Effet des acides gras oméga-3 sur l’inflammasome NLRP3 et les facteurs de risque de diabète de type 2 chez l’humain : modèles in vivo et ex vivo
310
0
0
Texte intégral
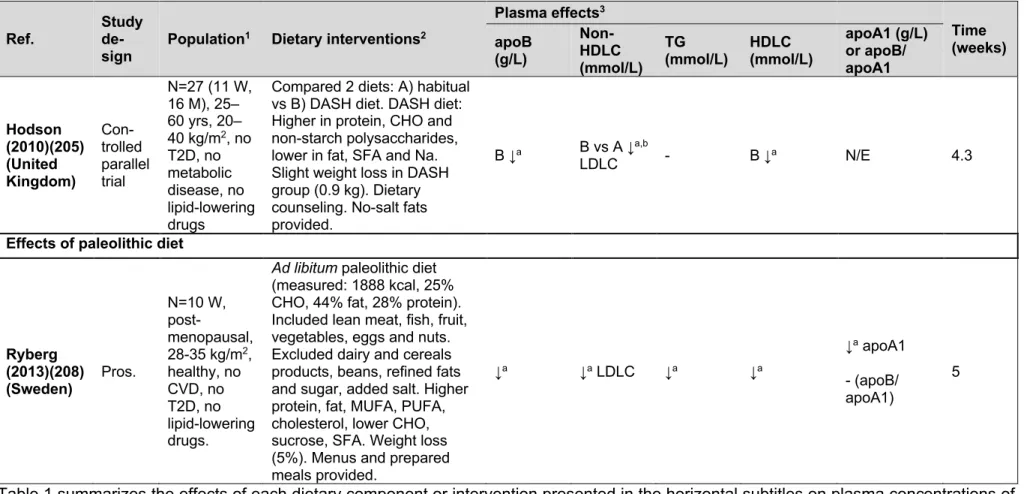
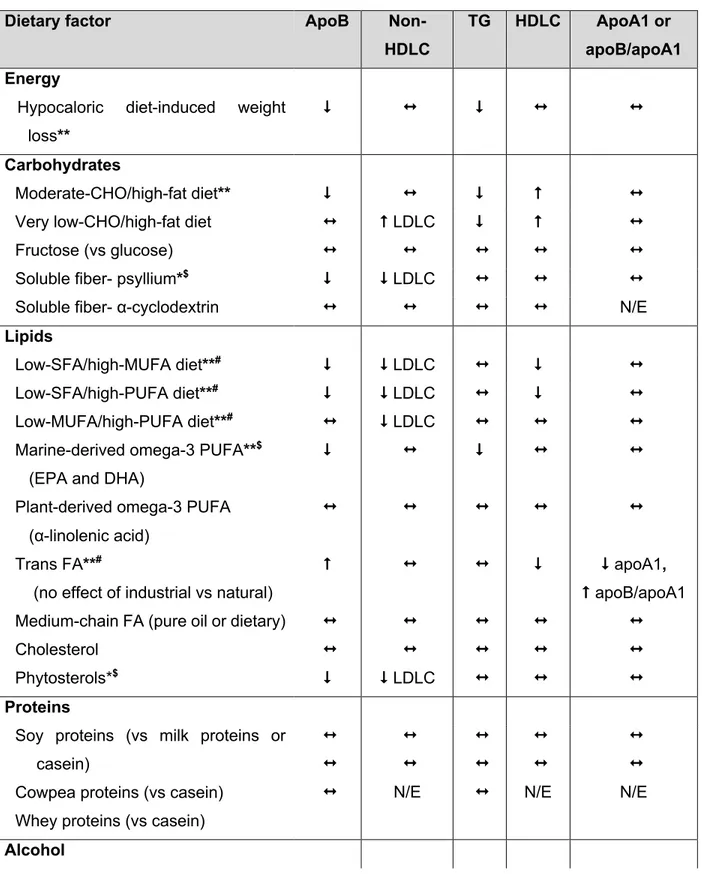
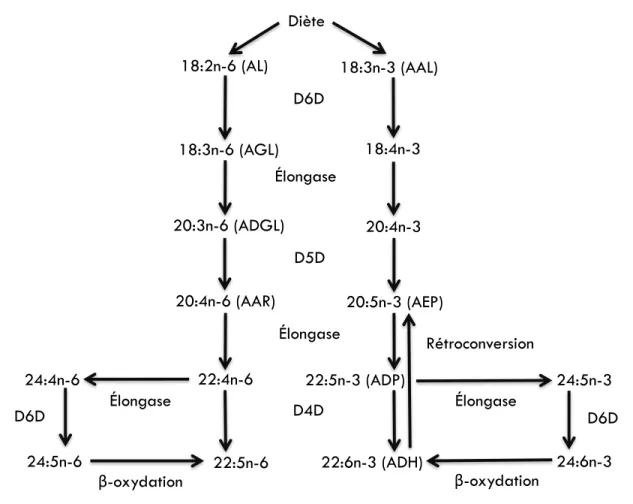
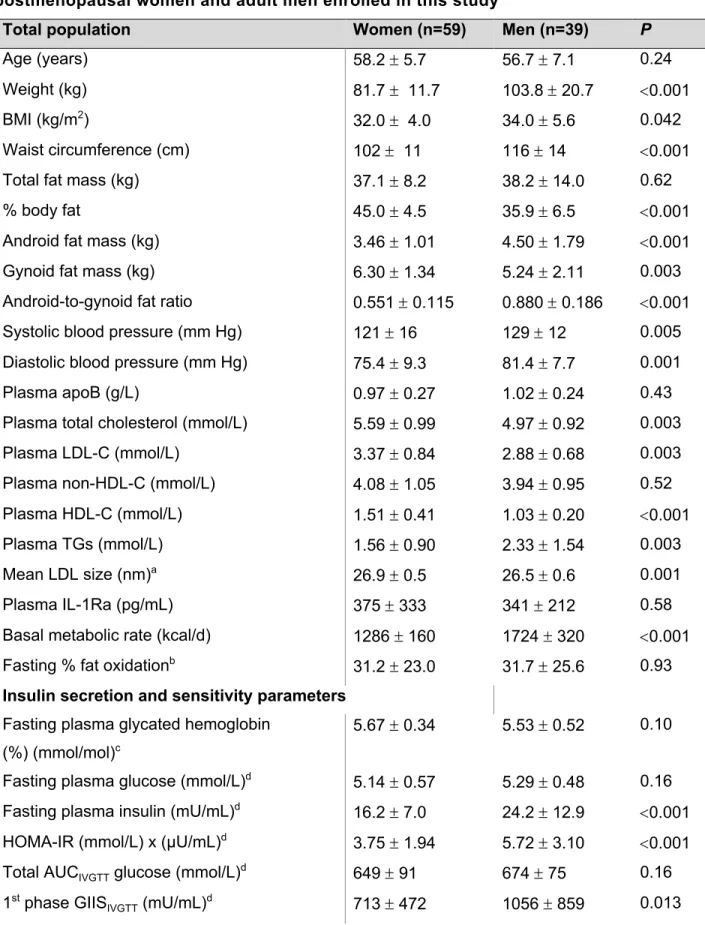
Figure



+7

Documents relatifs