulbecco’s Modified Eagle’s Medium
SD standard deviation
DNS dansyl
“If you are not willing to look stupid, nothing great is ever going to happen to you”
REMERCIEMENTS
Mes premiers remerciements vont à mon directeur de recherche Éric Biron. Merci de m’avoir soutenu durant tant d’années, à travers mon processus d’apprentissage et de maturation professionnelle. C’est grâce à tes enseignements, tes bons conseils, ton esprit dynamique et ta créativité que j’ai pu réaliser un processus académique aussi complet et diversifié. Un merci aussi à mes collègues et amis du labo du passé ou du présent, Marie-Pier, Anick, François, Marie-Claude, Xinxia, Amélie, Antoine, Marie, Cassandre, Sophie, Steve et j’en passe, vous avez tous contribué de différentes façons à mon succès.
Je veux remercier aussi les membres de ma famille, qui m’ont soutenu depuis le début à la réalisation de mes projets de vie et d’étude. Un merci à ma mère Paule Vézina et son mari Alain Jacques, pour m’avoir accompagné et orienté depuis mes jeunes jours vers un avenir meilleur. Merci à mon père Nagi Dawod pour ton important soutient depuis mon entrée aux études universitaires. Merci à ma tante Johanne et à ma grand-maman de faire partie de ma vie et de m’apporter du bonheur chaque fois que je vous vois. Un important merci à tous mes amies et amis (vous êtes trop nombreux, je ne vous nommerai pas), pour m’avoir aidé à décrocher et à relaxer lorsque j’en avais besoin. Enfin un merci à toi, qui se reconnaîtra peut-être, tu as joué un rôle important durant mes premières années à l’université et ta présence dans ma vie aura sans doute contribué à la motivation et à l’élan nécessaire pour entrer aux études graduées.
Chapitre 1
Introduction
1.1 Les interactions protéine-protéine
La vie comme nous la connaissons aujourd’hui se définit par un ensemble de molécules interagissant entre-elles pour jouer un rôle structural ou fonctionnel au niveau cellulaire, tissulaire ou de l’organisme. Parmi les biomolécules constituant les cellules, les protéines sont les plus abondantes et représentent environ 50 % du poids des cellules.1 Cette
abondance reflète le rôle ubiquitaire des protéines dans pratiquement tous les aspects structuraux et fonctionnels de la cellule. Pour réaliser pleinement leurs rôles biologiques, une majorité de protéines doit interagir physiquement avec d’autres molécules qui sont, la plupart du temps, des protéines.2 Comme la majorité des fonctions cellulaires est assurée
par des protéines, les interactions protéine-protéine (IPP) jouent un rôle crucial dans les processus cellulaires (Figure 1).3-5 Plusieurs maladies chez l’homme, incluant le cancer,
peuvent résulter d’une IPP inappropriée ou déréglée au sein d’une voie de signalisation.4-6
Étant donné leur rôle de premier plan dans les processus cellulaires, les IPP constituent un point stratégique d’intervention pour la conception de nouveaux agents thérapeutiques.7-12 L’industrie pharmaceutique se tourne d’ailleurs de plus en plus vers ces
cibles thérapeutiques qui sont au cœur du métabolisme cellulaire et dont notre compréhension va sans cesse grandissante.8-19 Une interaction entre deux protéines est le
résultat de plusieurs petites interactions entre différents acides aminés au sein d’une zone spécifique appelée site de reconnaissance ou domaine d’interaction protéique («Protein
Interaction Domain»).20 Parmi ces interactions, on y retrouve notamment des liaisons
hydrogènes, des interactions ioniques, des interactions dipolaires, des interactions hydrophobes et des interactions aromatiques mais aussi des contraintes spatiales et tridimensionnelles.2, 20 Les acides aminés impliqués dans la reconnaissance et
responsables de l’IPP sont par ailleurs positionnés dans l’espace de façon à optimiser les interactions chimiques nécessaires. On les retrouve souvent au sein de structures secondaires à conformation bien définie, tels des tournants β, des hélices α ou des arrangements en feuillet. C’est alors sans surprise que les IPP sont généralement très sélectives et présentent de bonnes affinités de liaison. Malgré tous les investissements des dernières années et la demande croissante de l’industrie pharmaceutique pour des inhibiteurs d’IPP, le développement de ces molécules est, entre autres, grandement ralenti par la nature plane et grande (1500-3000 Å2) de la surface d’interaction et par la
localisation intracellulaire de plusieurs IPP (Figure 2).20
Figure 2. Représentation de l’interaction entre deux protéines (A et B) et de la très grande
surface d’interaction.
Protéine A Protéine B
Cependant, il a été récemment démontré qu’au travers de cette surface d’interaction, de petites zones de hautes affinités appelées « hot spots » (300-1000 Å2) seraient
responsable de la majeure partie de l’interaction.21, 22 Ces petites zones clés sont
habituellement de dimensions plus raisonnables (300-1000 Å2) pour le développement
d’agents thérapeutiques. Il a alors été proposé qu’il suffit d’occuper une ou des petites zones de haute affinité pour empêcher l’interaction entre les protéines cibles. Une étude provenant d’une base de données regroupant des IPP connues présente des résultats encourageants pour le développement de petites molécules inhibitrices (Figure 3). En effet, de ces complexes protéine-peptide, la majorité se retrouve avec une surface d’interaction inférieure à 500 Å, ce qui est considéré comme « druggable » par l’industrie pharmaceutique.21, 22
Figure 3. Distribution de ligands peptidiques connus en fonction de leur taille (gauche) et de leur
surface d’interaction avec leur protéine partenaire (droite).22
1.2 Développement d’agents thérapeutiques
Un ligand peut moduler une IPP soit en se liant directement sur une zone cruciale de la surface d’interaction, soit en se liant à un site allostérique, induisant alors un changement conformationnel qui empêche subséquemment l’IPP. Il n’est donc pas obligatoire de se lier directement sur la surface d’interaction pour observer une régulation positive ou négative. L’approche plus classique du développement d’inhibiteurs enzymatiques menait souvent à des molécules riches en centres sp2, contrairement aux composés bioactif naturels qui
présentent souvent un caractère sp3 plus riche, une abondance de centres chiraux et des
structures 3D impressionnantes.23 C’est donc dans une optique de haute sélectivité et
haute affinité que les chimistes médicinaux devront revoir leurs approches pour découvrir des agents thérapeutiques vis-à-vis des cibles de plus en plus complexes.
