THESE
PRESENTEE ET PUBLIQUEMENT SOUTENUE DEVANT LA FACULTE DE PHARMACIE DE MARSEILLE
Le Jeudi 13 Septembre 2018
PAR
M
elleRouaud Margot
Née le 14 août 1992 à Schœlcher
EN VUE D’OBTENIR
LE DIPLOME D’ETAT DE DOCTEUR EN PHARMACIE
TITRE :
Le rôle des microdomaines dans l’activation plaquettaire
JURY :
Président : Pr Laurence CAMOIN-JAU Membres : Dr Sylvie COINTE
27 Boulevard Jean Moulin – 13385 MARSEILLE Cedex 05 Tel. : 04 91 83 55 00 – Fax : 04 91 80 26 12
ADMINISTRATION :
Doyen : Mme Françoise DIGNAT-GEORGE
Vice-Doyens : M. Jean-Paul BORG, M. François DEVRED, M. Pascal RATHELOT
Chargés de Mission : Mme Pascale BARBIER, M. David BERGE-LEFRANC, Mme Manon CARRE, Mme Caroline DUCROS, Mme Frédérique GRIMALDI
Conseiller du Doyen : M. Patrice VANELLE
Doyens honoraires : M. Jacques REYNAUD, M. Pierre TIMON-DAVID, M. Patrice VANELLE
Professeurs émérites : M. José SAMPOL, M. Athanassios ILIADIS, M. Jean-Pierre REYNIER, M. Henri PORTUGAL
Professeurs honoraires : M. Guy BALANSARD, M. Yves BARRA, Mme Claudette BRIAND, M. Jacques CATALIN, Mme Andrée CREMIEUX, M. Aimé CREVAT, M. Bernard CRISTAU, M. Gérard DUMENIL, M. Alain DURAND, Mme Danielle GARÇON, M. Maurice JALFRE, M. Joseph JOACHIM, M. Maurice LANZA, M. José MALDONADO, M. Patrick REGLI, M. Jean-Claude SARI
Chef des Services Administratifs : Mme Florence GAUREL Chef de Cabinet : Mme Aurélie BELENGUER
Responsable de la Scolarité : Mme Nathalie BESNARD
DEPARTEMENT BIO-INGENIERIE PHARMACEUTIQUE
Responsable : Professeur Philippe PICCERELLE
PROFESSEURS
BIOPHYSIQUE M. Vincent PEYROT
M. Hervé KOVACIC
GENIE GENETIQUE ET BIOINGENIERIE M. Christophe DUBOIS
PHARMACIE GALENIQUE, PHARMACOTECHNIE INDUSTRIELLE,
MAITRES DE CONFERENCES
BIOPHYSIQUE M. Robert GILLI
Mme Odile RIMET-GASPARINI Mme Pascale BARBIER
M. François DEVRED Mme Manon CARRE M. Gilles BREUZARD Mme Alessandra PAGANO
GENIE GENETIQUE ET BIOTECHNOLOGIE M. Eric SEREE-PACHA
Mme Véronique REY-BOURGAREL PHARMACIE GALENIQUE, PHARMACOTECHNIE INDUSTRIELLE,
BIOPHARMACIE ET COSMETOLOGIE M. Pascal PRINDERRE M. Emmanuel CAUTURE Mme Véronique ANDRIEU Mme Marie-Pierre SAVELLI
NUTRITION ET DIETETIQUE M. Léopold TCHIAKPE
A.H.U.
THERAPIE CELLULAIRE M. Jérémy MAGALON
ENSEIGNANTS CONTRACTUELS
ANGLAIS Mme Angélique GOODWIN
DEPARTEMENT BIOLOGIE PHARMACEUTIQUE
Responsable : Professeur Philippe CHARPIOT
PROFESSEURS
BIOCHIMIE FONDAMENTALE, MOLECULAIRE ET CLINIQUE M. Philippe CHARPIOT
BIOLOGIE CELLULAIRE M. Jean-Paul BORG
HEMATOLOGIE ET IMMUNOLOGIE Mme Françoise DIGNAT-GEORGE
Mme Laurence CAMOIN-JAU
Mme Florence SABATIER-MALATERRE Mme Nathalie BARDIN
MICROBIOLOGIE M. Jean-Marc ROLAIN
M. Philippe COLSON PARASITOLOGIE ET MYCOLOGIE MEDICALE, HYGIENE ET
MAITRES DE CONFERENCES
BIOCHIMIE FONDAMENTALE, MOLECULAIRE ET CLINIQUE Mme Dominique JOURDHEUIL-RAHMANI M. Thierry AUGIER
M. Edouard LAMY
Mme Alexandrine BERTAUD Mme Claire CERINI
Mme Edwige TELLIER M. Stéphane POITEVIN
HEMATOLOGIE ET IMMUNOLOGIE Mme Aurélie LEROYER
M. Romaric LACROIX Mme Sylvie COINTE
MICROBIOLOGIE Mme Michèle LAGET
M. Michel DE MEO
Mme Anne DAVIN-REGLI Mme Véronique ROUX M. Fadi BITTAR
Mme Isabelle PAGNIER Mme Sophie EDOUARD
M. Seydina Mouhamadou DIENE PARASITOLOGIE ET MYCOLOGIE MEDICALE, HYGIENE ET
ZOOLOGIE Mme Carole DI GIORGIO M. Aurélien DUMETRE
Mme Magali CASANOVA Mme Anita COHEN
BIOLOGIE CELLULAIRE Mme Anne-Catherine LOUHMEAU
A.H.U.
HEMATOLOGIE ET IMMUNOLOGIE M. Maxime LOYENS
DEPARTEMENT CHIMIE PHARMACEUTIQUE
Responsable : Professeur Patrice VANELLE
PROFESSEURS
CHIMIE ANALYTIQUE, QUALITOLOGIE ET NUTRITION Mme Catherine BADENS CHIMIE PHYSIQUE – PREVENTION DES RISQUES ET
NUISANCES TECHNOLOGIQUES M. Philippe GALLICE
CHIMIE MINERALE ET STRUCTURALE – CHIMIE THERAPEUTIQUE
M. Pascal RATHELOT M. Maxime CROZET
CHIMIE ORGANIQUE PHARMACEUTIQUE M. Patrice VANELLE
M. Thierry TERME PHARMACOGNOSIE, ETHNOPHARMACOLOGIE, HOMEOPATHIE Mme Evelyne OLLIVIER
MAITRES DE CONFERENCES
BOTANIQUE ET CRYPTOGAMIE, BIOLOGIE CELLULAIRE Mme Anne FAVEL
Mme Joëlle MOULIN-TRAFFORT CHIMIE ANALYTIQUE, QUALITOLOGIE ET NUTRITION Mme Catherine DEFOORT
M. Alain NICOLAY Mme Estelle WOLFF Mme Elise LOMBARD Mme Camille DESGROUAS CHIMIE PHYSIQUE – PREVENTION DES RISQUES ET
NUISANCES TECHNOLOGIQUES
M. David BERGE-LEFRANC M. Pierre REBOUILLON
CHIMIE THERAPEUTIQUE Mme Sandrine FRANCO-ALIBERT
Mme Caroline DUCROS M. Marc MONTANA Mme Manon ROCHE CHIMIE ORGANIQUE PHARMACEUTIQUE
HYDROLOGIE M. Armand GELLIS M. Christophe CURTI
Mme Julie BROGGI M. Nicolas PRIMAS M. Cédric SPITZ M. Sébastien REDON PHARMACOGNOSIE, ETHNOPHARMACOLOGIE, HOMEOPATHIE M. Riad ELIAS
Mme Valérie MAHIOU-LEDDET Mme Sok Siya BUN
Mme Béatrice BAGHDIKIAN
MAITRES DE CONFERENCE ASSOCIES A TEMPS PARTIEL (M.A.S.T.)
