HAL Id: dumas-01708509
https://dumas.ccsd.cnrs.fr/dumas-01708509
Submitted on 13 Feb 2018HAL is a multi-disciplinary open access
archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
La mucoviscidose : traitements actuels et perspectives
thérapeutiques
Norman Bideau, Benjamin Boucherle
To cite this version:
Norman Bideau, Benjamin Boucherle. La mucoviscidose : traitements actuels et perspectives thérapeutiques. Sciences pharmaceutiques. 2005. �dumas-01708509�
AVERTISSEMENT
Ce document est le fruit d'un long travail approuvé par le
jury de soutenance et mis à disposition de l'ensemble de la
communauté universitaire élargie.
Il n’a pas été réévalué depuis la date de soutenance.
Il est soumis à la propriété intellectuelle de l'auteur. Ceci
implique une obligation de citation et de référencement
lors de l’utilisation de ce document.
D’autre part, toute contrefaçon, plagiat, reproduction illicite
encourt une poursuite pénale.
Contact au SID de Grenoble :
bump-theses@univ-grenoble-alpes.fr
LIENS
LIENS
Code de la Propriété Intellectuelle. articles L 122. 4
Ill lllllÎIÏ i1Uffllil 111111 .
Thèse d'exerciceA
~ "-X~~
re
~
D 11 s os7136 s . .d · 1 · h , ·Li:1 mu1.:uvisci ose : traitements actue set perspectives t erapeutiques
Année: 2005
UNIVERSITE JOSEPH FOURIER FACULTE DE PHARMACIE DE GRENOBLE
LA MUCOVISCIDOSE :
TRAITEMENTS ACTUELS ET PERSPECTIVES THERAPEUTIQUES.
THESE
PRESENTEE POUR L'OBTENTION DU TITRE DE DOCTEUR EN PHARMACIE DIPLÔME D'ETAT
BIDEAU NORMAN
BOUCHERLE BENJAMIN
THESE SOUTENUE PUBLIQUEMENT A LA FA CULTE DE PHARMACIE DE GRENOBLE
Le : 07 DECEMBRE 2005
DEVANT LE JURY COMPOSE DE
Président du jury : M. CHRISTOPHE RIBUOT Membres
M. JEAN-LUC DECOUT M. JEAN-PIERRE GOUT M. PATRICE FAURE
La Faculté de Pharmacie de Grenoble n 'entend donner aucune approbation ni improbation aux opinions émises dans les thèses ; ces opinions sont considérées comme propres à leurs auteurs.
[Données à caractère personnel]
Remerciements
Nous tenons
àremercier: notre directeur de thèse, Jean-Luc Décout, pour
nous avoir guidé tout au long de ce travail, Jean-Pierre Gout pour ses remarques
et conseils au cours de la rédaction, Patrice Faure pour sa présence dans notre
jury et Christophe Ribuot pour avoir accepté la présidence de ce jury de thèse.
Nous remercions également le DPM et ses personnels pour le soutien logistique
notamment pour la recherche bibliographique. Merci à l'association « Vaincre la
mucoviscidose
»
pour les documents gracieusement fournis. Et enfin merci à la
faculté de pharmacie de Grenoble pour les belles années d'études que nous
y
1. Introduction 1.1. Historique 1.2. Définition 2. Physiopathologie 2.1. La Protéine CFTR 2.1.1 Structure 2.1.2. Biosynthèse et maturation 2.1.3 Localisation 2.1.4. Fonction 2.1.4.1. Canal ionique
2.1.4.2. Régulation d'autres canaux ioniques 2.1.4.3. Autres fonctions de CFTR
2.1.5. Régulation 2.2. Le gène CF
2.2.1. Description
2.2.2. Les différentes mutations 2.3. Les différentes classes de mucoviscidose 2.4. Le transport des ions
Cl-3. Clinique 3 .1. Symptômes 3 .1.1 Manifestations respiratoires 3 .1.2. Manifestations digestives 3 .1.3. Manifestations hépatiques 3 .1.4. Manifestations génitales
3 .1.5. Autres manifestations et cause de décès 3.2. Corrélations génotype/phénotype
4. Epidémiologie 4.1. Données 4.2. Dépistage
4.2.1. Les tests prénataux 4.2.2. Tests néonataux 4.3. Diagnostics page 1 page 1 page2 page 3 page 3 page 12 page 15 page 18 page 19 page 19 page 24 page 25 page 25 page 27 page 30
5 .1. Modifications pharmacocinétique au cours de la mucoviscidose 5.2. Traitement de l'inflammation
5 .3. Produit agissant sur la viscosité du mucus
5.4. Traitements antibiotiques au cours de la mucoviscidose 5.4.1. Principes généraux de !'antibiothérapie
page 32 page 33 page 35 page 36 5.4.2. Stratégie de !'antibiothérapie de Staphylococcus aureus 5.4.3. Stratégie de !'antibiothérapie de Pseudomonas aeruginosa 5.4.4. Conclusion
5.5. Autres thérapeutiques à visée respiratoire 5.5.1. Corticothérapie par voie orale 5.5.2. Corticothérapie inhalée 5.5.3. Bronchodilatateurs
5.5.3.1. Les anticholinergiques 5.5.3.2. Les B-2-mimétiques 5.6. Aérosols d'antibiotiques
5.6.1. Les générateurs d'aérosols
5.6.1.1. Les générateurs pneumatiques 5.6.1.2. Les générateurs ultrasoniques
page 43
page 45
5.6.2. Paramètres influençant l'activité des aérosols d'antibiotiques 5. 7. Kinésithérapie respiratoire page 4 7 5.8. La transplantation pulmonaire
5.9. Mucoviscidose et diabète 5.10. Extraits pancréatiques
5.11. Notions de diététique chez l'enfant atteint de mucoviscidose 5 .12. Grossesse et traitement de la mucoviscidose
6. Perspectives thérapeutiques
6.1. Amélioration des traitements symptomatiques 6.2. Thérapie génique
6.2.1. Définition et principe
6.2.2. Les différents types de vecteurs
page 48 page 49 page 50 page 50 page 50 page 53 page 53 page 54
6.2.2.3. Les méthodes n'utilisant pas de vecteurs 6.2.3. Les essais de thérapie génique
6.2.4. Les limites de la thérapie génétique 6.2.5. Mucoviscidose et thérapie génétique 6.3. Thérapie protéique ou pharmacologique
6.3.1. Poursuite de la traduction à travers un codon stop 6.3.2. Rôle des macrolides
6.3 .3. Activation de CFTR 6.3.3.1. Les benzoquinoliziniums 6.3.3.2. Les n-alcanols 6.3.3.3. La génistéine 6.3.3.4. Les xanthines 6.3.3.5. Les benzoaminothiophènes 6.3.3.6. Autres molécules activatrices 6.3.3.7. Inhibiteurs des phosphodiestérases 6.3.3.8. Conclusion sur les activateurs 6.3.4. Activation d'autres transporteurs ABC 6.3.5. Autres modulateurs du transport ionique 6.3.6. Maturation et adressage
6.4. Conclusion sur les perspectives thérapeutiques Annexes
A : Le test de la sueur B : Traitements
C : Essais de thérapie génique pour la mucoviscidose D : Index des abréviations
E : Index des figures et des tableaux Note sur la bibliographie
Bibliographie Conclusion
Serment des apothicaires
page 66 page 84 page 87 page 91 page 99 page 104 page 106 page 108 page 110
1. Introduction
1.1. Historique
La mucoviscidose est la plus fréquente des maladies génétiques létales. Cette maladie serait apparue en Turquie et en Irak, il y a environ 5000 ans, avant de se répandre sur l'ensemble des continents (http://fr.wikipedia.org/wiki/Mucoviscidose ).
Dès le XVIIème siècle, la littérature fait mention de cette maladie, mais c'est en 1936 que la première description clinique est réalisée par le professeur Guido Fanconi (Roussey, 2000). Elle est basée sur l'association d'une fibrose kystique congénitale du pancréas et d'une bronchectasie.
Il lui donne le nom de mucoviscidose en référence aux mots mucus et visqueux, auxquels s'ajoute le suffixe ose classiquement utilisé pour définir une maladie. La dénomination anglaise « Cystic Fibrosis » est basée sur l'autre manifestation principale de la maladie : la fibrose kystique du pancréas.
Il faut attendre 1953 pour que soit proposé le premier diagnostique spécifique de cette maladie par le professeur Di Sant Agnese par l'intennédiaire du test de la sueur, encore utilisé actuellement. Ce test est basé sur l'observation d'un excès de chlorure de sodium dans la sueur des enfants atteints. Cette anomalie fut précisée dans les années 1980 : elle correspond à un défaut de perméabilité aux ions chlorures (Cr) des cellules épithéliales.