Les deux stratégies les plus couramment utilisées pour le développement d’agents thérapeutiques sont mal adaptées pour le développement d’inhibiteurs d’IPP. D’abord, l’étude de la relation structure-activité du ligand endogène n’est pas une approche appropriée puisque le ligand, qui correspond à une protéine, est beaucoup trop gros. Ensuite, l’utilisation des produits naturels comme composés de départ n’est pas adéquate puisqu’il y a très peu de produits naturels connus pour inhiber les IPP. Deux autres stratégies sont donc majoritairement utilisées pour découvrir des inhibiteurs d’IPP : la cartographie épitopique (« epitope mapping » ou « PepScan ») d’une région de la protéine partenaire et le criblage de chimiothèques combinatoires. La première stratégie consiste en la synthèse de chimiothèques de peptides constitués de différentes sections superposées de la séquence de la protéine partenaire. La chimiothèque permet de couvrir toute la séquence de la protéine, afin d’identifier quelle région est nécessaire à l’interaction (Figure 4).
Figure 4. Exemple d’une chimiothèque pour le développement d’inhibiteur d’IPP par la stratégie de
la cartographie épitopique.
Cela permet ainsi de connaître les séquences peptidiques capables d’inhiber l’interaction protéine-protéine ciblée. Il y a toutefois quelques désavantages associés aux peptides comme agents thérapeutiques, qui seront abordés à la section suivante. Par contre, ces ligands peptidiques constituent un excellent point de départ dans le processus
Protéine de départ
d’optimisation en chimie médicinale par une approche de design rationnel à partir du ligand (« ligand-based drug design ») pour le développement d’un médicament.
La deuxième stratégie consiste à cribler une chimiothèque combinatoire sur la protéine d’intérêt pour identifier directement des molécules possédant une certaine affinité pour la cible. Les collections de molécules criblées sont, la plupart du temps, composées de familles de molécules possédant des propriétés pharmacologiques et pharmacocinétiques intéressantes du point de vue pharmaceutique. Cette stratégie a l’avantage de générer des composés nécessitant moins d’optimisation que les ligands peptidiques issus de la cartographie épitopique ce qui accélère considérablement le développement d’un inhibiteur d’IPP efficace.
Par contre, bien souvent les chimiothèques criblées possèdent d’importantes limitations pour la découverte d’inhibiteurs d’IPP. Certaines chimiothèques démontrent une faible diversité moléculaire, car un seul type de prototypes moléculaires (« scaffolds ») a été utilisé et décoré de manière aléatoire. D’un autre côté, certaines chimiothèques possèdent une grande diversité de molécules, mais avec des structures inappropriées pour cibler des protéines. Par conséquent, le design et la synthèse de la chimiothèque à cribler sont extrêmement importants dans cette stratégie. L’objectif lors du design d’une chimiothèque ciblant les IPP est de générer en un nombre minimal d’étapes une chimiothèque possédant une diversité moléculaire maximale tant au niveau des groupements fonctionnels que du squelette (diversité fonctionnelle et structurale) avec des prototypes moléculaires adaptés pour la liaison aux protéines.
1.2.1 Développement d’inhibiteurs d’IPP : l’exemple du complexe p53-MDM2
La protéine p53, alias le suppresseur tumoral p53, est un facteur de transcription qui joue un rôle majeur au niveau du cycle cellulaire, de l’apoptose et de la réparation de l’ADN via la régulation de l’expression de gènes clés.24-27 La protéine MDM2 (Mouse Double Minute2 ou l’équivalent chez l’homme HMD2) est l’E3 ubiquitine ligase responsable de la régulation négative de p53. Composée de 491 acides aminés, elle se lie au domaine d’activation de la transcription du p53 pour inhiber son rôle chez la cellule saine. Pratiquement toutes les cellules cancéreuses présentent un mauvais fonctionnement du p53, notamment par mutation du gène p53 ou par une surexpression de la protéine MDM2.28-30 Étant donné le rôle primordial de l’interaction MDM2-p53 dans la survie des
cellules cancéreuses, le développement d’inhibiteurs de cette IPP était extrêmement prometteur pour le traitement d’un grand nombre de cancers. La stratégie était donc de développer une molécule capable de se lier à MDM2 et d’empêcher son interaction avec la protéine p53.
Figure 5. Schéma montrant le rôle biologique de la protéine pro-apoptotique p53.
L’analyse de la structure cristallographique de la protéine MDM2 avec le segment peptidique responsable de la transactivation de p53 a démontré la présence d’une petite surface d’interaction bien définie. Ce segment peptidique a ensuite servi de point de départ pour le développement de plusieurs inhibiteurs de cette IPP.31-33 À partir de cette
structure, les chercheurs ont observés que les chaines latérales Phe19, Leu22, Trp23 et
Leu26, qui sont contenus dans une section en hélice α de p53, vont interagir avec une
poche hydrophobe importante sur MDM2 (Figure 6).
A) B)
Figure 6. Site d’interaction entre la protéine MDM2 et un fragment peptidique de p53.
MDM2
Ces observations ont mené au développement de plusieurs inhibiteurs de l’interaction p53-MDM2 (Figure 7). Des chercheurs ont notamment découvert en 2001 la chlorofusine (1), un peptide cyclique naturel découvert durant le criblage d’extraits bactériens.34 Par la
suite, Robinson et ses collaborateurs ont découvert un macrocycle peptidique inhibiteur (2) démontrant un IC50 de 140 nM. Ce macrocycle présente une conformation en β-hairpin
dont la section bioactive est spatialement une mimétique de Phe19 et Trp23 de la section
hélicoïdale de p53.35 C’est alors que fit l’apparition de petites molécules non-peptidiques.