CHIMIE ANALYTIQUE, QUALITOLOGIE ET NUTRITION Mme Anne-Marie PENET-LOREC CHIMIE PHYSIQUE – PREVENTION DES RISQUES ET
NUISANCES TECHNOLOGIQUES M. Cyril PUJOL
DROIT ET ECONOMIE DE LA PHARMACIE M. Marc LAMBERT
GESTION PHARMACEUTIQUE, PHARMACOECONOMIE ET ETHIQUE PHARMACEUTIQUE OFFICINALE, DROIT ET COMMUNICATION PHARMACEUTIQUES A L’OFFICINE ET GESTION DE LA PHARMAFAC
Mme Félicia FERRERA
A.H.U.
CHIMIE ANALYTIQUE, QUALITOLOGIE ET NUTRITION M. Mathieu CERINO
ATER
CHIMIE ANALYTIQUE M. Charles DESMARCHELIER
DEPARTEMENT MEDICAMENT ET SECURITE SANITAIRE
Responsable : Professeur Benjamin GUILLET
PROFESSEURS
PHARMACIE CLINIQUE Mme Diane BRAGUER
M. Stéphane HONORÉ
PHARMACODYNAMIE M. Benjamin GUILLET
TOXICOLOGIE GENERALE M. Bruno LACARELLE
TOXICOLOGIE DE L’ENVIRONNEMENT Mme Frédérique GRIMALDI
MAITRES DE CONFERENCES
PHARMACODYNAMIE M. Guillaume HACHE
Mme Ahlem BOUHLEL M. Philippe GARRIGUE
PHYSIOLOGIE Mme Sylviane LORTET
Mme Emmanuelle MANOS-SAMPOL
TOXICOCINETIQUE ET PHARMACOCINETIQUE M. Joseph CICCOLINI
Mme Raphaëlle FANCIULLINO Mme Florence GATTACECCA TOXICOLOGIE GENERALE ET PHARMACIE CLINIQUE M. Pierre-Henri VILLARD
Mme Caroline SOLAS-CHESNEAU Mme Marie-Anne ESTEVE
A.H.U.
PHARMACIE CLINIQUE M. Florian CORREARD
CHARGES D’ENSEIGNEMENT A LA FACULTE
Mme Valérie AMIRAT-COMBRALIER, Pharmacien-Praticien hospitalier M. Pierre BERTAULT-PERES, Pharmacien-Praticien hospitalier
Mme Marie-Hélène BERTOCCHIO, Pharmacien-Praticien hospitalier Mme Martine BUES-CHARBIT, Pharmacien-Praticien hospitalier M. Nicolas COSTE, Pharmacien-Praticien hospitalier
Mme Sophie GENSOLLEN, Pharmacien-Praticien hospitalier M. Sylvain GONNET, Pharmacien titulaire
Mme Florence LEANDRO, Pharmacien adjoint M. Stéphane PICHON, Pharmacien titulaire
M. Patrick REGGIO, Pharmacien conseil, DRSM de l’Assurance Maladie Mme Clémence TABELE, Pharmacien-Praticien attaché
Mme TONNEAU-PFUG, Pharmacien adjoint
M. Badr Eddine TEHHANI, Pharmacien – Praticien hospitalier M. Joël VELLOZZI, Expert-Comptable
Remerciements
A mon jury de Thèse
A Madame le Professeur Laurence CAMOIN-JAU
Professeur de l’Université de Pharmacie Aix-Marseille Praticien Hospitalier
Service d'Immunologie et d'Hématologie
Qui m’a fait l’honneur de présider mon jury de thèse et de la diriger, Pour son temps accordé ainsi que sa confiance,
Hommages respectueux.
A Madame le Docteur Christilla BACHELOT-LOZA Chargé de Recherche à l’INSERM
Innovation thérapeutique en hémostase
Sans qui cette thèse n’aurait existé,
Pour l’enseignement apporté tout au long de ma présence à l’INSERM 1140, Pour ses conseils avisés et sa confiance,
J’exprime toute ma reconnaissance.
A Madame le Docteur Sylvie COINTE Assistant hospitalier universitaire
Laboratoire d’hématologie hôpital la Conception
Qui m’a fait l’honneur et le plaisir d’accepter de participer à mon jury de thèse, Pour sa réelle sympathie,
A l’unité INSERM 1140
Tout d’abord, je voudrais remercier le Professeur Pascale Gaussem pour m’avoir accueilli au sein de son laboratoire de recherche ainsi que pour ses nombreux conseils.
Je tiens également à remercier le Docteur Tiphaine Belleville-Roland pour sa bonne humeur, son implication et ses conseils qui m’ont permis d’avancer dans la réalisation de mon projet. Je remercie Sonia Chassac pour ses précieux conseils et son aide qui m’ont permis d’améliorer grandement mes résultats. Merci également à Keirththana pour m’avoir assisté dans la réalisation des Western Blot.
Merci à tous les autres membres de l’équipe « Plaquette » pour votre implication dans mon projet. Merci notamment à Benoit pour le partage de ses connaissances qui m’a permis de développer mon esprit critique,
Je remercie également Elise pour m’avoir soutenue et aidée tout au long de ce projet mais aussi pour tous les instants agréables que nous avons passés ensemble.
Enfin, je remercie toute l’unité 1140 pour leur accueil et leur disponibilité. Une pensée particulière à mes camarades de M2 et notamment Elodie et Giulia pour votre soutient et votre amitié qui ont rendu ce stage agréable. Merci aux doctorants et en particulier Alison pour tous les bons moments passés ensemble et pour les prochains…
A mes parents, pour m’avoir tant donné, pour votre soutien tout au long de ces années et
pour tout votre amour, Merci du plus profond de mon cœur
A ma sœur, pour m’en avoir fait voir de toutes les couleurs, pour tous ces moments partagés,
pour être mon modèle dans la vie et m’avoir ouvert la voie
A Blaise, pour tout le bonheur que tu m’apportes au quotidien, pour tous ces moments
partagés et tous les suivant
A Sophie, Charlotte, Marine, Aurélie, Camille, Cécile, Clara, Merci de m’avoir soutenu tout
au long de ces années, d’avoir toujours été là pour moi dans les bons comme les mauvais moments, d’être des amies extraordinaires au quotidien
A Marine et Justine, Merci d’avoir partagé avec moi ces 6 années d’aventures que sont les
études de Pharmacie et d’avoir fait de ces années, des moments inoubliables
A tant d’autres
Merci de partager ma vie tout simplement...