Le caractère héréditaire de la maladie étant supposé, la recherche s'est orientée vers l'identification du locus du gène impliqué. Cela fut réalisé en 1985 par clonage positionne! : le locus fut localisé sur le bras long du chromosome 7.
Quatre ans plus tard, une étape importante est franchie lorsque l'équipe du professeur Lap Chee Tsui découvre l'anomalie génétique qui est à l'origine de la maladie. Il s'agit d'une mutation en 7q31, au niveau du gène qui sera appelé gène Cystic Fibrosis (CF) codant pour une protéine nommée « Cystic Fibrosis Transmembrane Conductance Regulator » (CFTR) (Riordan et al, 1989).
La mucoviscidose : traitements actuels et perspectives thérapeutiques
1.2.
Définition
La mucoviscidose est une maladie polymorphe ; en effet, les différentes mutations entraînent diverses manifestations cliniques. C'est une maladie autosomique récessive qui s'exprime uniquement pour les sujets homozygotes à une mutation et les hétérozygotes composites à deux mutations.
Af508 Af 508 Af508 homozygote aut r e Af508 hété rooz ygote composite autre Af508 mutation en Cis
Figure 1 : Exemple de combinaisons de mutations entraînant la mucoviscidose, Lif 508 étant la principale mutation de la protéine CFTR (Cf. 2.2.2.)
2
.
Phys
iopatho
log
ie
2
.1
.
La
Proté
ine
CFTR
2
.1
.1
.
Structure
La protéine CFTR fait partie de la super famille des transporteurs ABC (ATP Binding Cassettes) (ATP: Adénosine TriPhosphates), cette famille est retrouvée dans toutes les espèces, tantprocaryotes qu'eucaryotes.
Ces transporteurs réalisent le transfert àtravers les membranes cellulaires, grâce à
!'hydrolysede l'ATP, d'éléments très variables, tels que des acides aminés, des peptides, des protéines, des ions organiques et inorganiques, certaines toxines, voire même des antibiotiques (Akabas et al,2000).
Outre CFTR, la P-gP (glycoprotéine P), produit du gène MDR (multidrug resistance), la MRP (multiresistance protéine) et les récepteurs aux sulfonylurées (SUR) font partie de cette super famille. Une des caractéristiques de cette famille est la présence au sein de lastructure de laprotéine de domaines NBD (Nucleotides Binding Domains).
embrane
Cy
top
lasm
Figure 2: Schéma de structure de la protéine CFTR
NBD-2
~
EBP-50La mucoviscidose : traitements actuels et perspectives thérapeutiques
La protéine CFTR est composée de 1480 acides aminés répartis en deux séquences homologues, comportant chacune six domaines trans-membranaires et un domaine de fixation de nucléotides (NBD-1 et -2). Ces deux séquences sont reliées entre elles par un domaine régulateur (R). Les douze domaines trans-membranaires délimitent un canal ionique dont l'activité est déterminée par la phosphorylation du domaine R. L'énergie nécessaire à ce transport est fournie par l'hydrolyse de molécules d'ATP fixées dans les NBDs.
Ce canal possède une grande sélectivité pour les anions, et notamment pour les ions chlorure, due au résidu arginine en position 352 (R352). Les domaines Net C terminaux de la protéine sont cytoplasmiques.
Le domaine R correspond aux résidus 590 à 831 codés par l'ex on 13. Il est très spécifique du canal CFTR puisque les autres protéines de la famille ABC ne possèdent pas de domaine régulateur. La phosphorylation de résidus sérine (Ser) de ce domaine par les protéines kinases A et C (PKA, PKC) est un prérequis à l'ouverture du canal par l' ATP complexé par l'ion magnésium (Riordan et al., 1989). Le canal est maintenu fermé lorsque le domaine Rest déphosphorylé (Cotten et al, 1997).
Dans la séquence des NBD se trouvent les sites de liaison à l'ATP (Walker A et B) ams1 qu'une séquence commune à tous les transporteurs ABC appelée motif C (LSGGQXQR). Les limites des domaines NBDl et NBD2 varient selon les auteurs. Pour NBDl, la limite inférieure varie du résidu F434 (codé par l'exon 9) à L441, et la limite supérieure est comprise entre 1586 (codée par l'exon 12) et K684 avec une extension possible jusqu'à F650. Le domaine NBD2 est formé des résidus Ll 127 à Ll480 (Bianchet et al, 1997).
2
.1
.2
.
B
iosynthèse
et
maturat
ion
apical plasma membrane
'uJ
'uJ
V
\
.
~
~
~
1
/a
te
endosome
-i-
early/recycling \endosome
Q
vesicle~
lysosome proteosQme - CFTRFigure 3 : Biosynthèse de laprotéine CFTR sauvage
Peu de protéines CFTR (moins de 1000 copies par cellules) sont synthétisées (Chang et al, 1998). Leur adressage àla membrane apicale dépend d'une séquence signal, non-clivable, (Chen et al, 1996). En positions 894 et 900 se trouvent deux sites potentiels de liaison d'oligosaccharides. Leur liaison donne la forme mature de la protéine de 168 kDa.
Durant sa maturation dans le réticulum endoplasmique (RE), la protéine CFTR est associée aux protéines HSP 70 (Heat Shock Protein). La maturation de la protéine CFTR sauvage est peu efficace, seulement 30% des précurseurs nouvellement synthétisés seront transformés en molécules matures. En effet, 70 % du précurseur sauvage et 100% des précurseurs mutés dont le repliement est incorrect sont rapidement dégradés (demi-vie de 20 à 40 minutes) par le protéasome avant d'atteindre l'appareil de Golgi (Jensen et al, 1995). La durée de la liaison entre CFTR et les protéines HSP, ainsi que l'étape de
La mucoviscidose : traitements actuels et perspectives thérapeutiques
libération des ribosomes, module la dégradation cytosolique du canal CFTR (Ward et al, 1995). Le blocage des protéines immatures au niveau du réticulum est un phénomène sensible à la température et aux mutations dans le domaine NBD 1 (Kopito et al, 1999). Le faible taux de maturation de la protéine CFTR n'est pas une caractéristique des transporteurs ABC dont certains peuvent atteindre un degré de maturation de 100%.
Après la dissociation du complexe protéine CFTR-protéines chaperonnes, la protéine CFTR acquiert sa structure tridimensionnelle et transite alors vers l'appareil de Golgi. Puis elle est adressée à la membrane cytoplasmique dans laquelle elle s' insert.
2.1.3. Localisation
Le canal CFTR fonctionnel est principalement localisé dans la membrane apicale des cellules épithéliales polarisées. Le rôle de l'épithélium est de former une barrière continue et asymétrique séparant deux milieux distincts. Soit l'épithélium sépare le compartiment externe du milieu interne, soit il délimite un espace spécialisé (glande exocrine).
Les cellules épithéliales présentent une organisation polarisée selon un axe baso-latéral apical ; la face apicale étant en contact avec le compartiment muqueux et la face basale reposant sur un support (lame basale). Elles assurent un transport sélectif des ions, des solutés et des macromolécules.
canal chlorure
Pôle ba:solatéral Pôle apical (lumière)
Figure 4 : Cellules épithéliales présentant la protéine CFTR sur leur face apicale
2.1.4. Fonctions
2.1.4.1. Canal ionique
La protéine CFTR est un canal à spécificité anionique résultant de la présence du résidu arginine R352, (le
cr
est 10 à 20 fois plus perméant que le Na+) (Anderson et al, 1991 ; Bear et al, 1991 ). La pennéance est supérieure pour les ions facilement déshydratés par rapport aux ions qui retiennent plus fortement l'eau d'hydratation. La partie du pore la plus resserrée a un diamètre d'environ 5,3A
(Hanrahan et al, 1998). Le classement de la perméabilité du canal CFTR à différents halogénures est le suivant : Bf >cr
>r
> F.Cette séquence distingue CFTR des autres canaux chlorure épithéliaux, pour lesquels la perméabilité de l'iodure est plus importante que celle du chlorure (Sheppard et al, 1999). La protéine CFTR est également considérée comme un canal à A TP par certains auteurs (Fanen 2001).
iv!b
apicale
ORCC
Mb basolatérale
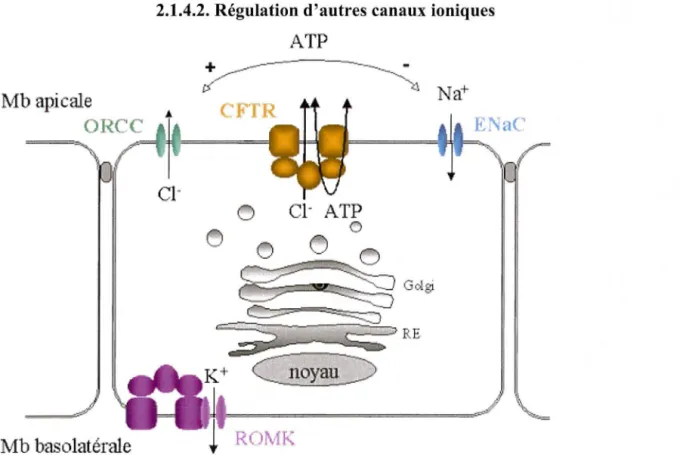
2.1.4.2. Régulation d'autres canaux ioniques
c1-0
0
Cl- ATP
00
ROtvfKLa mucoviscidose : traitements actuels et perspectives thérapeutiques
La protéine CFTR n'est pas seulement un canal ionique, mais également, comme son nom l'indique, un puissant régulateur de nombreux canaux et transporteurs influant sur la conductance (Schwiebert et al, 1999).