En 2004, l’équipe de Vassilev ont découvert par criblage de chimiothèques synthétiques des dérivés cis-imidazoline appelés Nutlins (3).36 Ces molécules inhibent efficacement la
formation du complexe p53-MDM2 avec des IC50 entre 100 et 300 nM. Un groupement
bromophényl se retrouve dans la poche du Trp23, un autre groupement bromophényl se
retrouve dans la poche de Leu22 et la chaîne éthyléther dans la poche de Phe19.
Finalement, c’est en 2005 que des dérivés 1,4-benzodiazépine-2,5-diones (4) ont été rapportés comme inhibiteurs de l’interaction p53-MDM2.37 À partir d’une méthode hybride
de chimiothèques virtuelle et combinatoire, des composés inhibiteurs démontrant des IC50
variant entre du bas µM au haut nM ont été identifiés.
1.3 Les ligands peptidiques et le peptidomimétisme
Une approche très prometteuse pour le développement d’inhibiteurs d’IPP consiste en la conception de molécules capables de mimer des structures secondaires de protéines.8-10
En effet, en se basant sur la nature du domaine d’interaction protéique, les structures secondaires de protéines jouent un rôle primordial dans la reconnaissance moléculaire et l’interaction entre les protéines. Avec une grande capacité à mimer des structures secondaires de protéines, les peptides représentent donc un prototype moléculaire de choix pour moduler les IPP.38, 39 Les peptides sont alors d’excellents outils pour étudier ou
moduler les IPP in vitro. Comme agents thérapeutiques, ils démontrent une forte activité biologique et une importante sélectivité. Ils possèdent également une faible toxicité, s’accumulent peu dans les tissus et présentent peu d’interaction médicament-médicament.40 De surcroît, avec un nombre important d’acides aminés disponibles, ils
offrent une immense diversité fonctionnelle ainsi qu’une grande diversité structurale étant donné les différentes structures secondaires qu’ils peuvent adopter.
Par contre, les peptides linéaires sont généralement vulnérables face aux protéases ce qui leur confère une faible stabilité in vivo et donc une faible demi-vie. Ils présentent également une faible biodisponibilité orale dû à leur hydrophilicité, leur poids moléculaire élevé et leur susceptibilité à l’inactivation enzymatique dans le tractus gastro-intestinal.41
Effectivement, les peptides se heurtent généralement à la membrane cellulaire, étant incapables de la traverser pour accéder à la cellule. Puisque la majorité des IPP se produisent à l’intérieur des cellules, il y a un besoin manifeste de molécules pouvant mimer les structures secondaires de protéines tout en étant capable de pénétrer à l’intérieur des cellules et ayant une stabilité in vivo améliorée.
Figure 8. Le peptidomimétisme pour le développement d’inhibiteurs d’IPP.
Agent thérapeutique
Peptides
Peptidomimétisme
Développement d’inhibiteurs d’IPP
• Dégradation protéolytique • Faible pénétration cellulaire
• Conservation de la diversité fonctionnelle et
conformationelle des peptides
• Augmentation de la stabilité métabolique • Meilleure pénétration cellulaire
Le peptidomimétisme permet de contourner ce problème jusqu’à un certain niveau. Une molécule peptidomimétique est une molécule dont la structure de base est non peptidique, mais dont les motifs structuraux reproduisent les caractéristiques spatiales (hélice α, feuillet , tournants, épingle à cheveux, etc.) et fonctionnelles des structures peptidiques.42-45 Ces molécules permettent donc de conserver les caractéristiques
avantageuses reliées aux peptides tout en éliminant certains désavantages. Effectivement, elles ne sont pas reconnues ni dégradées par les protéases, ce qui augmente significativement leur stabilité et leur biodisponibilité. Par exemple, la morphine (5), un alcaloïde opiacé, et ses dérivés représente un exemple classique de composés peptidomimétiques. La morphine et ses dérivés comme la phénazocine (6) reproduisent l’effet biologique des endorphines, encéphalines et dynorphines sur les récepteurs opioïdes (Figure 9).46
Figure 9. Comparaison des structures de la morphine (5), phénazocine (6) et Met-encéphaline.
Plusieurs stratégies ont été mises au point pour développer des molécules peptidomimétiques dont : 1) l’utilisation d’acides aminés modifiés (acides aminés D, acides aminés cycliques, C-alkylés, Cβ-alkylés ou N-alkylés); 2) la restriction conformationnelle
(cyclisation, résidu rigidifiant); 3) l’imitation du squelette peptidique par des structures moléculaires organiques.47 Le but ultime de ces stratégies est de : 1) augmenter la
stabilité en réduisant la reconnaissance moléculaire par les protéases; 2) accroître l’affinité envers la cible en diminuant les coûts entropique de liaison via une rigidification du squelette et 3) améliorer la sélectivité via une restriction conformationnelle plus grande pour exposer les groupements fonctionnels essentiels de manière optimale.
1.3.1 Prototypes moléculaires mimant le squelette peptidique
La mise au point de molécules capables de mimer des structures secondaires de protéines a été largement utiliser dans le développement d’agents thérapeutiques. Ces
recherches ont mené au développement d’un très grand nombre de prototypes moléculaires mimant des structures en hélice- et feuillet-β en plus d’une grande variété de coudes et tournants qui sont des composantes essentielles au sein des protéines. Plusieurs prototypes moléculaires («scaffolds») peptidomimétiques existent afin de développer des composés tête-de-série comme les benzodiazépines, qui selon la position et la nature des substituants, peuvent adopter des mimétiques d’hélice α ou de tournants.
48-50
Figure 10. Exemples de structures moléculaires mimant une conformation en hélice-.