« L’Université n’entend donner aucune approbation, ni improbation aux opinions émises dans les thèses. Ces opinions doivent être considérées comme propres à leurs auteurs. »
Table des matières
LISTE DES ILLUSTRATIONS ... 7
ABRÉVIATIONS ... 9 INTRODUCTION ... 11 I. Les plaquettes ... 11 1. Production ... 11 2. Structure ... 13 a. Zone Périphérique ... 13 b. Le cytoplasme ... 15
i. Les granules alpha ... 15
ii. Les granules denses ... 15
iii. Les lysosomes ... 16
3. L’hémostase primaire : principale fonction plaquettaire ... 17
a. Adhésion plaquettaire... 19
b. Activation plaquettaire ... 20
c. Agrégation ... 20
II. Régulation de l’activation plaquettaire médié par les nucléotides cycliques ... 22
1. Synthèse des nucléotides cycliques ... 22
2. Voie de signalisation de l’AMPc ... 24
3. Régulation du taux d’AMPc au sein des plaquettes... 25
III. Les microdomaines ... 26
1. Structure ... 26
2. Microdomaines et voie de signalisation de l’AMPc ... 27
3. Microdomaines dans les plaquettes ... 28
IV. But de l’étude ... 29
MATÉRIELS & MÉTHODES ... 30
I. Matériels ... 30
1. Solutions et Tampons ... 30
3. Kit et matériel ... 31
4. Anticorps ... 31
II. Méthodes ... 32
1. Préparation des plaquettes lavées ... 32
2. Isolement des radeaux lipidiques ... 33
a. Séparation des fractions ... 33
b. Récupération des fractions ... 33
c. Précipitation des protéines ... 33
c. Western-blot ... 34
d. Révélation ... 35
4. Test d'agrégation plaquettaire ... 35
5. Immunofluorescence ... 36
a. Préparation de la lame et incubation des anticorps ... 36
b. Révélation ... 36 6. Technique du Duolink® ... 37 a. Principe ... 37 b. Méthode ... 37 7. Statistiques ... 38 RÉSULTATS ... 39
I. Rôle des microdomaines dans l’activation plaquettaire ... 39
1. Rôle des microdomaines sur l’agrégation plaquettaire ... 39
2. Rôle des microdomaines dans la voie de l’AMPc... 40
II. Caractérisation des microdomaines ... 43
1. Isolement des microdomaines : mise au point et validation de la technique ... 43
2. Microdomaines et effecteurs de la voie de l’AMPc ... 48
III. Visualisation et co-localisation in situ des différents effecteurs de la voie de signalisation de l’AMPc .. 50
1. Immunofluorescence ... 50
2. Mise en évidence d’une co-localisation des protéines d’intérêts ... 51
DISCUSSION ... 53
Liste des illustrations
Figure 1 : Les différentes étapes de la mégacaryocytopoièse humaineFigure 2 : Principaux récepteurs plaquettaires des protéines adhésives et des agonistes
solubles ainsi que leur ligand.
Figure 3 : Structure de la plaquette sanguine
Figure 4 : La cellule endothéliale : rôle inhibiteur (cellule au repos) ou activateur (cellule
activée) de l’hémostase.
Figure 5 : Les 3 étapes de l’hémostase primaire Figure 6 : Les mécanismes de l’hémostase primaire
Figure 7 : Inhibition plaquettaire induite par la prostacycline et l’adénosine Figure 8 : Activation et inhibition plaquettaire régulées par la voie de l’AMPc Figure 9 : Régulation du taux d’AMPc au sein des plaquettes sanguines Figure 10 : Les microdomaines
Figure 11 : Etapes successives de la technique Duolink
Figure 12 : Pourcentage d'agrégation au PAR1-ap à 5 µM en fonction de la concentration en
MCD en mM
Figure 13 : Pourcentage d'agrégation au PAR1-ap à 5 µM en fonction de la concentration en
forskoline
Figure 14 : Comparaison des pourcentages d’inhibition en présence (bleu) ou en absence
(noir) de forskoline 0,1 µM à différentes doses de MCD (n=6)
Figure 15 : Etude de l’effet inhibiteur de différentes doses de MCD, en présence (rouge) ou en absence (noir) de forskoline 0,25 µM, sur l’agrégation induite par PAR1-ap (5 µM)
Figure 16 : Isolement des microdomaines dans le gradient de sucrose après
ultracentrifugation
Figure 17 : Isolement des microdomaines (Raft) des plaquettes au repos
Figure 18 : Isolement des microdomaines d’une suspension de plaquettes activées Figure 19 : Isolement des microdomaines (Rafts) des plaquettes au repos préalablement
incubées au MCD (0,5mM)
Figure 22 : Mise en évidence des fractions contenant différents acteurs de la voie de l’AMPc
après activation plaquettaire
Figure 23 : Mise en évidence des fractions contenant différents acteurs de la voie de l’AMPc
suite à l’incubation des plaquettes au MCD
Figure 24 : Mise en évidence d’une co-localisation entre différents acteurs de la voie de
signalisation de l’AMPc
Figure 25 : Localisation de MRP4 après activation plaquettaire
Figure 26 : Hypothèse de la localisation de MRP4 dans les plaquettes au repos
Figure 27 : Hypothèse de la voie de signalisation de l’AMPc après désorganisation des
microdomaines par du MCD dans les plaquettes au repos
Abréviations
AC : adénylate cyclase ADP : adénosine diphosphate
AKAPs : “A-kinase anchoring protein” AMPc : adénosine monophosphate cyclique ATP : adenosine triphosphate
AVC : accident vasculaire cérébral COX : cyclo-oxygénase
DO : densité optique DTT : diThioThreitol
EFS : établissement français du sang Fg : fibrinogène
Fk : forskoline
FP4 : facteur plaquettaire 4 GAG : glycoaminoglycanes GC : guanylate cyclase
GMPc : guanosine monophosphate cyclique GP : glycoprotéine
GPI : glycosylphosphatidylinositol LAT : “linker for activated T-cells” MCD : methyl--cyclodextrine MK : mégacaryocyte
MRP4 : “multidrug resistance protein 4” NO : monoxide d’azote
PDE : phosphodiestérase PGI2 : prostacycline PKA : protéine kinase A PKG : protéine kinase G
PT : lysat plaquettaire total TCA : acide trichloroacétique TpL : tampon de lavage
TpR : tampon de re-suspension TTBS : Tampon Tris avec Tween 20
VASP : “vasodilator-stimulated phosphoprotein” vWF : factor willebrand
Introduction
I.
Les plaquettes
Les plaquettes sanguines (ou thrombocytes) sont de petits fragments cellulaires anucléés de forme discoïde au repos qui possèdent de nombreux organelles : granules , granules denses et lysosomes.
Elles ont pour fonction principale le maintien de l’intégrité vasculaire et, à ce titre, jouent un rôle majeur dans l’hémostase.
Ces petites cellules de 2 à 5 µm de diamètre ont une durée de vie de 8 à 10 jours puis sont éliminées par la rate. Leur taux normal dans le sang circulant se situe entre 150 et 450.109/L. Les plaquettes sont issues de la fragmentation cytoplasmique de leur précurseur médullaire : les mégacaryocytes (Patel, Hartwig, and Italiano 2005).