- Régulation du canal ORCC (Outwardly Rectifying Chloride Channel): Le canal chlorure rectifiant sortant (ORCC) est une protéine dont on a longtemps pensé qu'elle était impliquée dans le défaut de conductance membranaire retrouvé dans la mucoviscidose. Des études d'interactions ont montré que la présence de CFTR fonctionnelle était indispensable à l'activation du canal ORCC par la Protéine Kinase A. En effet, l' ATP extrudé par CFTR activée permettrait l'activation des canaux ORCC par un mécanisme autocrine : il se fixerait sur un récepteur purinergique, qui lui-même activerait l'ouverture du canal ORCC (Schwiebert et al, 1999).
- Régulation d'autres canaux chlorure:
CFTR activerait également les canaux chlore calcium dépendants par l'intermédiaire del' ATP. Ceci provoque une sécrétion d'ions chlorure supplémentaires.
- Régulation du canal ENaC (Epithelium Sodium Channel) :
Ce canal de transport du Na+, aussi appelé canal sodique sensible à l'amiloride, est responsable de l'absorption d'ions sodium au pôle apical de la cellule épithéliale. L'absorption accrue de sodium, dans les épithéliums respiratoires des patients atteints de mucoviscidose, montre un contrôle négatif du canal ENaC par la protéine CFTR normale (Mallet al, 1998).
- Interaction avec ROMK :
Identifiés sur la membrane basolatérale des cellules des épithéliums respiratoires, les canaux KIR interviennent dans le recyclage du potassium. Dans certaines cellules, il a été démontré que la présence du canal CFTR influence l'activation par l'AMPc (Adénosine MonoPhosphate cyclique) des canaux potassium épithéliaux (Loussouam et al, 1996). Un canal potassium a été identifié sur la membrane apicale des cellules épithéliales des voies respiratoires : ROMKl. La protéine CFTR pourrait être couplée avec ROMK dans la membrane cellulaire, procurant un domaine manquant à ces canaux, entraînant ainsi une sensibilité aux sulfonylurées. L'interaction entre ROMK et la protéine CFTR nécessite la présence du domaine NBDl du canal CFTR fonctionnel (McNicholas et al, 1996). CFTR
serait ainsi capable de transférer les effets d'agonistes et d'antagonistes sur d'autres protéines modulant de ce fait leur activité.
2.1.4.3. Autres fonctions de CFTR
La physiopathologie de la mucoviscidose qui dépasse les simples désordres des mouvements ioniques, ainsi que le faible taux d'expression de CFTR au niveau pulmonaire ont conduit à penser que CFTR possédait de multiples autres fonctions.
- Rôle dans l'inflammation :
Le nombre important de polynucléaires neutrophiles (PNN) dans le liquide de lavage broncho-alvéolaire ams1 qu'un déséquilibre entre les cytokines pro et anti-inflammatoires, tendent à montrer que CFTR possède un rôle dans le contrôle de l'inflammation.
- Rôle dans la défense anti-infectieuse :
Il a été montré par Goldman et al (1997) que les B-défensines, petits peptides bactéricides actifs sur P. aeruginosa et S. aureus, seraient inhibés par la concentration en NaCl du liquide de surface bronchique. Cette propriété est directement liée à l'activité de CFTR, car elle est restaurée après transfection cellulaire du gène CFTR normal.
- Fonction de canal autre que chlorure:
Des auteurs rapportent que CFTR est capable de réaliser le transport actif d'autres molécules que les ions chlorures. Ainsi Lindsdell et al, (1998) rapportent que CFTR serait capable de transporter l 'ATP ainsi que des conjugués du glucuronate et du glutathion.
- Fonctions en cours d'investigation:
La protéine CFTR est supposée être impliquée dans d'autres activités comme la sécrétion des composés du mucus, les processus de recyclage des membranes cellulaires ...
La mucoviscidose : traitements actuels et perspectives thérapeutiques
2.1.5. Régulation
Figure 6 : Représentation schématique du canal CFTR et de son domaine régulateur
La phosphorylation du domaine R conditionne la probabilité d'ouverture du canal CFTR, en effet une phosphorylation importante favorise le maintien d'une conformation ouverte. Cette phosphorylation est réalisée par la PKA et la PKC. À l'inverse, la Protéine Phosphatase 2 A (PP2A) déphosphoryle le domaine R.
Lorsque le canal est ouvert, le transport de
cr
est sous la dépendance de l' A TP, dont l'hydrolyse au niveau des NBD fournit l'énergie nécessaire à ce transport. La concentration intracellulaire en A TP, ainsi que le rapport des concentrations en ATP, ADP (Adénosine DiPhosphates) et AMP (Adénosine MonoPhosphate) régulent le transport chlornre via CFTR. L 'AMP et l'ADP sont inhibiteurs puisqu'ils peuvent se lier au niveau des NBD, sur le même site de fixation que l'ATP, mais ne permettent pas, de part leur hydrolyse, l'apport d'énergie nécessaire au transport (Schultz et al, 1995).Les protéines kinases sont sous l'influence directe du taux d'AMPc: plus ce taux est élevé, plus leur activité est importante. L'AMPc est produite par l'adénylate cyclase et sera dégradée par les phosphodiestérases, régulant de ce fait sa concentration.
Une autre hypothèse de fonctionnement a été récemment proposée par Hanrahan et al (2004). D'après ces auteurs, le canal CFTR pourrait s'ouvrir de deux façons différentes. La première décrite précédemment est ATP dépendante et nécessite la présence d'ions magnésium. Le deuxième mode, appelé ouverture non hydrolytique (non hydrolytic gating) ne fait pas intervenir l'hydrolysede l'ATP.En effet, une seule molécule d'ATP fixée sur le domaine NBDl pourrait interagir avec le domaine NBD2 et permettre le maintien d'une conformation ouverte du canal CFTR.
~-
----:
OJ
1 :ADP/Mg AIP ADP/Mg :1-
_Z,
'<
PO, 1.:41
OJ
1
-
r
D
:i 1l
MgATP ' ~ 1ATPi
~ ATP/MgI
r
-
-
-
-r
-
-
-
-
-
-
-
-
-
-
-
'
Î
'
l
'
Hydrolytic gating C> Tm1 Tm2 NBD1 NBD2 ()~
D
~ ..c:C:
~ 0 tf.' iz
iooou••••-••••••••••••••• JFigure 7 Mécanisme supposé d'ouverture hydrolytique et non hydrolytique du canal CFTR
La mucoviscidose : traitements actuels et perspectives thérapeutiques
2.2. Le gène CF
2.2.1. Description
Figure 8 : Le gène CF sur le chromosome 7
...
Le gène CF (CF pour Cystic Fibrosis) a été découvert en 1989 (Riordan et al, 1989). Il contient 27 exons s'étendant sur 250 kb du chromosome 7, en 7q31, et code pour un ARNm (Acide RiboNucléique messager) de 6,5 kb. L'identification directe du gène et de ses différentes mutations, réalisable depuis 1989, a permis de décrire les diverses mutations responsables de la mucoviscidose. À l'initiative des découvreurs du gène, le "Consortium International d'Analyse Génétique de la Mucoviscidose" (http://www.genet.sickkids.on.ca/cftr/) a été créé. Ce réseau d'échange d'informations a permis d'établir rapidement le spectre des mutations rencontrées au sein des différentes populations étudiées dans le monde.
2.2.2. Les différentes mutations
Les défauts moléculaires du gène CF sont répartis comme suit : Type Nombre Fréquence%
Mutation faux-sens 562 40,5 Décalage du cadre de lecture 228 16,4 Grandes variations de séquence 199 14,3 Mutation d'épissage 181 13,0 Mutation non-sens 143 10,3 Mutation du promoteur 8 0,6 Tableau I : Répartition des mutations du gène CF
La majorité des mutations sont des mutations ponctuelles, cependant quelques grandes délétions sont également reportées. Au 10 octobre 2005, 1402 mutations étaient
rapportées sur le site du Consortium International d'Analyse Génétique de la Mucoviscidose.