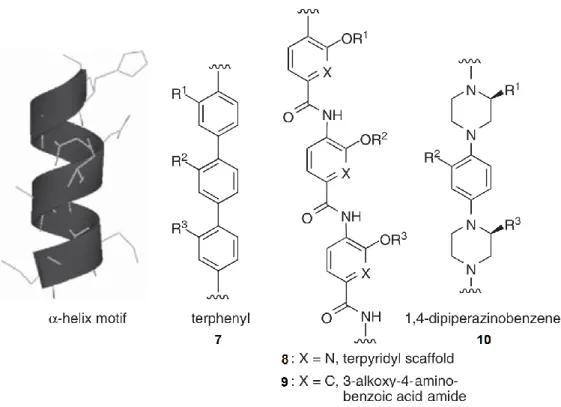
L’hélice- est la structure secondaire la plus commune et représente environ la moitié des structures polypeptidiques retrouvées dans les protéines. Les premiers prototypes moléculaires mimant les hélice- proposés par Hamilton et al. étaient basés sur des structures terphényle (7) et terpyridyle (8) (Figure 10).51, 52 Par la suite, Boger et al. ont
décrit des structures oligoamides à base d’acide 3-alkoxy-4-aminobenzoïque (9) ayant une longueur d’environ 3 résidus (Figure 10).53 Le trimère constitue une structure rigide
permettant aux groupements substituants de mimer les chaînes latérales aux positions i,
i+4 et i+7 d’une hélice- et d’introduire des éléments de diversités moléculaires. Un autre exemple de prototypes moléculaires mimant les hélice- est une 1,4-dipipérazinobenzène
(10) proposé par Koenig et al. Cette molécule capable de mimer de courtes hélices contient les isostères de chaînes latérales sur les deux pipérazines (Figure 10).54
Figure 11. Exemples de structures moléculaires mimant une conformation en feuillet-β.
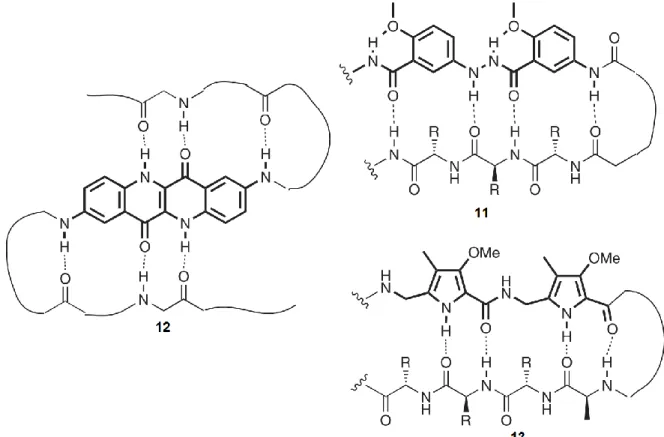
Les feuillets β sont aussi des éléments structuraux très importants chez les protéines et ils sont caractérisés par une série régulière de liaisons hydrogène intramoléculaires connectant les brins β ensemble. Il y a d’ailleurs un intérêt croissant pour ce genre de structures car de plus en plus d’études démontrent qu’elles sont impliquées dans le développement et la progression de certaines neuropathies via l’aggrégation d’oligomères adoptant des structures en feuillets β.55 La majorité des prototypes moléculaires capables
de mimer des feuillets β ont été développé en tirant avantage d’entités spécifiques possédant une structure plane et pouvant établir des liaisons hydrogènes parallèlement et comme accepteur et donneur.56 C’est le cas des mimétiques
5-amino-2-méthoxybenzamides et hydrazides (11) pour stabiliser un feuillet β antiparallèle,57, 58 de
l’épindolidione (12) et des acides aminés méthoxypyrole (13) qui peuvent servir au cœur d’un brin β pour orienter adéquatement les fonctionnalités formeuses de liaisons hydrogènes (Figure 11).59, 60
Les coudes β («β-turns») sont la structure secondaire de protéines qui a été le plus fréquemment minés. Un coude β est une séquence tétrapeptidique où la distance entre le Ci et Ci+3 est ≤ 7Å.47 Le coude peut être stabilisé par chélation d’un cation ou liaisons
hydrogène intramoléculaire (Figure 12). Un prototype mimétique idéal devrait être rigide et orienter les chaînes latérales comme le peptide naturel tout en augmentant la solubilité et la résistance à la dégradation protéolytique. Plusieurs stratégies ont été utilisées pour le design de prototypes mimant les coudes β dont l’utilisation de petites molécules hétérocycliques (14) ou l’incorporation d’isostères dipeptidiques pour induire le coude dans un motif peptidique (15) (Figure 12). De plus, une grande variété de contraintes ont été développé pour stabiliser la structure en coude β dans une molécule macrocyclique (16) (Figure 12).
Figure 12. Exemples d’approches pour mimer les structures en coude β.
1.3.2 Modifications du squelette peptidique
Une des approches les plus conservatrices pour introduire une restriction conformationelle et stabiliser la molécule face aux protéases est la modification du squelette peptidique. Plusieurs études ont été réalisées pour remplacer les liens peptidiques par des groupements capables de mimer le lien au niveau structural, électronique et de la capacité à former des liaisons hydrogènes.61-63 Ces remplaçants du lien amide sont aussi connus
sous le nom d’isostères peptidiques (Figure 13). Par exemple, la substitution du lien amide par un groupement aliphatique augmente la flexibilité localement tandis qu’un isostère oléfine n’affecte pas la conformation. De plus, la capacité à former des liens hydrogènes
peut être modulée en utilisant divers isostères peptidiques comme un groupement sulfonamide, phosphinique ou peptoïde (Figure 13). Un grand nombre d’isostères peptidiques a été développé et étudié mais la grande majorité sont difficilement accessibles car leur synthèse requiert plusieurs étapes et beaucoup de ressources. Parmi ces groupements nous nous sommes particulièrement intéressés aux peptoïdes car ils offrent plusieurs avantages au niveau peptidomimétique. Dans un premier temps, leur synthèse est très accessible et rapide avec une possibilité de diversité moléculaire phénoménale. Les peptoïdes ont démontré des capacités peptidomimétiques très variées offrant ainsi une flexibilité au niveau des structures à mimer. Finalement, il a été démontré qu’ils possèdent significativement une plus grande capacité à franchir les membranes cellulaires que les peptides.
Figure 13. Exemples d’isostères du lien peptidique.