1. Production
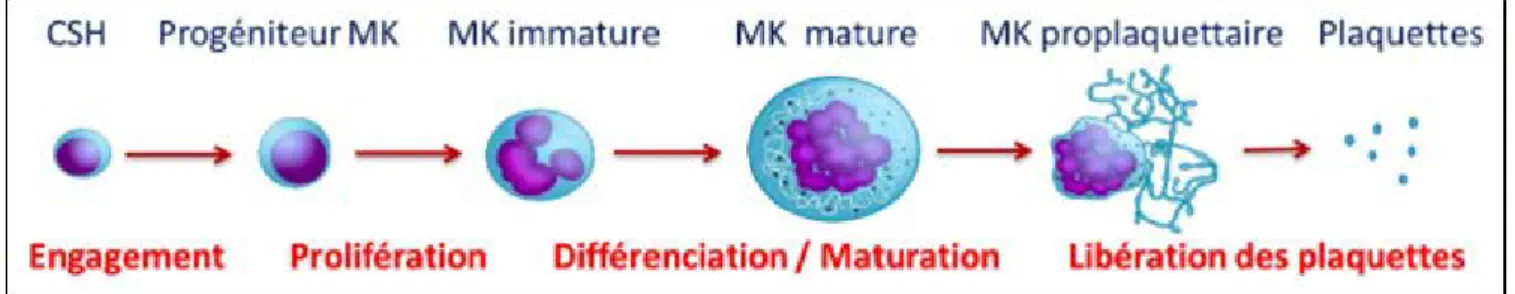
Chaque jour, environ 1011 plaquettes sont produites suite à la fragmentation du cytoplasme des mégacaryocytes (MK). Ces derniers sont des cellules géantes myéloïdes dont le diamètre varie entre 50 et 120 µm.
Les mégacaryocytes se retrouvent principalement dans la moelle osseuse mais sont également retrouvés dans le sang périphérique et les poumons.
Lors de La mégacaryopoïèse, qui est l’ensemble des processus de production des plaquettes, les progéniteurs mégacaryocytaires vont subir différentes étapes de prolifération et de différentiation pour aboutir aux mégacaryocytes matures puis aux plaquettes (Figure 1).
Au cours de ces étapes, le mégacaryocyte devient de moins en moins prolifératif et va subir des endomitoses qui va le rendre polyploïde (de 4N à 64N). La différentiation mégacaryocytaire s’accompagne également de la maturation du cytoplasme. Cette maturation se traduit par une augmentation du volume cytoplasmique ainsi que par la formation des futurs granules plaquettaires et des membranes de démarcation qui sont un
réservoir membranaire nécessaire à la formation des futures plaquettes (Corash and Levin 1990)
Les mégacaryocytes matures migrent jusqu’au flux sanguin à travers les sinusoïdes médullaires. Cette migration contraint les mégacaryocytes à se déformer et à former des protubérances pour passer dans le compartiment vasculaire (Avecilla et al. 2004). Sous l’effet du flux sanguin, les protubérances des mégacaryocytes se fragmentent alors en « proplaquettes », état intermédiaire de la formation des plaquettes, puis en plaquettes (Junt et al. 2007).
Toutefois, si l’observation des fragments de mégacaryocytes et de plaquettes au niveau de la sortie des sinusoïdes médullaires est largement rapportée dans la littérature (Behnke and Forer 1998 ; Junt et al. 2007 ; Becker and De Bruyn 1976), un autre site de formation des plaquettes a été identifié.
En effet, suite à la mise en évidence de mégacaryocytes et de proplaquettes au niveau des poumons (Levine et al. 1993), des équipes ont démontré que le fort taux de cisaillement au sein des artérioles pulmonaires serait à l’origine de la formation d’environ 50% des plaquettes (Lefrançais et al. 2017 ; Zucker-Franklin and Philipp 2000).
Figure 1. Les différentes étapes de la mégacaryocytopoièse humaine
Ce schéma représente les étapes de la mégacaryocytopoièse à partir de la cellule souche hématopoïétique (CSH), présente dans la moelle osseuse, jusqu’aux plaquettes libérées dans le sang.
2. Structure
Les plaquettes sont les plus petits éléments figurés du sang circulant. A l’état non activé, elles ont une forme discoïde. Cependant, suite à leur activation, la morphologie des plaquettes change. Elles deviennent sphériques et émettent des pseudopodes.
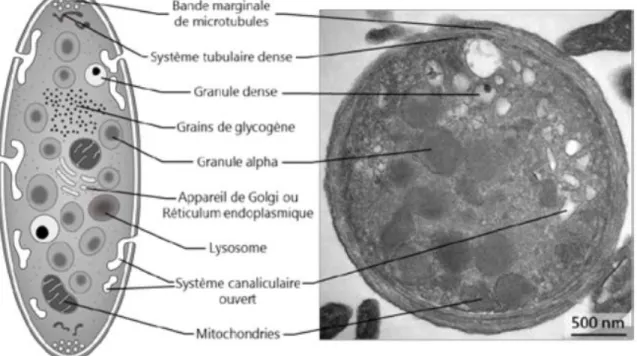
Les plaquettes sont composées d’une zone périphérique qui comprend le glycocalyx, la membrane plasmique ainsi que les systèmes membranaires dont le système canaliculaire ouvert et d’une zone interne le cytoplasme qui en plus d’être constitué d’un cytosquelette comprend différents types d’organelles.
a. Zone Périphérique
Le glycocalyx est un revêtement de surface situé à l’extérieur de la membrane plasmique. C’est une couche irrégulière dont l’épaisseur varie de 10 à 50 nm et qui est constitué de glycosaminoglycanes (GAG). C’est le premier site d’interaction des plaquettes avec l’environnement extérieur.
Nous retrouvons ensuite la membrane plasmique constituée d’une bicouche lipidique. Cette bicouche est constituée principalement de phospholipides (80%) distribués de façon asymétrique.
Sont également insérés dans cette bicouche différents récepteurs dont les glycoprotéines impliquées dans la fonction plaquettaire (Figure 2).
Figure 2. Principaux récepteurs plaquettaires des protéines adhésives et des agonistes solubles ainsi que leur ligand.
Au niveau de la membrane plasmique des plaquettes on retrouve des récepteurs des protéines adhésives dont les principaux sont les suivant : GPVI, GPIV, GP Ib-IX-V, IIbainsi que des récepteurs des agonistes solubles : TP, 5HT2a, PAR1, PAR4, IP, P2Y12, P2Y1. Ce schéma représente également les principaux ligands de ces récepteurs.
Thromboxane A2 (TXA2), sérotonine (5HT), facteur willebrand (vWF), prostacycline (PGI2), adénosine diphosphate (ADP).
La membrane plasmique présente également de nombreuses invaginations ouvertes sur l’extérieur formant ainsi le système canaliculaire ouvert. Ce système joue un rôle dans l’absorption de molécules par la plaquette et participe, lors de l’activation des plaquettes, à la libération du contenu granulaire.