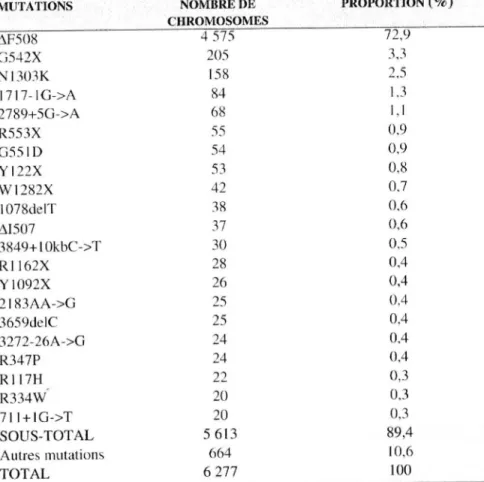
MUTATIONS NOrvIBREDE PROPORTION(%)
CHROMOSOMES Af508 4 575 72,9 G542X 205 3J N 1301K 158 2,5 17 17- IG->A 84 1.3 2789+50->A 68 1, 1 R55 3X 55 0.9 G55 1D 54 0,9 Yl 22X 53 0,8 Wl282X 42 0.7 1078de1T :rn 0,6 t.1507 37 0,6 3849+ 1 OkbC->T 30 0.5 RI 162X 28 0,4 Y 1092X 26 0,4 2 183AA->G 25 0,4 3659delC 25 0,4 3272-26A->G 24 0,4 R347P 24 0,4 RI 17H 22 0 ,3 R334W 20 0,3 711+10->T 20 0,3 SOUS-TOTAL 5 613 89,4 Autres mutations 664 10,6 TOTAL 6277 100
Tableau II: Principales mutations retrouvées en France (Observatoire National de la Mucoviscidose (ONM) 2001)
- La mutation LiF508
La mutation la plus fréquente est une délétion de trois nucléotides aboutissant à l'élimination de la phénylalanine en position 508 (LiF508).
Les observations des systèmes d'expression exogène transfectés suggèrent que la protéine mutée produite est incapable de se replier correctement au niveau du domaine NBDl (domaine de fixation des nucléotides). En effet, une protéine non glycosylée est formée et se localise dans le cytoplasme, ce qui tend à démontrer que la protéine n'entre pas dans l'appareil de Golgi, lieu de glycosylation de la protéine nonnale. La protéine mutée est retenue par les protéines chaperonnes de type HSP 70 et la calnexine dans le RE où elle est
La mucoviscidose : traitements actuels et perspectives thérapeutiques
Des études in vitro ont montré qu'une diminution de quelques degrés Celsius
permettait de restaurer un adressage correct à la membrane cellulaire, la protéine CFTR-L'lF508 fonctionnant comme un canal chlorure activé par l'AMPc (Frizzell et al, 1995). Ceci serait dû à la diminution de l'interaction entre CFTR/L'lF508 et les HSP, permettant ainsi la maturation de CFTR mutée.
Cette mutation correspond à une mucoviscidose de classe II et rend compte de 70% des allèles CF en Europe. Une étude collaborative européenne (EWGCFG 1990) portant sur 4871 chromosomes CF et 3539 chromosomes normaux a montré la grande hétérogénéité de répartition de cette anomalie.
Il existe un gradient nord-ouest/sud-est avec, par exemple 88% de L'lF508 au Danemark et 50% en Italie. Dans la population française, cette mutation représente approximativement 65-70% des chromosomes CF, avec de fortes variations régionales allant de 64 % en Languedoc-Roussillon à 81 % en Bretagne occidentale.
La fréquence élevée de cette mutation au sein des populations nord européennes suggère l'existence possible d'un mécanisme de sélection des hétérozygotes et d'un important effet fondateur. L'analyse de marqueurs intra-géniques associés à L'lF508 suggère qu'un seul événement mutationnel est survenu dans le passé (Morral et al, 1994).
De plus, pour expliquer la dispersion de la mutation dans les populations européennes, l'hypothèse d'un avantage sélectif des hétérozygotes a été avancée. En effet, les hétérozygotes semblent mieux protégés de la déperdition hydrique et saline induite au cours des diarrhées dues à des entérotoxines, comme Escherichia coli et Vibrio cholerae
(Baxter et al, 1988). Les toxines produites par ces bactéries augmentent la concentration d'AMPc ou de GMPc (Guanosine MonoPhosphate cyclique) dans les entérocytes et entraînent une augmentation de la sécrétion d'ions chlorure. Si on admet que les hétérozygotes ont 50% d'activité globale pour la protéine CFTR, leur sécrétion de chlorure en réponse aux toxines bactériennes sera moins élevée que celle des sujets non transmetteurs : la déperdition hydrique sera donc moins importante et la survie prolongée.
En outre, il a été suggéré récemment que la première boucle extracellulaire de la protéine CFTR est nécessaire à l'internalisation de S typhi dans les cellules épithéliales intestinales (Pier et al, 1998). La protéine liF508, associée à une diminution de l'expression de CFTR à la surface des épithéliums, diminuerait l'entrée du pathogène dans l'épithélium intestinal assurant une protection vis-à-vis de l'infection.
2.3. Les différentes classes de mucoviscidose
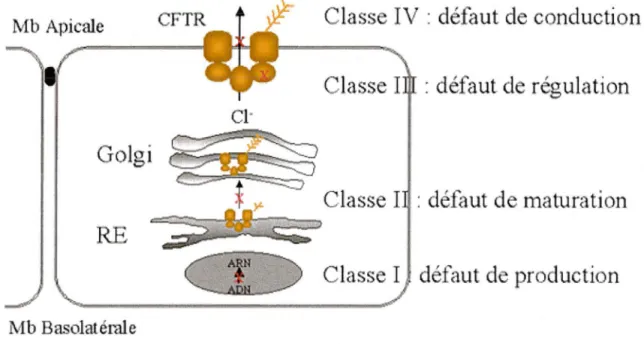
Il existe un grand nombre de mutations qui ont été classées en fonction de leurs conséquences au niveau biochimique. Elles ont tout d'abord été divisées en quatre classes différentes définies comme suit (Welsh et al, 1993) :
Mb Apicale
Classe IV : défaut de conduction
•
Classe I : défaut d . régulation
Golgi
Classe I : défaut de 1naturation
Classe I léfaut d production
Mb Basolatérale
Figure 9 : Classification des mutations du gène CFTR, d'après Welsh & Smith, 1993
• Classe I : Défaut de production de la protéine.
Ce type de mutation entraîne une absence totale ou partielle de la protéine. Ceci inclut les mutations non-sens et les mutations produisant un codon stop prématuré, ainsi que les mutations entraînant un ARNm muté instable.
La mucoviscidose : traitements actuels et perspectives thérapeutiques • Classe II : Défaut de maturation
La protéine issue de l' ARNm muté subit une maturation incorrecte et/ ou un défaut d'adressage. Ce type de mutation entraîne un déficit de protéines fonctionnelles dans la membrane apicale.
Exemple : 11F 508 (Délétion de la phénylalanine en position 508)
• Classe III : Défaut de régulation du canal
cr
La protéine est située dans la membrane, mais ne peut être activée du fait, soit d'une mutation sur le domaine R empêchant les phosphorylations nécessaires à son activation, soit d'une mutation située sur les domaines NBD ne permettant pas la fixation ou l'hydrolyse de l'ATP.
Exemple: G 551 D (Remplacement de la glycine en position 551 par l'acide aspartique) • Classe IV : Défaut de conductance
La protéine est correctement située dans la membrane, elle peut être activée et régulée, cependant le canal transmembranaire définissant le pore ionique est anormal. Ceci entraîne un défaut de conductance ionique par diminution du flux d'ions ou par modification de la sélectivité.
Exemples: Rll 7H (Remplacement de l'arginine en position 117 par l'Histidine), R334W (Remplacement de l'arginine en position 334 par le triptophane)
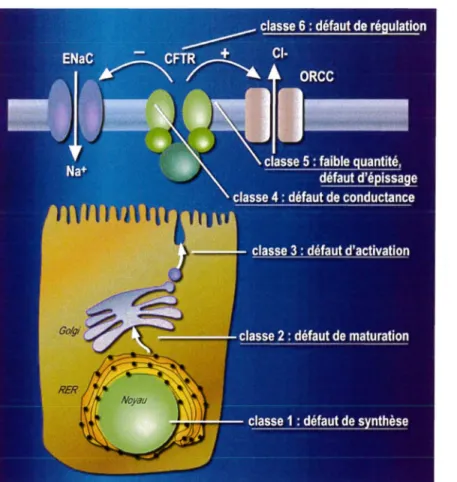
D'autres auteurs ont par la suite proposé une classification selon six critères. La classe I a ainsi été subdivisée en classe I et en classe V, cette dernière comprenant des mutations altérant la stabilité de l 'ARN m CFTR et la classe VI a été ajoutée (Haardt et al, 1999 ; Pilewski et al, 1999 ; Mickle et al, 1998).
