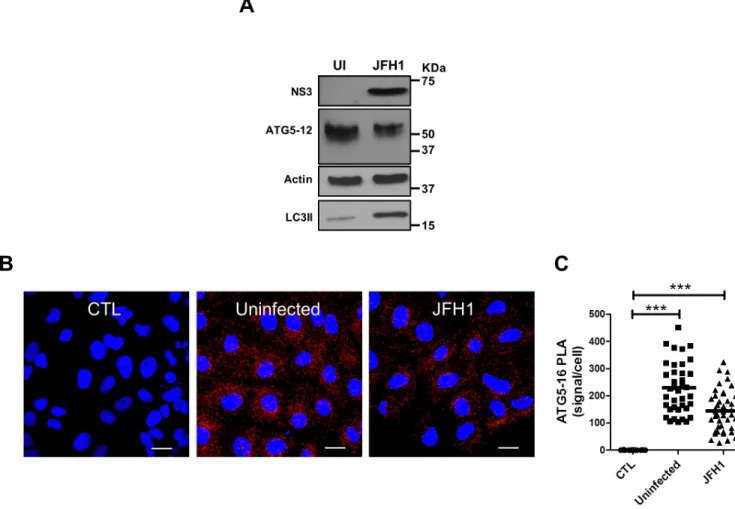
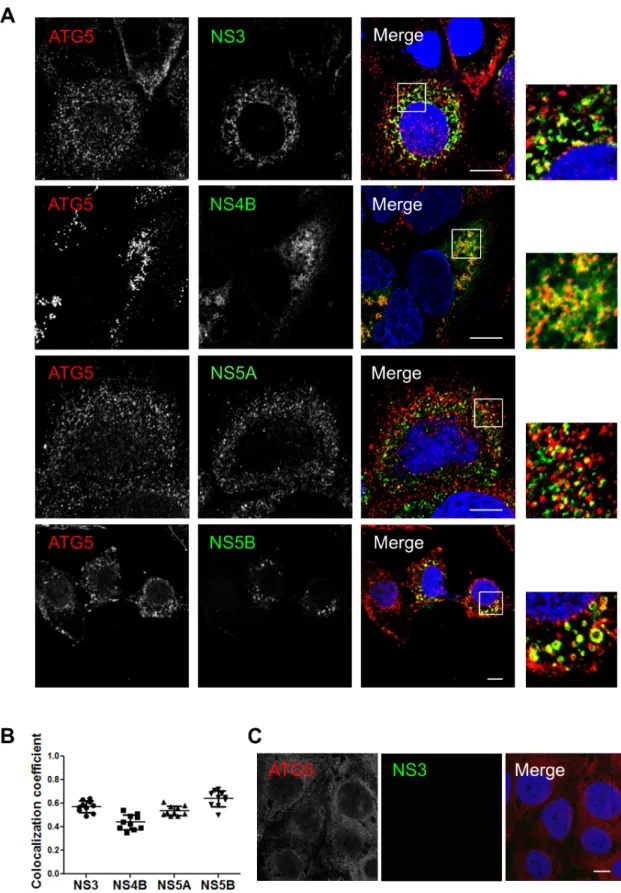
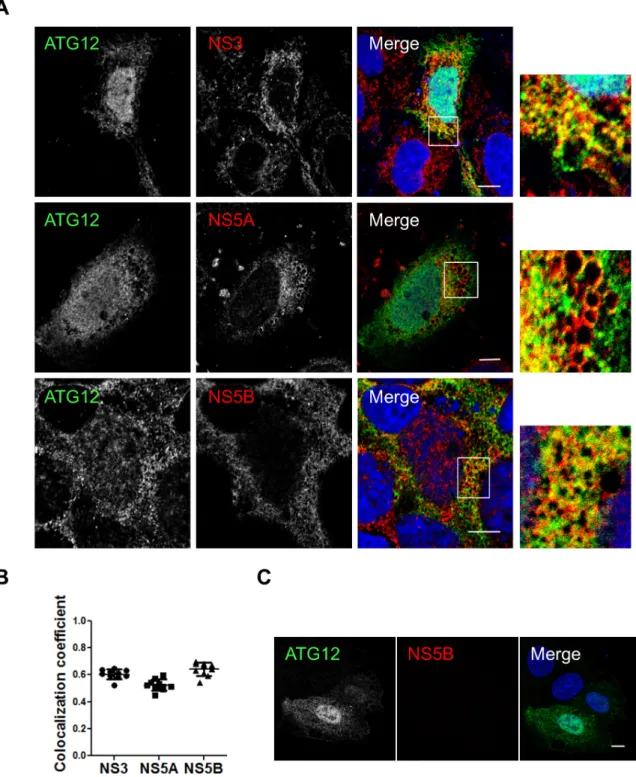
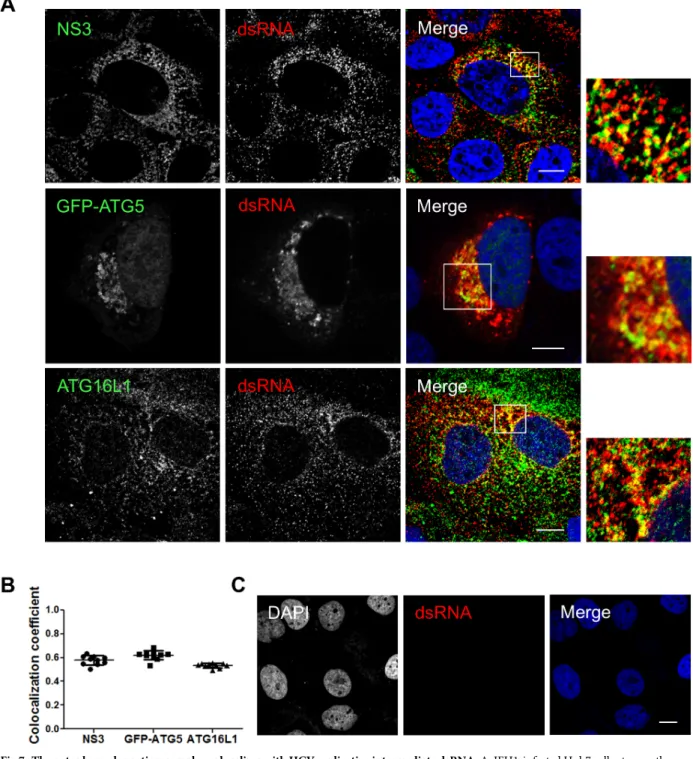
LC3B is not recruited along with the autophagy elongation complex (ATG5-12/16L1) at HCV replication site and is dispensable for viral replication
22
0
0
Texte intégral
Figure

+2
Documents relatifs