De la même manière au niveau du cytoplasme plaquettaire, nous pouvons distinguer le système tubulaire dense, réseau de membrane issu du réticulum endoplasmique lisse d’origine mégacaryocytaire. Ce dernier a pour fonction la régulation de la concentration du calcium (Ca2+) plaquettaire et joue ainsi un rôle important dans l’activation (Ebbeling et al. 1992).
b. Le cytoplasme
Le cytoplasme plaquettaire est constitué à sa périphérie d’un cytosquelette. Ce dernier fournit à la membrane plasmique une charpente solide et flexible. Il est composé principalement de microtubules, responsables de la forme discoïde des plaquettes au repos et de microfilaments d’actine qui lors de l’activation plaquettaire, se réorganisent pour permettre le changement de forme des plaquettes (Hartwig 1992).
En plus de son rôle d’échafaudage cellulaire, le cytosquelette se révèle être un lieu capital pour la relocalisation de nombreux complexes protéiques.
Les plaquettes renferment également dans leur cytoplasme de nombreux organelles tel que des mitochondries, des grains de glycogène et plusieurs types de granules (les granules alpha, denses, et les lysosomes).
Lors de l’activation plaquettaire, la fusion des membranes des granules avec la membrane plasmique induit la sécrétion de molécules qui participent aux différents mécanismes de l’hémostase.
i. Les granules alpha
Prépondérantes par leur nombre (environ 100 par plaquettes humaines) et par leur taille (0,2 à 0,5 µm), les granules alpha contiennent de nombreuses protéines dont le facteur plaquettaire 4 (PF4), des facteurs de coagulation et des protéines adhésives (FV, FXI, FXIII, Fg, vWF…) et des facteurs de croissance (PDGF, TGF-β, EGF…).
ii. Les granules denses
Nommées ainsi car elles apparaissent denses aux électrons en microscopie électronique, les granules denses sont le lieu de stockage de molécules de nature non protéique. Elles contiennent de fortes concentrations d’adénosine triphosphate (ATP), d’adénosine diphosphate (ADP) et de nucléotides cycliques mais aussi des acides aminés comme la sérotonine ou l’histamine ainsi que cations bivalents comme le Ca2+, Mg2+ (Magnésium) et le pyrophosphate.
iii. Les lysosomes
Représentant la 3ème catégorie de granules plaquettaires, les lysosomes, dont la taille est intermédiaire aux granules et denses, contiennent des hydrolases acides capables d’éliminer l’agrégat plaquettaire circulant (Rendu and Brohard-Bohn 2001).
Bien que la libération du contenu lysosomale présente des mécanismes analogues aux granules présentées ci-dessus, la sécrétion du lysosome nécessite une très forte stimulation plaquettaire (Greenberg-Sepersky and Simons 1985).
Figure 3. Structure de la plaquette sanguine
A gauche schéma d’une plaquette en coupe transversale (B. Boneu et J_P Cazenave « Introduction à l’étude de l’hémostase et de la thrombose » deuxième édition (1997).
A droite photographie d’une coupe frontale d’une plaquette sanguine en microscopie électronique à transmission (Image : Fabien Pertuy, UMR_S949, EFS-Alsace).
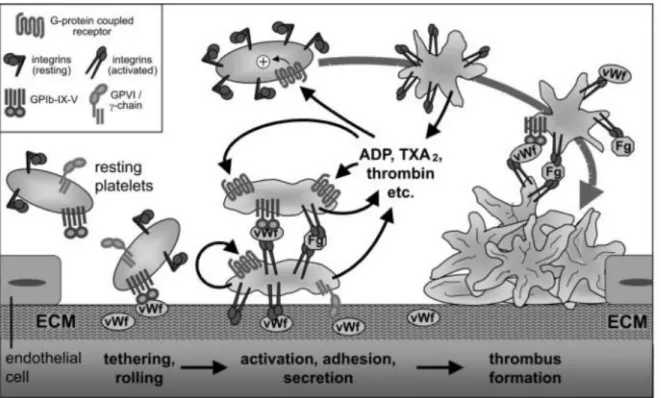
3. L’hémostase primaire : principale fonction plaquettaire
Les plaquettes ont pour fonction principale le maintien de l’intégrité vasculaire et, à ce titre, jouent un rôle majeur dans l’hémostase. Il s’agit du phénomène physiologique activé lors d’une brèche vasculaire et qui aboutit à l’arrêt du saignement grâce à la formation d’un thrombus occlusif.
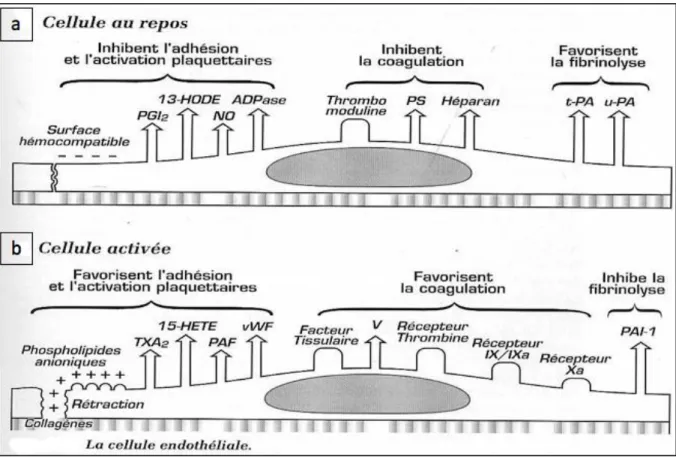
En condition physiologique, plusieurs facteurs permettent de maintenir les plaquettes à l’état de repos dans la circulation : le maintien du flux sanguin ainsi que les cellules endothéliales. Le sang circule effectivement en contact permanent avec une monocouche de cellules endothéliales dont un des rôles majeurs est de faciliter l’écoulement sanguin en fournissant une surface antithrombogène. En effet, elles expriment à leur surface du CD39 qui possède une activité ADPase, permettant la dégradation de l’ADP, amplificateur de la réponse plaquettaire. De plus, les cellules endothéliales sécrètent de la prostaglandine I2 (prostacycline, PGI2) ainsi que du monoxyde d’azote (NO), deux molécules activatrices de deux voies de signalisations inhibitrices des plaquettes (figure 4a). La voie de l’AMPc est principalement activée par la PGI2 et celle du GMPc par le NO.
Lorsque l’endothélium vasculaire est lésé, les cellules endothéliales s’activent et acquièrent un phénotype prothrombotique. Elles sécrètent alors des molécules favorisant l’adhésion et l’activation plaquettaire comme le thromboxane A2 (TXA2) et le facteur willebrand (vWF) (figure 4b).
Figure 4. La cellule endothéliale : rôle inhibiteur (cellule au repos) ou activateur (cellule activée) de l’hémostase.
Lorsque les cellules endothéliales sont au repos, elles sécrètent des substances ou expriment à leur surface des protéines favorisant l’inhibition de la formation du caillot, l’hémostase primaire et la coagulation, et favorisant sa destruction, la fibrinolyse (a).
Suite à une lésion vasculaire, les cellules endothéliales vont s’activer et devenir prothrombogène. Elles sécrètent alors des substances favorisant l’hémostase primaire et la coagulation et favorisant l’inhibition de la fibrinolyse (b).