Figure 10 : Classification de la mucoviscidose en six classes
• Classe I (redéfinie): Défaut de synthèse
La mutation entraîne une absence de l' ARNm ou la synthèse d'un ARNm instable. Cette classe comprend donc les mutations non-sens et les mutations produisant un codon stop prématuré (décalage du cadre de lecture ou ARNm tronqué).
• Classe V: Défaut d'épissage et quantité d'ARNm
La mutation se situe au niveau du promoteur entraînant une protéine fonctionnelle, mais dont la quantité est anormalement faible, ou alors la mutation perturbe l'épissage alternatif aboutissant à une protéine non fonctionnelle.
• Classe VI : Défaut de régulation
Ce type de mutation provoque une modification de la régulation des autres canaux ioniques par la protéine CFTR.
La mucoviscidose : traitements actuels et perspectives thérapeutiques
classe III. La prévalence des autres classes est difficile à estimer car les atteintes semblent plus faibles, le diagnostic étant donc plus difficile à établir.
2.4. Le transport des ions Cr
L'ion chlorure est le principal amon plasmatique puisqu'il y est présent à la concentration normale de 1 OO à 110 mmol/L.
On peut classer les canaux chlorures selon deux critères : en fonction de leur structure (récepteurs canaux, canal CFTR et famille des canaux CLC) ou en fonction de leur conductance unitaire (canaux de conductance élevée, canaux ORCC et canaux de faible conductance).
Les ions chlorure passent à travers la protéine CFTR normale et activée selon un gradient électro-chimique. Au niveau pulmonaire, ce passage se fait de l'intérieur vers la lumière. Dans les cellules sudoripares, c'est l'inverse : la sécrétion se fait de la lumière vers la cellule. Ainsi, la conséquence d'un défaut de la protéine CFTR est la rétention d'ions
cr
dans les cellules épithéliales bronchiques entraînant une rétention passive d'eau. Cela se traduit par une déshydratation du film hydrique de surface et donc par une diminution de la clairance muco-ciliaire avec finalement une obstruction des petites voies aériennes par des sécrétions peu mobiles créant ainsi un milieu favorable au développement d'agents infectieux (Wine, 1999). Inversement, au niveau sudoripare, ceci se solde par le maintien d'une sécrétion sudorale riche en chlorure de sodium et en eau permettant le diagnostic par le test de la sueur.3
.
C
l
in
ique
3
.1
.
Symp
tômes
L'âge d'apparition des premiers symptômes est très variable, les poumons sont généralement normauxàla naissance, alors que les lésions pancréatiques et les anomalies du mucus intestinalsont fréquemment présentes dès laphase fœtale.
Peau / Cf', >60 tnrnoVliter Puu.auu.u. Broncruecta.s1s Pneumothonix Hemoptysis Cor pulmonale Foie
Destruction des voies bilaifes
Pancreas
Insuffisance enzymal:!qUe
Diabète insulino-d!pcndant Petitln:
Mecomwnileus
S 'e~
Infertilité masculine
Absence congémtalc de Vllideferens
D'après Ackerman et Clapham,1997.
Figure 11 :Les différentes manifestations de lamucoviscidose
3
.1
.1
.
Man
ifes
ta
t
ions
resp
ira
to
ires
Ces manifestations restent la cause majeure de morbidité et de mortalité de cette maladie. Elles sont liéesàl'obstructiondes bronches,plus précisément des bronchioles, par
La mucoviscidose :traitements actuels et perspectives thérapeutiques
un mucus épais et visqueux qui favorise la croissance des microorganismes et engendre des détresses respiratoires.
Figure 12 : Représentation de mucus, semblable à celui retrouvé chez les patients atteints. On peut observer différents virus, des bactéries, des cellules et des brins d'ADN (Acide DésoxyriboNucléique) qui restent englués, épaississant encore ce mucus, déjà anormalement épais à l'origine.
(http://tecfa.unige. ~ lombardf/calvin/TM/02/mucoviscidose/laure-vieux.html).
Les signes respiratoires et les infections des voies aériennes sont, d'une manière générale, les plus fréquents. Ils conditionnent le pronostic vital et la qualité de survie. Ils se caractérisent par une toux chronique, sèche, quinteuse, rapidement productive, ainsi que des bronchites infectieuses et ou asthmatifonnes, marquées par leur caractère récidivant. Ces manifestations surviennent dans 80% des cas avant l'âge d'un an. On note l'apparition d'une dystrophie thoracique associée à une cyphose dorsale et un thorax en carène, ainsi qu'un hippocratisme digital témoignant de l'évolutivité de cette bronchopathie chronique obstructive.
Une pathologie rhinosinusienne s'ajoute souvent avec l'âge, faisant intervenir une sinusite chronique, ainsi qu'une polypose nasale.
Les infections bronchiques sont attribuables à trois principaux germes pathogènes : le Staphylocoque doré (Saureus), l'Haemophilus influenza, et surtout le Bacille pyocyanique(P aeruginosa). Ce dernier constitue le principal souci infectieux et marque le plus souvent un tournant évolutif péjoratif de la maladie. Les conséquences de ces infections chroniques sont l'apparition de kystes et d'abcès pulmonaires. La fibrose pos t-inflamrnatoire aboutit à une insuffisance respiratoire majeure. En effet, le système immunitaire détecte ces bactéries et envoie des cellules dans le but de les détruire. Ces
cellules sont par la suite engluées au sein du mucus. En tentant de détruire les bactéries, elles endommagent également le tissu pulmonaire, par l'intermédiaire d'un syndrome inflammatoire sévère.
Certaines souches dites "mucoïdes" se développent au sem de microcolonies entourées d'une matrice exopolysaccharidique (slime). Ce caractère mucoïde est pratiquement spécifique de l'infection. L'incidence des souches mucoïdes s'élève avec l'âge et l'évolutivité de la maladie respiratoire. Il est probable que le slime augmente l'adhésion des bactéries aux structures contaminées et gêne la pénétration des antibiotiques (Roussey, 2000). De plus, il peut apparaître des infections virales, aspergillaires ou mycobactériennes.
Ces atteintes pulmonaires évoluent par poussées et conduisent, à plus ou moins long terme, à une insuffisance respiratoire. Ces poussées sont synonymes de fortes dépenses énergétiques, elles sont souvent accompagnées d'anorexie, elles peuvent donc aboutir à une dénutrition ou bien à un retard pubertaire. Elles seront tenues pour responsables de complications telles que les hémoptysies, des pneumothorax récidivants, ainsi qu'un reflux gastro-oesophagien lié à l'emphysème pulmonaire et aux quintes de toux.
En résumant de façon simpliste, les poumons sont triplement touchés : ./ Leurs canaux sont obstrués par le mucus .
./ Ils sont la cible d'infections bactériennes récidivantes .
../ Les tissus pulmonaires sont attaqués par le système immunitaire avec inflammation et apparition de lésions.
3.1.2. Manifestations digestives
Les lésions pancréatiques font intervenir une obstruction des canaux proximaux par des sécrétions visqueuses, des acinis peu développés voire détruits, une surcharge graisseuse, une fibrose et progressivement la destruction du tissu pancréatique. De plus, l'accumulation d'enzymes pancréatiques peut aboutir à un processus d'autodigestion exposant les patients
à des pancréatites aigues (Atlas et al, 1992).
L'iléus méconial, occlusion aiguë néonatale par le méconium insuffisamment liquéfié, est la conséquence d'une insuffisance de sécrétion protéolytique, par le pancréas et les gîandes intestinales au cours de la vie fœtale. Dans 10% des cas, il constitue la
La mucoviscidose : traitements actuels et perspectives thérapeutiques
manifestation initiale de la maladie. Se situant au niveau de l'iléon terminal, il entraîne vomissements et ballonnements dès la 48ème heure après la naissance, mais sans émission de méconium.
L'insuffisance pancréatique, présente dans 90% des cas, entraîne une maldigestion responsable d'une diarrhée chronique avec émission de selles volumineuses graisseuses et nauséabondes. Cette mauvaise digestion sera responsable d'une carence secondaire en vitamines liposolubles (A, D, E et K) et en oligoéléments conduisant à un retard de croissance staturale et pondérale. La fibrose pancréatique peut s'étendre aux îlots de Langherans et, dans 5 à 10% des cas, induire à l'adolescence un diabète insulino-dépendant. Enfin, le pancréas sera attaqué lui-même par l'excès d'enzymes et peut être pratiquement détruit.
Les sécrétions bilio pancréatiques sont épaisses, appauvries en eau, en bicarbonate et en enzymes pancréatiques. La diminution de la sécrétion bicarbonatée retentit sur la digestion.