1.4 Les peptoïdes
Les peptoïdes ou oligomère de glycine N-substituée (OGN) forment une classe de molécules peptdidomimétiques particulièrement intéressante. Cette section fait un survol
des propriétés, des aspects synthétiques et conformationnels reliés aux peptoïdes. Ces oligomères ont prouvé leur utilité à travers une vaste gamme d’application comme le développement d’agents antibactériens,64 d’agents thérapeutiques,65 de catalyseurs
énantiosélectifs66 et même dans le domaine des matériaux nanostructurés.67 Dans une
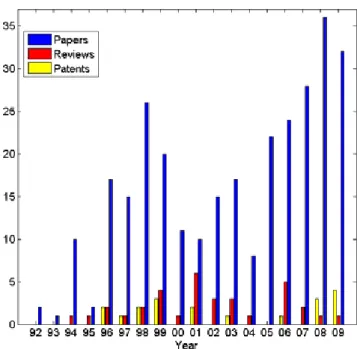
optique de développement de prototypes moléculaires peptidomimétiques optimisés pour le développement d’inhibiteurs d’IPP, il a été démontré que les peptoïdes sont d’excellents candidats pour une approche combinatoire ou de design rationnel. Une recherche faite par le Atlantic Cancer Research Institute à Moncton montre qu’entre 1992 et 2009, les recherches sur les peptoïdes se sont accentués mais restent relativement modestes avec un maximum de parution de 35 articles en 2008 (Figure 14).68 Avec un énorme potentiel
pour l’industrie pharmaceutique et les sciences biomédicales, de plus en plus de chercheurs se mettront à l’œuvre sur ce fascinant et prometteur sujet que sont les peptoïdes.
Figure 14. Évolution des articles, des revues et des brevets portant sur les peptoïdes.68
1.4.1 Structure, synthèse et généralités
Comparativement aux peptides, la chaîne latérale se retrouve branchée sur l’azote, faisant ainsi disparaître la chiralité sur le carbone α (Cα) (Figure 15). Ceci s’accompagne notamment par une augmentation de la flexibilité et donc une perte de définition structurale. Aussi, l’absence de liaisons hydrogènes intramoléculaires (LHI) empêche les
peptoïdes de s’organiser par des interactions longues distances. Par contre, les liens amides tertiaires ainsi formés confère aux peptoïdes une grande résistance à la dégradation enzymatique.69 La disparition du lien polaire N-H fait aussi diminuer la polarité
globale de la molécule, provoquant une augmentation de l’hydrophobicité et de la pénétration membranaire.70 Ce type d’oligomère est donc un candidat de choix comme
prototype moléculaire peptidomimétique à biodisponibilité améliorée.
Figure 15. Comparaison entre les structures générales d’un peptide et d’un peptoïde. Une excellente revue de littérature a été produite en 2010 sur les différentes approches de synthèse ainsi que sur les méthodes utilisées pour l’analyse des peptoïdes.68 La synthèse
des peptoïdes est généralement effectuée sur support solide avec d’excellentes puretés brutes. Les approches se regroupent en 2 catégories principales : l’approche submonomérique et l’approche monomérique (Figure 16).
L’approche monomérique utilise des monomères de glycine N-substitués avec la chaine
latérale et N-protégés avec le groupement fluorénylméthyloxycarbonyl (Fmoc). Le
monomère est ensuite incorporé sur l’oligomère croissant sur support solide en utilisant les protocoles standard de synthèse peptidique («solid phase peptide synthesis», SPPS) (Figure 16a).71-79 Plusieurs approches submonomériques ont aussi été exploitées comme
l’amination réductive (Figure 16b)80-82 et l’activation d’alcool par approche photochimique
(Figure 16c),83, 84mais la méthode la plus couramment utilisée reste l’approche
submonomérique développée par Zuckermann et al. (Figure 16d).8485Cette méthode passe
par la bromoacétylation sur support solide de l’amine secondaire libre de l’oligomère croissant suivi par le déplacement nucléophile du brome avec une amine (R-NH2). En plus
d’être extrêmement efficace, cette dernière méthode est très rapide et compatible avec les micro-ondes. De plus, cette approche permet de générer une immense diversité moléculaire car un nombre phénoménal d’amines diverses est disponible commercialement. Cette importante diversité constitue un avantage majeur dans une optique de développement d’agents bioactifs et de génération de chimiothèques combinatoires à haute diversité moléculaire.
Figure 17. Exemple d’amines utilisées (protégés s’il y a lieu) dans l’approche par submonomère de
Zuckermann et al. Entre parenthèse la nomenclature du monomère correspondant au sein d’un oligomère.
Un certain consensus existe concernant la nomenclature des peptoïdes. Pour nommer les monomères, un code à 3 lettres peut être utilisé avec comme préfixe « N » pour N-substitution (Figure 17).85 Par la suite, si l’acide aminé correspondant existe, le code déjà
existant est ajouté. Par exemple, le monomère N-isopropyl-glycyl sera appelé NVal puisqu’il mime parfaitement la valine. Dans le cas où il y a un méthylène de plus par entre la chaîne latérale existante et le squelette, on ajoute le terme homo ou « h ». Par exemple, la N-(2-hydroxyéthyl)-glycyl peut aussi être appelé NhSer. Enfin, pour les chaînes latérales sans équivalence avec les acides aminés, un code à trois lettres peut être utilisé avec la chiralité R ou S en premier pour les N-Cα tertiaires. En exemple, la N-(S-phényléthyl)glycyl sera appelé Nspe et la N-phénylglycyl la Nphg.
Figure 18. Comparaison entre le protocole à température ambiante et celui sous irradiations
micro-ondes.