Par ailleurs, suite à cette lésion, le sous-endothélium est exposé au flux sanguin et les plaquettes adhèrent à cette surface riche en protéines adhésives, dont le collagène et le vWF. Il s’en suit alors des étapes d’activation, de sécrétion puis d’agrégation plaquettaire, dont l’ensemble de ces processus correspond à l’hémostase primaire (Picker 2011).
Figure 5. Les 3 étapes de l’hémostase primaire
GPIIb-IIIa (intégrine IIb3), GPIa IIa (intégrine 21)
a. Adhésion plaquettaire
Suite à la mise à nue du sous-endothélium, les protéines d’adhésion, alors en contact avec la circulation sanguine, sont soumises à des forces de cisaillement élevées dans la microcirculation et les artères.
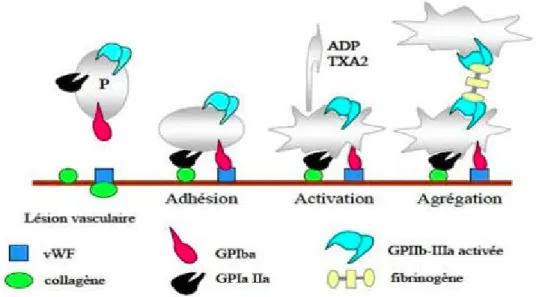
L’une d’elle appelée facteur Willebrand va alors changer de conformation ce qui va augmenter son affinité pour son récepteur plaquettaire la GPIb (Bergmeier et al. 2006). Cette adhérence permet le recrutement des plaquettes au niveau de la brèche vasculaire. Cependant, cette interaction n’aboutit pas à elle seule à une adhérence totale des plaquettes mais à un phénomène de roulement sur l’endothélium suivit d’un ralentissement. C’est ce ralentissement qui va permettre, par l’intermédiaire d’intégrines plaquettaires, l’adhésion irréversible des plaquettes : interaction du vWF avec l’intégrine IIb3 et du collagène avec l’intégrine 21 et la glycoprotéine GPVI
L’adhésion irréversible des plaquettes déclenche alors une signalisation cellulaire entrainant l’activation plaquettaire.
b. Activation plaquettaire
L’activation des plaquettes est caractérisée par deux phénomènes principaux : leur changement de forme et leur activation métabolique.
Cette activation est un processus actif nécessitant de l’énergie sous forme d’ATP, et la disponibilité cytosolique de calcium.
En effet, L’augmentation du Ca2+ est indispensable à l’activation et à l’agrégation plaquettaire car de nombreux mécanismes sont dépendants de la présence et de la liaison aux ions calciques.
Lors de l’activation, les plaquettes initialement discoïdes deviennent sphériques et s’étalent sur la surface d’adhésion formant des filopodes puis des lamellipodes, témoignant de la réorganisation du cytosquelette.
Parallèlement à ces modifications morphologiques, l’activation entraîne une sécrétion plaquettaire par deux mécanismes distincts : la sécrétion du contenu granulaire par fusion des granules avec la membrane plasmique ou le système canaliculaire ouvert (Rendu and Brohard-Bohn 2001) et la sécrétion de molécules néosynthétisées.
Ces phénomènes de sécrétion libèrent de nombreuses substances proagrégantes (ADP, thromboxane A2 (TXA2), sérotonine), mais aussi procoagulantes (ex : facteur V) ou vasomotrices (sérotonine, NO, TXA2) contribuant à l’élaboration d’un environnement favorable à l’agrégation plaquettaire et à la réalisation de la coagulation.
En effet, la fixation d’agonistes plaquettaires comme l’ADP (contenu dans les granules denses) et le TXA2 (provenant de l’hydrolyse des phospholipides membranaires (Hamberg, Svensson, and Samuelsson 1975)) à leurs récepteurs plaquettaires respectifs, P2Y12 et P2Y1 pour l’ADP et le TP pour le TXA2, entrainent l’activation de voies de signalisation à l’origine de l’amplification de l’activation plaquettaire et du recrutement des plaquettes circulantes nécessaire à l’agrégation.
c. Agrégation
L’agrégation plaquettaire, formée par des plaquettes adhérentes les unes aux autres, résulte de la formation d’un pont de fibrinogène entre deux récepteurs IIb3
préalablement activés à la surface de deux plaquettes (Bennett 2005)
En effet, suite à l’activation des plaquettes, l'intégrine IIb3 (ou GPIIb-IIIa) subit des changements conformationnels qui lui permettent de fixer le fibrinogène. Ces changements sont dus à une voie de signalisation appelée « inside-out ».
Cette voie « inside-out » correspond aux mécanismes de signalisation responsables de diverses modifications d’interaction intra et extra moléculaire du domaine intracellulaire et consécutivement d’un changement de conformation du domaine extracellulaire (Shattil, Kim, and Ginsberg 2010).
L’intégrine IIb3 acquière alors une conformation de forte affinité pour ses ligands, lui permettant de fixer le fibrinogène pour former, en présence de Ca2+, des ponts interplaquettaires qui permettront la formation de l'agrégat.
Figure 6. Les mécanismes de l’hémostase primaire.
Le sous-endothélium pro-thrombogène est exposé lors d’une brèche vasculaire. Le vWF interagit avec les plaquettes provoquant leur
ralentissement, leur arrêt et leur activation. Les plaquettes sécrètent et activent les plaquettes environnantes permettant leur recrutement, leur activation, et la formation d’un thrombus (Offermanns 2006).
II.
Régulation de l’activation plaquettaire médié par les
nucléotides cycliques
En situation physiologique, il existe un équilibre entre les signaux qui activent les plaquettes et ceux qui les maintiennent au repos (Nolte et al. 1994). Ainsi, pour éviter toute activation illégitime des plaquettes à l’origine de thrombose, les cellules endothéliales sécrètent des substances inhibitrices de l’activation plaquettaire comme la PGI2 et le NO. Ces derniers sont à l’origine de la production de puissants inhibiteurs de l’agrégation plaquettaire : les nucléotides cycliques.
1. Synthèse des nucléotides cycliques
La découverte de l’adénosine monophosphate cyclique (AMPc) en 1957 a valu à Earl W. Sutherland le prix Nobel de physiologie et de médecine en 1971. Ces travaux ont permis de développer le concept de second messager dont l’AMPc reste prépondérant. L’AMPc est le premier nucléotide cyclique décrit comme étant un puissant inhibiteur plaquettaire. Il a rapidement été montré que sa production est associée à la sécrétion de la prostacycline par les cellules endothéliales (Radomski, Palmer, and Moncada 1987; Schwarz, Walter, and Eigenthaler 2001).
Les plaquettes possèdent également des récepteurs à l’adénosine (récepteurs A2A et A2B) qui, lorsqu’ils sont activés, sont également capable d’induire la synthèse d’AMPc (Johnston-Cox, Yang, and Ravid 2011).
La fixation de la prostacycline et de l’adénosine à leurs récepteurs respectifs couplés à une protéine G (RCPG) (récepteur IP pour la prostacycline et récepteurs A2A/A2B pour l’adénosine), entraine la dissociation de la sous-unité avec les sous-unités de la protéine G associée (Gs). La sous-unité libérée, devient alors capable d’activer l’adénylate cyclase qui synthétise l’AMPc à partir de l’ATP (figure 7).