3.1.3. Manifestations hépatiques
Un ictère rétentionnel dû à une bile trop épaisse peut être révélateur en période néonatale. En effet, l'atteinte hépatobiliaire est fréquente, mais ne conduit à la cirrhose que dans 5 à 10% des cas. Elle est précédée par une augmentation des transaminases et des yGT (gamma Glutamyl Transférase), et peut se compliquer par une hypertension portale responsable de varices oesophagiennes et/ou gastriques avec risque d'hémorragies.
On notera aussi une vésicule biliaire souvent atrophiée chez les malades, avec un risque de lithiase important, augmentant avec l'âge. On retrouve également une hépatomégalie dans 30% des cas et une insuffisance hépatique dans 9% des cas. Ces éléments sont liés à l'obstruction des voies biliaires intra- ou extra-hépatiques par compression au niveau du pancréas.
3.1.4. Manifestations génitales
Les hommes atteints sont généralement stériles par atrésie des canaux déférents entraînant une azoospermie ou une oligospermie sévère avec moins de 5 millions de spermatozoïdes par mL. On retrouve fréquemment des lésions de la prostate et des vésicules séminales.
Chez les femmes, il existe une hypofertilité par modification de la glaire cervicale. Toutefois, de nombreux cas de grossesse ont été rapportés. Cette modification de la glaire peut provoquer la formation d'un bouchon de mucus qui empêche la pénétration du spermatozoïde dans l'utérus.
Normal Mucoviscidose
pH >8 <7
Acide citrique L > 2000 mg/lOOmL
Phosphatase 140-290 µg/ml 760-1140 µg/mL
Fructose 250-720 mg/1 OOmL 30-80 mg/lOOmL Tableau III : Caractéristiques du sperme des hommes atteints de mucoviscidose
On notera que 98% des hommes atteints sont stériles, alors que 80% des femmes atteintes sont fertiles.
3.1.5. Autres manifestations et causes de décès
Dans certains cas de mucoviscidose, on peut retrouver des myocardiopathies non obstructives ou des arthropathies. Il est également à noter que la perte accrue de sel par la sueur peut entraîner une déshydratation aigüe.
La mucoviscidose : traitements actuels et perspectives thérapeutiques Population totale Cause cardiaque Cause respiratoire Cause hépatique Cause traumatique Suicide Autres causes
Cause non documentée ou non renseignée
NOl\:tlJRE DE DECES 53 2 40 l 1 2 4 3 PROPORTION ( % ) lOO,O 3,8 75,5 l ,9 1,9 3,8 7,5 5,7
Tableau IV: Cause de décès des patients atteints de mucoviscidose (ONM 2001)
L'atteinte pulmonaire est de loin la plus fréquente des causes de mortalité, représentant à elle seule les trois quarts des décès. Ceci est en accord avec la physiopathologie de cette maladie.
3.2. Corrélations génotype/phénotype
La multiplicité des symptômes au sein d'une même fratrie laisse penser que le phénotype ne peut être simplement corrélé au génotype (Zielenski, 2000). En effet, l'atteinte pulmonaire, l'âge de survenue de la maladie ainsi que le taux de chlorure sudoral ne peuvent être reliés à un génotype particulier. Toutefois, l'état de la fonction pancréatique exocrine est identique chez tous les membres atteints d'une même famille (Corey et al, 1989). Il est à noter que certaines mutations faux-sens permettent le maintien d'une fonction pancréatique exocrine normale (Kristidis et al, 1992).
La mutation A455E est fortement associée à l'état de la fonction pulmonaire. En effet, des études menées aux Pays-Bas et au Canada ont montré que les sujets hétérozygotes llF508 / A455E possédaient une fonction pulmonaire améliorée ainsi qu'un taux de colonisation par le bacille pyocyanique inférieur aux sujets homozygotes llF508 de la même population (Gan et al, 1995 ; De Braekeleer et al, 1997).
L'analyse génotypique est parfois compliquée du fait d'une deuxième mutation en cis sur le même allèle influençant le phénotype. Une étude suggère que la mutation R553Q en cis sur l'allèle portant llf 508 module l'expression de cette dernière (Dork et al, 1991 ).
De manière générale, la connaissance du génotype d'un individu ne permet pas de prévoir la symptomatologie et l'évolution de la maladie.
,
4. Epidémiologie
4.1. Données
La mucoviscidose est donc la plus fréquente de toutes les maladies génétiques, mais on note toutefois une forte variabilité inter-ethnique. On estime que, pour les populations d'origines européennes, cette maladie affecte 1 nouveau-né sur 2500 naissances, ce qm permet d'évaluer à 1 sur 25 la proportion d'individus transmetteurs (hétérozygotes).
La prévalence chez les populations noires et asiatiques n'est pas connue, mais elle semble très faible.
En France, la prévalence est de l'ordre de 1/3500 avec une forte disparité inter-régionale, 1/2000 en Bretagne et 1/10 000 en région Parisienne. Ramené à l'échelle de la population française, le nombre d'individus porteurs sains serait d'environ 2 400 000.
* Sotirce pour ta population wtale des dépo r1 e111e111s : lnsee,
RGP / 999.
INtU
O>J04
Figure 13 : Prévalence de la mucoviscidose en France ; Figure 14 : Répartition géographique des nouveaux cas rapportés en 2001 (Observatoire National de la Mucoviscidose ONM 2001)
Depuis la forte augmentation de 1999, le nombre de nouveaux cas recensés est relativement stable. Cependant, suite à la mise en place du dépistage systématique en France en 2002, on observe une hausse du nombre de nouveaux cas de 4,5% en 2003, avec 228 nouveaux diagnostics (Observatoire National de la Mucoviscidose (ONM) 2003).
La mucoviscidose :traitements actuels et perspectives thérapeutiques Nouveaux cas INED 07204 --~~~~~~~~~~~--~~~~~~~ Années
*Source pour la période1992-1998: ONM 1998,Rapp011 aruwel AFLA1 Unüéd'épi.démi.o!ogie génétiquelll/SER:W Ul 55, décembre 1999. ' ' Figure 15 :Nombre de nouveaux cas recensés par an (ONM 2001)
La population de patients atteints de mucoviscidose est très jeune, comme on pouvait s'y attendre. En 2001, l'âgemoyen des patients est de 15,2 ans contre 15,8 en 2003, cette augmentation est régulière depuis plusieurs années, rendant compte des progrès de la prise en charge. En 2003, l'espérancede vie des patients était supérieureà39 ans.
Âges
~~
~~~~~-Uge1ufe:
'· '/,Pop11la1io11 de l'ONM
Q Fru11œ mé1ropoliwi11e
INED ~
25 20 15 10 5 0 0 5 10 15 20 25
Effectifs moyens par année d'âge parpopulation ramenée à1 000 personnes
Figure 16: Pyramide des âges des patients comparéeàla population française (ONM 2001)
4.2. Dépistage
Les modes de dépistage varient en fonction de l'âge du patient lors de la suspicion de la maladie. 94 % des diagnostics sont posés avant l'âge de deux ans.
4.2.1. Les tests prénataux
Les tests de dépistages prénataux ne sont pas réalisés en pratique courante mais uniquement dans le cadre d'indications bien précises : soit lorsque l'échographie réalisée en cours de grossesse présente des signes pouvant orienter vers la mucoviscidose, soit lorsqu'il y a dans la fratrie un enfant atteint de mucoviscidose. Ces tests demandent une analyse du génotype. On dénombre deux tests possibles :
• L'amniocentèse:
Test le plus fréquent en routine, il est effectué aux environs de la quinzième semaine de grossesse. Il consiste à prélever des cellules du liquide amniotique.
• La choriocenthèse :
Elle est réalisable plus précocement, aux alentours de la dixième semame de grossesse, mais l'image du caryotype obtenue est moins bonne. Ici, on prélève des cellules du placenta. Ce dépistage prénatal ne sera effectué qu'en cas de forte suspicion.
La suite de la méthode est similaire pour les deux tests : on provoque l'explosion de la cellule puis on recueille l' ADN restant, qui est copié par Polymerase Chain Reaction (PCR), puis clivé. Par la suite, on choisit le gène à étudier (CFTR) et on fait migrer l' ADN (Acide DésoxyriboNucléique) par électrophorèse. Les parties mutées se positionnent particulièrement sur la plaque de gel et sont donc facilement identifiables.
Ce type de test ne permet pas d'assurer une efficacité de 100%, du fait de la multiplicité des mutations observées chez les malades atteints de mucoviscidose, mais permet néanmoins la détection de vingt à trente mutations majeures.
Thèse d'exercice
La mucoviscidose :traitements actuels et perspectives thérapeutiques
4
.2
.2
.
Tests
néonataux
Depuis septembre 2002, le dépistage systématique est mis en place sur l'ensemble de la France. Il s'effectue sur les nouveaux-nés d'un mois permettant ainsi une prise en charge précoce des nourrissons dépistés. Ce dépistage est accepté par les parents dans 99,6% des cas.
Déroulement du dépistage néonatal de la mucoviscidose.