L’utilisation de micro-ondes lors de la synthèse des peptoïdes a aussi prouvé son utilité. En effet, il a été démontré que l’utilisation d’irradiations micro-ondes permet d’augmenter la pureté brute et le rendement lors de la synthèse, en plus de réduire significativement les temps réactionnels jusqu’à 30 secondes pour l’étape d’acylation comparativement à 30 minutes à température pièce et jusqu’à 90 secondes pour l’étape d’amination (déplacement nucléophile) comparativement à 30 minutes à température pièce (Figure 18).86-90
Un autre avantage de la synthèse des peptoïdes sur support solide est la possibilité d’effectuer des modifications post-oligomérisation après une déprotection sélection ou via l’incorporation de résidus appropriés. En plus de modifications plus classiques comme une amidation, une estérification ou une substitution, des modifications plus complexes comme
la métathèse, des réactions multicomposante et la glycosylation ont été rapportées. De plus, la chimie «Click» («azide alkyne Huisgen cycloaddition») a été utilisée pour augmenter la complexité moléculaire post-oligomérisation (Figure 19).91 Ceci peut
constituer une excellente approche pour l’introduction de chiralité, de motifs tridimensionnels complexes ou de structures hétérocycliques privilégiées.
Figure 19. Modification post-oligomérisation par chimie click.
1.4.2 Aspects conformationnels
Le squelette peptidique et peptoïdique est constitué d’un enchaînement de liaisons amides séparés par un Cα. Ainsi, il est important de bien comprendre le comportement structural des amides pour poursuivre l’étude conformationnelle de ces oligomères. Une délocalisation électronique existe à partir des électrons de l’azote, ce qui donne à la liaison C-N un caractère rigide de double liaison (Figure 20). Ce phénomène rend ainsi la rotation C-N très difficile avec une barrière de rotation d’environ 105 kJ/mol. En résulte alors deux minima d’énergie à conformation planaire, le rotamère de forme cis-amide (ω = 0°) et un autre de forme trans-amide (ω = 180°). Chez les peptides, le rotamère trans est favorisé par environ 8 kJ/mol et est retrouvé majoritairement. Avec les peptoïdes cependant, l’absence de chiralité sur le Cα et de liens N-H mène à une perte de définition structurale. La différence énergétique entre le rotamère cis et trans est très faible ce qui permet aux liens amides de peupler équitablement les deux états. Aussi, le squelette de l’oligomère ne peut plus se replier par ponts hydrogène. Le tout résulte en une perte de définition structurale, provenant d’une grande flexibilité du squelette du peptoïde.
Figure 20. Tendances et isomérisation des liens amides secondaires et tertiaires retrouvés chez
les peptides et les peptoïdes.
Voyant le potentiel d’application et l’accessibilité synthétique des peptoïdes, certains groupes de recherche se sont alors intéressés à la rigidification des peptoïdes. C’est en 1997 qu’un premier article propose l’introduction de résidus avec un centre chiral en N-Cα pour induire une préférence énergétique pour le lien cis-amide.92 Un an plus tard, la
structure d’un pentapeptoïde à centre chiral en N-Cα fut étudiée par résonance magnétique nucléaire (RMN).93 Les résultats obtenus ont démontré qu’une hélice α
similaire à une hélice de polyproline type I est adoptée par ces d’oligomères. Par la suite, l’équipe de Kent Kirshenbaum proposa plutôt l’utilisation de groupements N-aryle pour induire une préférence énergétique vers le lien trans-amide.94 En effet, les résultats
obtenus d’abord par une approche computationnelle puis expérimentale ont démontré qu’il y a répulsion électronique importante entre l’oxygène du carbonyle puis l’anneau aromatique lorsque le lien amide est en cis (Figure 21).94, 95 La barrière énergétique
d’isomérisation serait variable en fonction de l’enrichissement ou de l’appauvrissement en électrons du cycle aromatique (Figure 21c).
Figure 21. Isomérisation d’une liaison N-arylamide. a) Nomenclature des atomes de carbone et
angles dihèdres pour un résidu peptoïde N-aryle i; b) Répulsion entre la densité électronique de l’oxygène du carbonyle et celle de l’anneau aromatique; c) Énergie d’isomérisation calculée pour les dérivés N-méthylacétamides.
L’analyse en RMN et en cristallographie des rayons-x d’oligomères de N-phénylglycine (Nphg) a démontré une conformation en hélice similaire à celle d’une hélice polyproline type II (Figure 22).94 Il a été également démontré que l’incorporation de Nphg à l’intérieur
de macrocycles force le lien en trans à la position arylée, ce qui ouvre la voie vers l’induction de tournants (Figure 23).94
Figure 22. Modèle de (Nphg)6 présentant une conformation d’énergie minimale ressemblant celle
d’une hélice polyproline type II.94
a)
b)
c)
Structure Decis-trans
(kJ/mol) Énergie d’isomérisation de dérivés
de N-méthylacétanilidea
aLes auteurs ont calculé par différentes approches
ab initio de mécanique quantique 13,40
3,85
13,85
Figure 23. Structure cristallographique d’un macrocycle possédant 2 résidus Nphg.94
Les résultats obtenus sur l’effet structurant de certains résidus peptoïdes sur la conformation a permis de générer des structures capables de mimer différents types d’hélices, de tournants et de coudes. Parallèlement plusieurs groupes de recherche se sont intéressés aux propriétés pharmacocinétiques et pharmacologiques des peptoïdes. Dans un premier temps, il a été démontré que les oligomères de peptoïdes étaient capables de résister aux protéases.69, 96 Par la suite Kodadek et al. ont démontré que les
peptoïdes ne généraient pas de réponse immunitaire.97 Finalement, la capacité des
peptoïdes à franchir les membranes cellulaires a été évaluée par différents groupes. Les résultats obtenus ont démontré que les peptoïdes franchissent plus facilement la membrane par diffusion passive que leur homologue peptidique.98, 99 La combinaison de
ces résultats nous a amené à nous intéresser aux peptoïdes cycliques comme prototypes moléculaires peptidomimétiques pour cibler les IPP intracellulaires. Mais tout d’abord, il était important de comprendre l’effet de la N-substitution sur la perméation cellulaire.