Figure 7. Inhibition plaquettaire induite par la prostacycline et l’adénosine
Dans les années 1980, un autre nucléotide cyclique, la guanosine monophosphate cyclique (GMPc), a été impliqué dans les mécanismes d’inhibition de l’activation plaquettaire. L’endothélium non-activé, anti-thrombogène, sécrète du NO qui pénètre dans les plaquettes par diffusion et induit l’activation de la guanylate cyclase (GC) et consécutivement la synthèse de GMPc à partir du GTP (Mellion et al. 1981; Radomski, Palmer, and Moncada 1987).
Ces nucléotides cycliques activent respectivement la protéine kinase A (PKA) et la protéine kinase G (PKG) qui possèdent de nombreuses protéines cibles. Leur activation permet de maintenir les plaquettes dans un état de repos.
Les agents pharmacologiques modulant les concentrations des nucléotides cycliques plaquettaires tel que les inhibiteurs de la voie de l’ADP, les analogues de la prostacycline ou encore les inhibiteurs des phosphodiestérases, se révèlent efficaces comme inhibiteurs de l’activation plaquettaire et sont utilisés dans la prévention des événements cardiovasculaires thrombotiques.
2. Voie de signalisation de l’AMPc
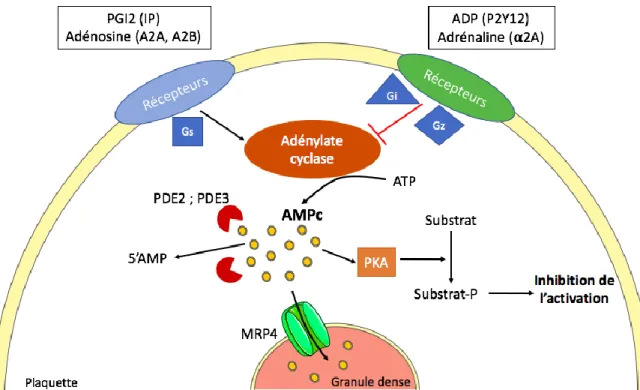
Comme décrit précédemment (chapitre Introduction II/1), l’activation de la voie de signalisation de l’AMPc au sein des plaquettes entraine l’inhibition plaquettaire. En effet, La PGI2 se fixe sur son récepteur plaquettaire IP pour entraîner, via la protéine Gs, une activation de AC à l’origine d’une augmentation de la concentration en AMPc, qui active la protéine kinase A. Cette dernière est formée de 4 sous-unités : 2 sous unités régulatrices (qui permettent de classer les PKA en deux catégories : de type I et de type II) et de deux sous unités catalytiques. La liaison de 4 molécules d’AMPc aux sous unités régulatrices libère les sous-unités catalytiques qui deviennent actives et peuvent alors phosphoryler de nombreux substrats comme la protéine VASP (vasodilator phosphoprotein) qui, sous forme phosphorylée (P-VASP), favorise le maintien au repos de l’intégrine αIIb3.
En revanche, lors d’une brèche vasculaire, les plaquettes s’activent et libèrent de l’ADP. Cet activateur plaquettaire se fixe sur ses récepteurs, P2Y1, couplé à une Gq, et P2Y12 couplé à une protéine Gi qui entraîne l’inhibition de l’AC. Cette inhibition est responsable de la diminution de la concentration en AMPc, ce qui inhibe l’activation de la PKA et permet l’activation d’αIIb3, la fixation du fibrinogène et finalement l’agrégation plaquettaire. Il existe par conséquent une balance entre état d’activation et état de repos des plaquettes régulée en partie par la voie de signalisation de l’AMPc (Smolenski 2012).
Figure 8. Activation et inhibition plaquettaire régulées par la voie de l’AMPc
L’ADP, agoniste de l’activation plaquettaire, entraine une diminution de synthèse de l’AMPc cytosolique et permet l’agrégation plaquettaire (en rouge, contrairement à la PGI2, inhibiteur plaquettaire, qui induit la synthèse d’AMPc cytosolique et ainsi le maintien des plaquettes au repos (en vert).
3. Régulation du taux d’AMPc au sein des plaquettes
La concentration plaquettaire d’AMPc joue donc un rôle crucial dans la balance « activation/inhibition » plaquettaire. Cette concentration est contrôlée par l’activité de synthèse des cyclases, mais également par l’activité des phosphodiestérases (PDE) qui hydrolysent les nucléotides cycliques et notamment l’AMPc en 5’AMP. En effet, Les plaquettes possèdent trois isoformes de PDE (PDE2, PDE3 et PDE5), avec une sélectivité différente pour l’AMPc et le GMPc (Burkhart et al. 2012).
La PDE3 et la PDE5 ont la capacité de dégrader respectivement l’AMPc et le GMPc.
La PDE2, contrairement aux deux autres, peut dégrader de manière équivalente l’AMPc et le GMPc.
Toutefois le GMPc exerce lui aussi une action sur les phosphodiestérases. En effet, il a la capacité de stimuler l’activité d’une enzyme qui le dégrade, la PDE2, et d’inhiber la PDE3.
Plus récemment, un rôle dans le contrôle de la concentration cytosolique de l’AMPc a également été attribué à la protéine d’efflux MRP4 par les travaux de notre équipe (Decouture et al. 2015). En effet, les transporteurs MRP4 (Multidrug Resistance Protein), membres de la famille des transporteurs ABC (ATP Binding Cassette), permettent la déplétion de l’AMPc du cytosol et limite ainsi l’action de l’AMPc sur ses effecteurs.
Figure 9. Régulation du taux d’AMPc au sein des plaquettes sanguines
Par ailleurs, un nouveau mécanisme a également récemment été proposé pour expliquer la régulation de la signalisation de l’AMPc : la compartimentation.
III. Les microdomaines
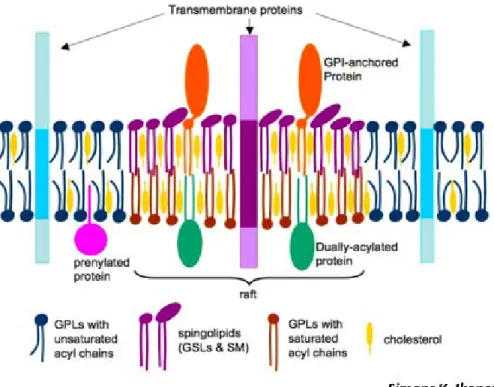
1. Structure
La membrane plasmique a longtemps été considérée comme une structure lipidique homogène. Il s’agit en fait d’une structure hétérogène dynamique marquée par la présence de radeaux lipidiques également appelés microdomaines (figure 10). Ces derniers sont des zones plus ou moins enrichies en lipides spécifiques comme le cholestérol, les sphingolipides et les phospholipides, qui se caractérisent par leur insolubilité dans les détergents doux (détergeant non ionique à faible concentration) ainsi que par leur faible densité (le terme « flottabilité » est utilisée lors de l’isolement par gradient de densité). Ils contrôlent et contiennent sélectivement certains acteurs de la signalisation plutôt que d’autres. Une
définition des rafts a été proposée en 2006 lors du congrès international "Keystone symposium on lipid rafts and cell function" : « Les radeaux lipidiques sont des nanodomaines de la membranes (10-200 nm), hétérogènes, très dynamiques, enrichis en stérols et sphingolipides, qui compartimentent les processus cellulaires. Ils sont capables de former des domaines microscopiques (>300nm) lors de leur regroupement induit par des interactions protéines-protéines ou protéines-lipides » (Pike 2006).