Nouveau.nés testésàJ3 (goutte de sang au talon)
!
l
1•dosage TIR <65 µgllI-
/
STOP jl
1°r
dosage deTIR>
~
-/
~
-~~~~~~-- ~~~~~
Biologiemoléculaire si consentement éclairé signé
(92 %des cas)
/
t
""
Pas de biologiemoléculaire
Refus ou consentementnon signé
CB %des cas)
l
2~j
1mut 1
1
o
mut ~ contrôleàJ21i
26dosage de TIR sur nouveauprélèvement
..
_
.
.
,
,
,
.
.
.
'
-
-
-
-
-
-
-
.
.
.
.
-
-
-
-
-
'
j2° ~ 40
µ&ïî]
~i
convocationau CRCMpour test de la sueur
J3 :a0Joµr de vie.
2n dosage TIR <40 µg/I
t
Pas deconvocation au CRCM
TIR :trypsine!mmunoréactive. Le seuil est égal à65 µgtl pour le 1"' dosage de TIR et 40 µg/I pour le28dosage de TIR, le taux de TIR diminuant au cours du 1ermois de vie.
Mut :mutailohde la protéine CPTR (cystfc f]brosis transmembrane conductance regulaton. CRCM :centre de ressources et de compétences de la mucoviscidose.
Figure 17 :Arbre décisionnel du dépistage néonatal (Soins pédiatrie -puériculture 2004)
• Test TIR (Trypsine Immuno Réactive):
Désormais, un nouveau-né subit automatiquement, en France, un test de dépistage de cinq maladies génétiques dont la mucoviscidose, les quatre autres étant la phénylcétonurie, l'hyperthyroïdie, la drépanocytose et l'hyperplasie des glandes surrénales. Le dépistage précoce permet une prise en charge rapide.
Ce test est très simple dans sa réalisation, cinq gouttes de sang sont prélevées au talon du nouveau-né, à partir du troisième jour de vie. Pour la mucoviscidose, on dose ensuite le taux de trypsine. Cette dernière est une enzyme pancréatique normalement excrétée dans la lumière de l'intestin. Les malades, du fait de leur atteinte pancréatique, ne peuvent pas excréter cette enzyme. On note ainsi une hypertrypsinémie due à une accumulation de trypsine au niveau sanguin. Les taux relevés chez les malades sont supérieurs à 900 µg/L. Un taux élevé de trypsine conduit à réaliser un diagnostic génétique fondé sur la détection d'allèles mutés.
Il est à noter que ce test n'est pas fiable à 100%. En effet, le nombre de faux positifs oscille entre 1 à 2%, alors que le nombre de faux négatifs s'élève à 10%.
• Tests génomiques (biologie moléculaire) :
Ils consistent à rechercher un panel de mutations à partir de l' ADN du patient issu de prélèvements sanguins. Les résultats obtenus sont très fiables, mais leur utilisation est limitée par le coût. Lors d'un test d'analyse d' ADN de routine, on ne dépistera qu'une trentaine de mutations différentes, ce qui correspond à 85% des mutations responsables de mucoviscidose en France.
Cependant, ces méthodes utilisées pour le dépistage systématique sont remises en cause par une étude récente (Sarles et al, 2005). En effet, la loi de bioéthique prévoit le consentement éclairé des deux parents pour réaliser les tests génomiques, or le nombre important de refus est un frein à cette technique. De plus, la recherche de mutation détecte un grand nombre d'hétérozygotes transmetteurs qui entraîne une information et éventuellement une recherche d'autre mutation au sein de la fratrie, ce qui dépasse les objectifs actuels du dépistage. Enfin, le surcoût généré par ces techniques d'analyse génétique n'est pas négligeable.
La mucoviscidose : traitements actuels et perspectives thérapeutiques
Les auteurs préconisent donc une stratégie « tout biologique » ou le dosage de TIR est associé à un dosage de la Protéine Associé à la Pancréatite (P AP), qui est un marqueur de pathologies pancréatiques. Les résultats de l'étude montrent que cette stratégie présente les mêmes perfonnances que l'analyse génétique pour un coût très inférieur. La Caisse Nationale d' Assurance Maladie des Travailleurs Salariés (CNAMTS) doit se prononcer prochainement sur cette alternative.
4.3. Diagnostics
Les tests suivants sont réalisés dans le cas de suspicion de mucoviscidose chez l'enfant ou chez l'adulte. En effet, dans le cas de mutation bénigne, le diagnostic peut aussi être évoqué chez un homme en azoospermie, au cours de recherches, par un urologue ou par imagerie médicale, d'une hypoplasie des vésicules séminales et/ou des canaux déférents.
• Test de la sueur :
Ce test est le moyen de dépistage le plus fiable, il consiste à mesurer les concentrations d'ions chlorure et/ou d'ions sodium dans la sueur.
En effet, chez les malades atteints de mucoviscidose, la sécrétion des glandes sudoripares possède une teneur particulièrement élevée en ions chlorure, potassium, et sodium. (Cf Annexe A)
• Différence de Potentiel de !'Epithélium Nasal (DDPTE ou NPD):
C'est une méthode simple et peu coûteuse, mais apparemment non réalisée en France en pratique clinique. Elle permet d'obtenir des résultats standardisés et reproductibles lorsqu'elle est utilisée après l'age de six ans. Elle consiste à mesurer la différence de potentiel entre la peau et la muqueuse nasale. Une valeur augmentée signale un patient atteint.
Cette méthode présente un intérêt dans trois types de situations :
1. Pour un diagnostic précoce chez le nouveau-né présentant une pathologie diagnostiquée suspecte.
2. En cas de diagnostic douteux ou de signes évocateurs, ou en cas de test à la sueur négatif.
3. Pour le suivi de l'évolution des patients, il semblerait qu'il y ait une corrélation entre DDPTE et le VEMS (Volume Expiratoire Maximum par Seconde) qui montre une gravité de l'atteinte respiratoire.
Cette méthode est fréquemment utilisée en recherche clinique pour évaluer l'efficacité d'un traitement.
• Test du méconium ou BM test:
La présence de protéines dans le méconium ou dans les premières selles peut évoquer la mucoviscidose, mais ce test souffre d'un manque de fiabilité. Il n'est plus réalisé en routine en France.
Si les tests biologiques s'avèrent négatifs pour la mucoviscidose, les diagnostics différentiels doivent s'orienter vers les pathologies suivantes:
Asthme,
Dysplasie broncho-pulmonaire, Déficit en alpha 1 antitrypsine, Tabagisme passif,
La mucoviscidose :traitements actuels et perspectives thérapeutiques
5
.
Tra
itements
actue
ls
5
.1
.
Mod
if
icat
ions
pharmacoc
inét
iques
au
cours
de
la
mucov
isc
idose
La biodisponibilité ne semble pas significativement différente entre les malades atteints de mucoviscidose et les sujets sains. Cependant il existe un retard à l'absorption. Malgré l'hypo-albuminémieexistante chez ces patients (Strober et al, 1969), lepourcentage de fraction libredes médicaments (cftableauV) varie peu par rapport aux individus sains. A l'inverse, le volume de distribution plasmatique semble augmenter significativement (cf tableau VI). Ceci pourrait être dûàl'apparition plus précoce du retard pondéral que du retard statural.
L'élimination tant rénale qu'hépatique semble variable en fonction des médicaments. n ~ n Suje1s sains IJicloxacillinc lO 11.6±7 .7 K 5.6±1.Y ~ 8'.l
'
85 Clox11dl/Îll1• S.2±S. I ~ U±2.I Ce(tmidime 10 %.9±6.1 IO 9iUl±5.6 Ge111t1111ü:h11' 12 85.7±2.6 8 ~ Tl11!opliylli11r Il 42-41 15 36-35 S1dj;1111éth1>.w1ml1· 7 5'.'±4 SH5 Trimfrltopriml.' 7 61±5 R 6.1±5 (R}-ll'fllJflrin 6 U.45±0.11 (l O.J7:tll.Ol1 111éopl1ylli11e 10 46±'1• [() J(d:(l+ (SJ-wm:f11ri11" (\ - ~ 0.55±0.0\J"' 11 Nnrnhn) dt! sujets ~ ~ ~ signilkatiwTableau V: Pourcentage de la fraction libre de certains médicaments dans le plasma au cours de lamucoviscidose.
