Combinaison de modèles phylogénétiques et longitudinaux pour l'analyse des séquences biologiques : reconstruction de HMM profils ancestraux
Texte intégral
Figure

![Figure 1.4. Molécules d’acides aminés représentées dans un modèle « boules et bâtons » et caractérisées par leurs différentes propriétés (classification issue des travaux de Dayhoff [Dayhoff et al., 1978] et de Taylor [Taylor, 1986])](https://thumb-eu.123doks.com/thumbv2/123doknet/7702053.245790/32.892.144.791.192.766/figure-molécules-représentées-caractérisées-propriétés-classification-dayhoff-dayhoff.webp)
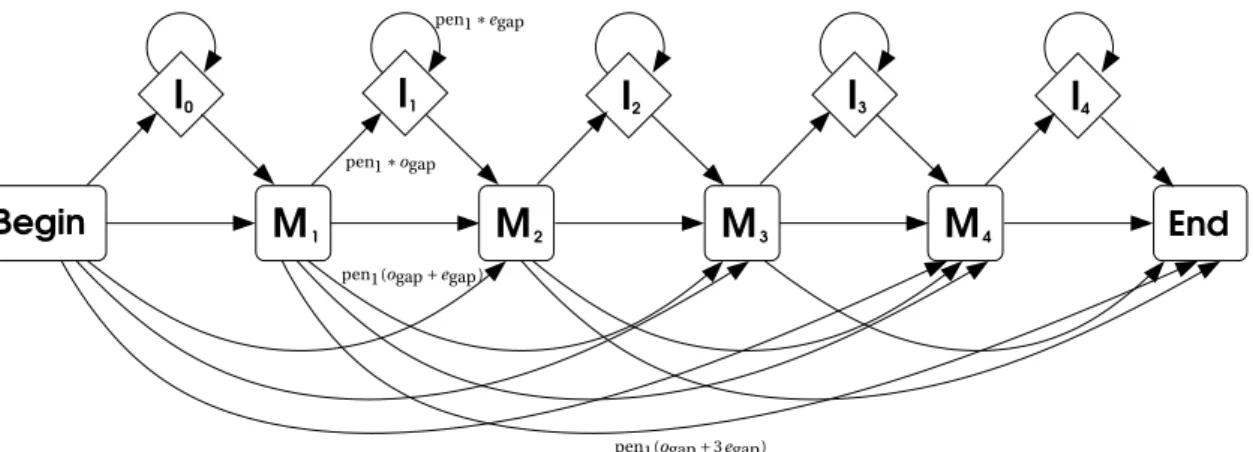
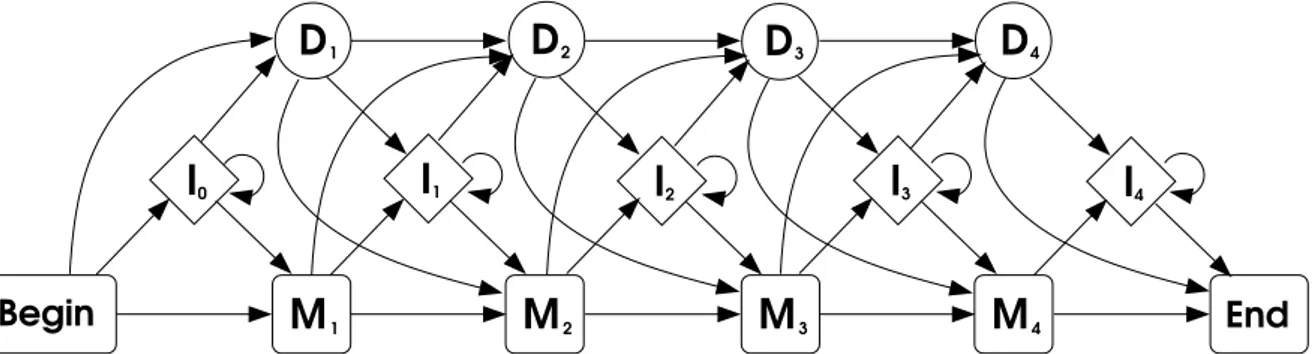
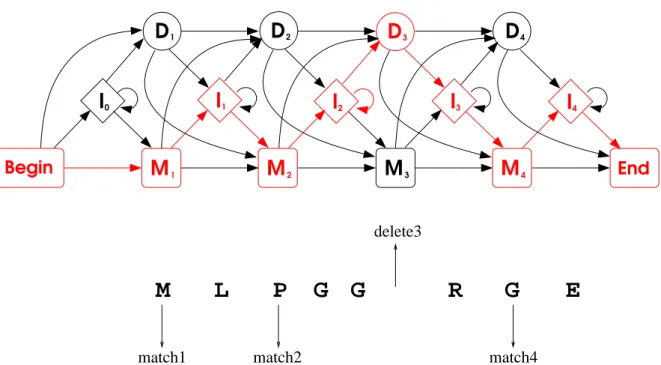
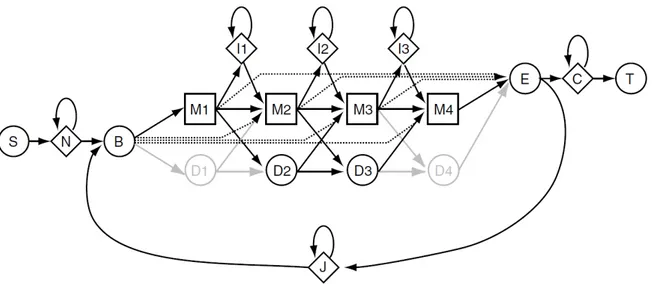
Outline
Documents relatifs
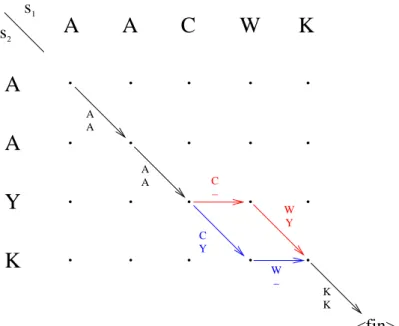
En notant str1 = prefixe1L et str2 = prefixe2L, une plus longue sous-chaine commune aux deux mots s’obtient en prenant une plus longue sous-chaine commune aux deux préfixes prefixe1
– Optimisation en temps: calculer la table autour d’une bande.. Temps et espace O(kn) où k est
• La plus grande valeur V(i,j) est le score du meilleur alignement local.. Alignement
• Alignement local: Plus long chemin entre n’importe quelles arêtes (i,j) et (i’, j’) du graphe d’édition.. • Quelle mesure
global, c'est-à-dire entre les deux séquences sur toute leur longueur local, entre une séquence et une partie d’une autre séquence.. Similarité global
En bioinformatique, la comparaison de séquences ADN deux à deux doit permettre de trouver des homologies c’est-à-dire comment les séquences ont muté à travers les espèces
Pour le tracing back (principe de la programmation dynamique, c'est par essence un processus off- line), on part de la case en bas à droite et on remonte le chemin d'alignement
• Procédez à un alignement des deux séquences ADN normale et mutée en visualisant les mutations avec l'outil EMBOSS proposé par EBI-EMBL : par