1.5 La pénétration cellulaire et les liaisons intramoléculaires
Lors du développement d’un agent thérapeutique, plusieurs facteurs thermodynamiques doivent être considérés.100 L’affinité de liaison (K
a) est principalement influencé par
l’énergie libre de Gibbs (ΔG) selon Ka = e(-ΔG/RT). L’énergie de Gibbs est elle-même dictée
par un terme enthalpique (ΔH) et un terme entropique (ΔS), selon (ΔG = ΔH - TΔS). Le terme enthalpique sera principalement lié aux forces d’attraction, comme les forces de
Van der Waals ou les liaisons hydrogène entre la molécule et la protéine cible ainsi qu’aux forces de répulsion comme l’effet hydrophobe qui pousse la molécule à se désolvater de l’eau pour se loger dans une poche hydrophobe. Un aspect conflictuel survient lorsque vient le temps d’optimiser l’affinité de liaison d’une molécule bioactive. D’un côté, il y a un aspect enthalpique favorable à introduire des groupements polaires au sein de la molécule qui feront une liaison hydrogène ou une interaction de Van der Waals avec la protéine. D’un autre côté, l’introduction de ces fonctionnalités s’accompagne avec des pénalités enthalpiques de désolvatation. Le chimiste médicinal doit peser les pours et les contres d’une modification chimique en ce sens. Le terme entropique, lui sera principalement influencé par le changement entropique lors de la désolvatation et lors des changements conformationnels. En effet, plus la protéine et la molécule doivent modifier leur conformation pour qu’il y ait liaison, moins bonne sera cette dite liaison. L’entropie conformationnelle sera pratiquement toujours défavorable, étant donné la perte en degré de liberté lors de la formation du complexe molécule-protéine, mais il est possible d’en réduire l’impact en s’assurant que la conformation de la molécule soit suffisamment rigide et suffisamment proche de la conformation qu’elle adoptera en se complexant.
Figure 24. Le repliement conformationnel d’une molécule grâce aux LHI encourage le passage en
milieu hydrophobe.
Pour qu’un agent thérapeutique se rende à son lieu d’action, il doit franchir plusieurs barrières biologiques. En exemple, bon nombre de médicaments administrés oralement doivent passer la membrane du tractus gastro-intestinal pour se rendre dans la circulation systémique. Mais plus généralement encore, tout médicament qui agit au niveau intracellulaire devra passer à travers la membrane lipidique des cellules, soit par transport actif ou passif. Il a été démontré que les liaisons hydrogènes intramoléculaires (LHI)
peuvent réduire dramatiquement le coût enthalpique de désolvatation et ainsi favoriser le passage passif vis-à-vis les membranes hydrophobes (Figure 24).101 Ceci a été démontré
notamment avec la cyclosporine A (CsA) (17),102-104 un immunosuppresseur
macrocyclique naturel ainsi qu’avec d’autres peptides cycliques synthétiques.105 La CsA
démontre des différences conformationnelles majeures selon la nature du milieu (aqueux ou hydrophobe) dans lequel elle se trouve. En effet, des études cristallographiques démontrent bien la présence des LHI dans les cristaux formés à partir du CCl4 avec les
divers groupements N-méthyl pointant vers le solvant. À l’opposé, la forme cristalline à partir de l’eau semble plus désordonnée et interagit avec plusieurs molécules d’eau (Figure 25). La molécule s’adaptera via ses LHI réduisant ainsi la pénalité de désolvatation.
Figure 25 Structure cristallographique de la CsA à partir de l’eau (gauche) ou à partir du CCl4
(droite).104
1.6 Les peptoïdes en chimie combinatoire
En plus de l’immense diversité moléculaire atteignable, la grande accessibilité synthétique des peptöides a propulsé leur utilisation en chimie combinatoire pour la génération de
Cyclosporine A
chimiothèques. Parmi les différentes approches combinatoires, la méthode «one-bead-one-compound» (OBOC) a certainement été la plus exploitée avec les peptoïdes. Initialement mise au point par le groupe du Dr Lam, elle a été grandement utilisée pour concevoir et cribler des chimiothèques de peptides linéaires.106-108 L’approche OBOC est
certainement une des méthodes combinatoires les plus accessibles et les moins coûteuses.
La stratégie OBOC consiste à utiliser la méthode de synthèse combinatoire
«split-&-pool»106, 109, 110 sur un support solide pour générer une chimiothèque contenant des milliers
à des millions de composés différents et dans laquelle chaque bille porte une entité moléculaire unique, soit un composé par bille. Comme les molécules à cribler sont attachées et exposées sur les billes, le criblage s’effectue sur bille avec la protéine cible en solution. Par la suite, les billes positives sont isolées et traitées individuellement afin de déterminer l’identité du composé sur la bille. Des chimiothèques OBOC ont permis d’identifier des ligands pour une grande variété de cible protéique comme des récepteurs membranaires, facteurs de transcription, enzymes, et autres protéines.111-122 L’utilisation
de prototypes moléculaires peptidomimétiques à base de peptoides macrocycliques rigidifiées dans l’approche OBOC pourrait considérablement accélérer la découverte de ligands pour des protéines cibles et d’inhibiteurs d’IPP. Ces molécules bioactives pourraient être utilisées directement comme outils moléculaires dans des essais cellulaires et en chimie biologique ou comme composés tête-de-série dans le développement d’agents thérapeutiques novateurs.
Chapitre 2
2.1 Hypothèses de travail
L’essentiel du projet repose sur l’exploitation du potentiel des macrocycles pseudopeptidiques à servir de vecteur pour le développement d’inhibiteurs d’IPP efficaces. Nous croyons que l’utilisation des peptoïdes cycliques permet de répondre à des critères essentiels pour le développement d’inhibiteurs d’IPP dans un contexte de développement thérapeutique :
Mimer la diversité fonctionnelle des peptides;
Incorporation facile d’une énorme diversité moléculaire;
Adoption de structures conformationelles rigides pouvant mimer les structures secondaires protéiques;
Résistance à la dégradation enzymatique; Meilleure pénétration membranaire.