~ Il rnalatb n SujcŒ snins Amikaâ!I(• l/1.7Jm2 Il 16.0±4.7 9 14.9±l8 Mérliic illin<' 11 J."?:1 m-' .., 1 41.J±lï..I (J '.'0.]±7.2 Crfw!nrliirt J/l.7Jm2 7 ~ 5 2:'i±5 ïàbramycii1<' 1/ 1.7 '\ ~ 12 15.5±-UI 8 14.4±5.2 Flimwrtint• l/."Okg§ u 1.6±0.):lJlSS 12 ~ 'l.nrss Tiwrâllim· Ilm2 1j 7.24±1.75 11 6.22±1.25 fS)·11w/1iri111· ml/kg 6 l5:1±18 8 138±22 Gmw111icine !!kg 19 O.J:l\.1;!;0,0JW 17 0.23610.0I(>+ Clnxr1dllb1e ml/ kg 12 91.2±34.2M 12 ~ n1iopltyfli111• liki; 10 ~ JO ~ Ct:fwzidimr likJ!
w
~ lO 0.197±0.o:H"'t\ Nombrede ~ s.s Al'é1a1 <l'équilibre
§ ~ ~ tÏliMImaigre ap Volume apparent ~ siatistiqucmcntsignilka11vc
Tableau VI : Volume de distributions de certains médicaments au cours de la mucoviscidose
5
.2
.
Tra
itement
de
l
'
inf
lammat
ion
Le processus inflammatoire est un moyen essentiel de protection de l'appareil respiratoire face aux agents bactériens. Il participe aussiàla réparation cellulaire via un équilibre entre cytokines pro (ILI, IL8 et TNFa) et anti-inflammatoires (ILlO).
Il existe deux voies d'inflammation, exogène et endogène. La voie exogène fait intervenir les PNN, qui sont activés suite à une infection. Ils provoquent la libération d'ADN responsable de l'hyperviscosité du mucus, la production d'oxydants, ainsi qu'un déséquilibre protéasesIanti-protéases. La voie endogène n'est pas secondaire à un stimulus infectieux. Chez les patients atteints de mucoviscidose, la diminution des cytokines ant i-inflammatoires observée notamment par Bonfield et al (1995), en particulier de la cytokine
La mucoviscidose : traitements actuels et perspectives thérapeutiques
diminution résulterait d'une baisse de régulation de la production par les cellules épithéliales respiratoires. Cette diminution de production aurait également lieu au niveau des lymphocytes T activés, ces derniers exprimant aussi CFTR.
Il semble nécessaire de privilégier un traitement anti-inflammatoire précoce afin de retarder la progression de l'atteinte.
Les anti-inflammatoires non stéroïdiens (AINS) seraient principalement dirigés contre l'activation des PNN. L'agent le plus couramment utilisé est l'ibuprofène, il semble permettre d'améliorer le VEMS et la capacité vitale (CV).
Les corticoïdes diminueraient l'adhésion et l'accumulation des PNN, inhiberaient la prolifération des lymphocytes T ainsi que la production d'IL2. Une dose de 1 à 2 mg/kg/jour de prédnisone par voie orale permet l'amélioration du VEMS et de la CV, mais de nombreux effets indésirables en découlent, avec notamment une intolérance glucidique et un retard de croissance. Les corticoïdes inhalés en aérosols doseurs à une posologie de 1500 µg deux à trois fois par jour semblent plus intéressants, mais il est difficile de déterminer une dose à administrer chez ces patients dont l'arbre bronchique est encombré par le mucus.
On peut aussi agir sur les modificateurs de l'inflammation, par exemple sur l'activité protéase. Ainsi des anti-protéases, tels que l'a-1-antitrypsine, permettent un traitement symptomatique de courte durée par aérosols, mais dont les résultats semblent modérés.
Enfin il est possible d'augmenter l'homéostasie anti-inflammatoire par instillation d'ILlO, mais cela n'aurait qu'un faible intérêt et uniquement lors de colonisation à
Pseudomonas aeruginosa.
En conclusion, de nombreuses questions subsistent sur le traitement de l'inflammation dans la mucoviscidose et de ses conséquences:
- un traitement précoce par AINS est-il le plus approprié?
- faut-il préférer la voie inhalée bien que l'on ne sache pas vraiment quelle quantité est absorbée par le patient ?
- quelle devrait être la durée de traitement, vraisemblablement un traitement permanent ?
5.3. Produits agissant sur la viscosité du mucus
Ces molécules agissent en cassant les liaisons covalentes qui permettent à la mucine de fonner de longues chaînes de polymère. Le N-acétylcystéine est l'agent mucolytique le plus largement utilisé par voie orale. Le Nacestyline®, ou lysinate de N-acétylcystéine, est un nouveau mucolytique qui présente une meilleure activité et une action activatrice sur les canaux chlorures (App et al, 1994).
Le Pulmozyme®, ou Domase a, a pour principe actif une désoxyribonucléase recombinante humaine. C'est une enzyme obtenue par génie génétique, similaire à l'enzyme humaine endogène qui hydrolyse l'ADN extracellulaire. Dans les sécrétions purulentes, les concentrations en ADN extracellulaire sont très élevées. En effet, les leucocytes s'accumulent en réponse à l'infection, leur altération entraîne la libération d' ADN qui est un polymère très visqueux. In vitro, la Domase a hydrolyse l'ADN du mucus et diminue la viscosité des expectorations. Cette Domase a est indiquée dans le traitement de l'encombrement bronchique afin d'améliorer la fonction respiratoire chez les patients âgés de plus de 5 ans, atteints de mucoviscidose, et dont la capacité vitale forcée (CVF) est supérieure ou égale à 40% de la valeur attendue. La posologie est la suivante : une nébulisation une fois par jour avec une ampoule de 2,5 mg (correspondant à 2500 UI) de désoxyribonucléase. Le contenu d'une ampoule (2,5 ml de solution) non dilué sera nébulisé à l'aide d'un système nébuliseur/compresseur pneumatique conformément aux recommandations (http://www. theriaque. org/InfoMedicaments/home). Son efficacité qualifiée de modeste vis-à-vis de l'amélioration de la fonction respiratoire à court terme, permettrait une moindre dégradation de la fonction respiratoire à plus long terme ainsi qu'une diminution des exacerbations respiratoires. La tolérance de cette spécialité est généralement bonne. Selon les données fournies, Pulmozyme® n'a pas démontré d'impact sur la mortalité ou sur la qualité de vie (Etude Z0507g). La diminution attendue en terme de morbidité, compte tenu des thérapeutiques actuelles, est faible. Il apporte une amélioration du service médical rendu de niveau III.
La mucoviscidose : traitements actuels et perspectives thérapeutiques
5.4. Traitements antibiotiques au cours de la mucoviscidose
(d'après la conférence de consensus sur la prise en charge du patient atteint de mucoviscidose du 18 et 19 novembre 2002 à Paris)La surinfection broncho-pulmonaire est le problème majeur auquel sont confrontés les patients. Elle est pratiquement constante. Elle est caractérisée par des exacerbations aigües intercurrentes responsables d'une détérioration des fonctions respiratoires. Deux germes prédominent, le Staphylococcus aureus et le Pseudomonas aeruginosa. D'autres germes sont aussi retrouvés , avec notamment Haemophylus injluenzae, et Burkholderia
cepacia.
5.4.1. Principes généraux de l'antibiothérapie
La posologie des antibiotiques ainsi que la durée du traitement doivent être adaptées à la sensibilité des germes aux antibiotiques et aux caractéristiques pharmacocinétiques des sujets atteints de mucoviscidose. Ainsi, dans cette affection, le volume de distribution par kilogramme de poids corporel est augmenté, tandis que la demi-vie d'élimination est raccourcie. L'augmentation de l'élimination rénale et non rénale nécessite des doses élevées d'antibiotiques. Ces caractéristiques sont encore modifiées par 1' état de nutrition du patient et par la gravité de la maladie (Cf. annexe B).
Les 13-lactamines ont une clairance et un volume apparent de distribution augmentés, justifiant une augmentation des doses et un rythme d'injection plus fréquent. Pour les aminosides, l'augmentation de leur clairance nécessite également une majoration de leur posologie. Du fait d'un index thérapeutique étroit de cette classe d'antibiotiques, l'adaptation des posologies justifie des dosages sériques en cours de traitement.
Les difficultés de pénétration des antibiotiques au sem du mucus et les caractéristiques des bactéries (résistance aux antibiotiques, Concentration Minimale Inhibitrice (CMI) élevée, présence d'un bio-film limitant l'accès des antibiotiques)
nécessitent l'obtention de fortes concentrations in situ et justifient de nouvelles stratégies : dose unique quotidienne pour les aminosides ou perfusion continue après une dose de charge pour la ceftazidime.
De ce fait, les posologies préconisées par la plupart des équipes spécialisées restent encore mal définies et correspondent rarement aux caractéristiques de l' Autorisation de mise sur le Marché (AMM) des produits.
5.4.2. Stratégie de
l'
antibiothérapie de Staphylococcus aureus
On distingue les souches de Staphylococcus aureus sensibles à la méticilline (SASM), les plus fréquentes, et les souches résistantes à la méticilline (SARM), parfois difficiles à traiter et dont la prévalence augmente.
Les principaux antibiotiques anti-staphylococciques utilisés dans la mucoviscidose sont présentés dans les tableaux VII et VIII.