L’induction de conformations fixes pourrait améliorer grandement leur affinité face aux protéines et du même coup induire une meilleure perméabilité membranaire via des considérations thermodynamiques. La stratégie est de tirer profit de la combinaison entre la macrocyclisation et l’introduction de résidus rigides pour restreindre au maximum la liberté conformationelle et ainsi créer des peptoïdes cycliques aux motifs structuraux bien définis. En faisant varier le type de résidu rigide, leur quantité et leur position relative dans le cycle, nous espérons déterminer des conditions spécifiques de rigidification structurale. Ces structures stables pourront ensuite être corrélées avec leur perméabilité membranaire et leur capacité à mimer des structures secondaires de protéines afin d’optimiser leur interaction avec des cibles protéiques. Le ou les prototypes moléculaires qui sortiraient vainqueurs de cette étude possèderaient donc une potentielle affinité de liaison supérieure et sélective en plus d’une biodisponibilité améliorée permettant l’atteinte d’IPP intracellulaires. Enfin, ces prototypes moléculaires seraient adaptés à la réalité du « beyond rule of 5 », avec des IPP définies par des surfaces d’interaction de plus en plus larges. En effet, les IPP deviennent de plus en plus difficiles à cibler avec de petites molécules respectant les règles dictées par Lipinsky.
L’approche OBOC permet de générer rapidement sur support solide des millions de composés différents. De plus, plusieurs essais biologiques peuvent se dérouler directement sur les billes. Ainsi, l’adaptation de cette méthode pour la préparation de chimiothèques basées sur nos prototypes macrocycliques rigidifiés pourrait permettre
d’accélérer significativement la découverte de ligands pour des protéines cibles et le développement d’inhibiteurs efficaces pour une panoplie d’IPP pertinentes autant en chimie biologique et qu’en sciences biomédicales et pharmaceutiques. Avec une plus grande capacité à pénétrer dans les cellules, ces molécules bioactives issues du criblage pourraient être utilisées directement comme outils moléculaires dans des essais cellulaires en chimie biologique ou biologie cellulaire ou comme composés tête-de-série dans le développement d’agents thérapeutiques novateurs.
Figure 1. Approche proposée pour le développement d’un prototype macrocyclique rigide à
pénétration cellulaire améliorée qui sera récupéré en chimie combinatoire.
Introduction de différents éléments rigidifiants à diverses positions du modèle Sélection d’un modèle de peptoïde flexible
Sélection d’une conformation rigide avec pénétration cellulaire optimale
Exploitation des zones variables du prototype pour l’adaptation à la technique OBOC
2.2 Objectifs
L’objectif principal de ce projet de recherche était de développer un prototype moléculaire peptidomimétique à conformation rigide et optimisé pour la pénétration cellulaire. Ensuite, les zones variables de ce prototype seraient exploitées pour l’introduction aléatoire de fonctionnalités chimiques dans une approche de chimie combinatoire pour la découverte rapide de nouveaux inhibiteurs d’IPP biodisponibles.
Des objectifs spécifiques ont été dégagés afin de bien diagnostiquer l’avancement du projet de recherche :
1. Synthétiser une collection de macrocycles pseudopeptidiques possédant des caractéristiques structurales diverses;
2. Évaluer la pénétration cellulaire relative de chaque macrocycle et identifier un ou plusieurs candidats avec perméation améliorée;
3. Établir des liens entre la structure et la pénétration cellulaire via des analyses conformationelles;
4. Exploiter les structures macrocycliques démontrant la meilleure pénétration cellulaire pour la génération de chimiothèques à haute diversité moléculaire et le criblage à haut débit.
Chapitre 3
Nouvelle méthode de synthèse de peptoïdes
avec des submonomères arylsulfonamide
Avant-propos
Ma contribution à cet article a été d’abord de synthétiser et de caractériser les différents submonomères N,O-diprotégés. Par la suite, j’ai aussi optimisé les conditions réactionnelles de substitution nucléophile et de déprotection du groupement arylsulfonamide sur support solide. Une partie appréciable du travail a été accompli par Antoine Derson qui m’a beaucoup aidé durant son stage d’été. J’ai finalement participé à la rédaction de cet article en collaboration avec mon directeur de recherche, Éric Biron. Cet article fut publié dans le journal Tetrahedron Letters (Elsevier) le 8 janvier 2015.
Résumé
Les peptoïdes, ou oligomères de glycines N-substitutées, sont des composés peptidomimétiques aux propriétés pharmacologiques et structurales intéressantes. Avec leur synthèse efficace par l’approche submonomérique, leur utilisation en chimie combinatoire est maintenant courante. Afin d’augmenter la diversité moléculaire et l’utilisation de résidus fonctionnalisés au sein de chimiothèques, nous présentons l’utilisation de dérivés arylsulfonamide N-substitués comme approche submonomérique alternative et complémentaire. La synthèse de submonomères amino-alcools N,O-protégés ainsi que les conditions d’incorporation sur support solide sont décrites. Cette méthode est compatible avec l’approche submonomérique classique et permet d’introduire facilement diverses chaînes latérales hydroxylées au sein des oligomères peptoïdes.
Tetrahedron Letters 2015, 56, 382-385
N-Substituted Arylsulfonamide Building Blocks as Alternative
Submonomers for Peptoid Synthesis
Simon Vézina-Dawoda,b, Antoine Dersona,b and Eric Birona,b,*
a Faculty of Pharmacy, Université Laval, Pavillon Ferdinand-Vandry, Québec, Québec,
G1V 0A6, Canada,
b Laboratory of Medicinal Chemistry, CHU de Québec Research Centre (CHUL Section), 2705 Boulevard Laurier, Québec, Québec, G1V 4G2, Canada
*Corresponding author :
Prof. Éric Biron, Laboratory of Medicinal Chemistry, CHU de Québec Research Centre (CHUL Section), 2705 Boulevard Laurier, Québec, Québec, G1V 4G2, Canada, Phone: 1
ABSTRACT
Peptoids (oligo N-substituted glycines) are peptidomimetic oligomers showing attractive structural and pharmacological properties. The efficiency of their synthesis has prompted the use of peptoids in combinatorial libraries. To increase the chemical diversity accessible in peptoid design and libraries, we demonstrate here that N-substituted o-nitrobenzenesulfonamide derivatives can be used as alternative building blocks in the synthesis of peptoids by the submonomer approach. The preparation of N,O-protected amino alcohol submonomers and the conditions for their incorporation into peptoid oligomers are reported. The described method is compatible with the submonomer approach and was applied to prepare peptoid oligomers bearing different hydroxylated side chains.