Figure 10. Les microdomaines
Les microdomaines sont des zones enrichies en sphingolipides (en violet) et en cholestérol (en jaune) et contiennent sélectivement certains acteurs de voie de signalisation comme les protéines ancrées au glycosylphosphatidylinositol (GPI) (en orange)
2. Microdomaines et voie de signalisation de l’AMPc
En maintenant certains acteurs de la signalisation plutôt que d’autres, les microdomaines ont la capacité de contrôler certaines voies de signalisation. Ainsi, la signalisation AMPc/PKA peut également être régulée de manière spatiale.
Des équipes ont montré dans divers types cellulaires l’importance de microdomaines dans la voie de signalisation de l’AMPc. Par exemple dans les cardiomyocytes, l’utilisation de
donc les microdomaines, a permis de mettre en évidence leurs importance. La perturbation de cette compartimentation de la voie récepteur β adrénergiques /AMPc, entraine une dérégulation de la signalisation à l’origine d’une hypertrophie cardiaque (Berthouze et al. 2011).
La capacité de ces microdomaines à contrôler ces voies de signalisation dépend essentiellement de protéines spécifiques qu’ils contiennent, comme les AKAPs (A-Kinase Anchoring Protein).
En effet, certaines de ces protéines, dont une cinquantaine ont actuellement été recensées dans de nombreux types cellulaires (Carnegie, Means, and Scott 2009), sont localisées au niveau de ces microdomaines.
D’un point de vue structural, les AKAPs ont un domaine leur permettant de cibler leur localisation cellulaire vers des structures subcellulaires définies tels que les microdomaines.
Elles sont également capables de fixer de nombreuses protéines impliquées dans la régulation de la signalisation de l’AMPc, telles que des PDEs, des phosphatases et/ou la PKA. Ces protéines permettent donc de créer un pool d’AMPc et ainsi de localiser et de contrôler sa signalisation. Les AKAPs sont donc le support de complexes protéiques de signalisation de l’AMPc.
3. Microdomaines dans les plaquettes
Dans les plaquettes, un rôle dans la régulation de voies de signalisation a été attribué aux microdomaines : Celle de la voie médiée par le récepteur P2Y12 (Quinton et al. 2005) et celle médiée par le récepteur FcRIIa (Bodin et al. 2003). De plus, une équipe s’est intéressée à la relation entre ces microdomaines et l’AMPc. Ces premières données sont en faveur de l’existence dans les plaquettes de structures équivalentes à celles décrites dans les autres types cellulaires avec ici une régulation spatiale d’un isoforme de PKA, la PKA I, assurée par la moesin une AKAPs, localisés aux sein des microdomaines (Raslan et al. 2015). Par cette étude, Raslan montre que cette régulation spatiale favoriserait l’inhibition plaquettaire par l’augmentation de la phosphorylation d’une protéine d’ancrage, la GPIb.
Ainsi, les microdomaines pourraient réguler la disponibilité et/ou la répartition de l’AMPc au niveau de la membrane des plaquettes. Ils pourraient favoriser le maintien de l’état de repos des plaquettes en rapprochant les différents acteurs de la signalisation de l’AMPc comme le propose Raslan, ou, au contraire, faciliter l’activation plaquettaire en éloignant ou en formant une barrière entre substrats et effecteurs de cette signalisation.
IV. But de l’étude
Le but de cette étude est, dans un premier temps, de mettre en évidence le rôle des microdomaines dans l’activation plaquettaire contrôlée par la voie de l’AMPc, puis dans un deuxième temps, de caractériser ces microdomaines.
Pour la première partie, des tests d’agrégations utilisant un perturbateur du cholestérol ont été réalisés en présence d’un activateur direct de la voie de l’AMPc afin d’établir le lien entre les microdomaines et la concentration cytosolique en l’AMPc.
Dans la deuxième partie de l’étude, suite à la mise au point de la méthode d’isolement des microdomaines plaquettaires, nous avons mis en évidence, par western blot, la présence ou l’absence d’effecteurs de la voie de signalisation de l’AMPc dans les microdomaines. Pour finir, ce travail a été complété par une étude permettant de confirmer et visualiser in situ une co-localisation des effecteurs de cette voie dans les plaquettes.
Matériels & méthodes
I.
Matériels
1. Solutions et Tampons
Tampon dénaturant : LDS Sample Buffer (NuPAGE® NP0008) Tampon de migration : MES SDS Run Buffer (20X) (NuPAGE®) Agent réducteur : DiThioThreitol DTT (NuPAGE® NP0009) Tampon phosphate salin (PBS) (Gibco®)
Fluorinerte.
Milieu de montage (immunofluorescence) : mowiol 4-88 (2,4g), glycérol (6g), Tris (0,2 M pH=8 12 mL), H2O (6mL), DABCO (1,4-diazabicyclo [2.2.2] octane ; 0,66g) Solution conservée à -20°C
Solution de saturation : Lait en poudre, écrémé, (Régilait®) (5 g/100mL)
Tris-buffered saline avec Tween 20 (TTBS) 10X : Tris 200 mM, NaCl 1,36 M, Tween 20 2%, pH ajusté à 7,5
Tampon de lavage plaquettaire (TpL) : 36 mM d’acide citrique, 5 mM de glucose, 5 mM de KCl, 2 mM CaCl2, 1 mM de MgCl2 et 103 mM de NaCl
Tampon de re-suspension plaquettaire (TpR) : 10 mM d’HEPES, 140 mM de NaCl, 3 mM de KCl, 5 mM de NaHCO3, 0,5 mM de MgCl2, 10 mM de glucose
Tampon de lyse 2X : 20 mM de Tris à pH 8, 100 mM de NaCl, 60 mM de pyrophosphate de sodium, 20 mM de glycerophosphate de sodium, 0,02% d’azide de sodium, 2 mM de vanadate de sodium, un cocktail inhibiteur de la protéase et différentes concentrations de Triton X-100 allant de 2% à 0,2% (w/v)
2. Réactifs
Inhibiteurs plaquettaires utilisés lors de la préparation des plaquettes lavées : o PGE1 (Sigma Aldrich® P5515) solution stock à 5 mM conservée à -20°C o Apyrase (AGROBIO®) solution stock à 170 Ui/mL conservé à -20°C
Activateur de l’adénylate cyclase : forskoline (Sigma Aldrich®) Cocktail inhibiteur de protéase : DMSO PICO2 100X (Novex®) Antioxydant : NP0005 (Novex®)
Rouge ponceau 0,4 % dans de l’acide acétique 5%
Peptide activateur de PAR1 (principal récepteur de la thrombine sur les plaquettes humaines) : PAR1-ap (Bachem®) solution stock à 1mM conservée à -20°C
Agent permettant la déplétion du cholestérol : Méthyl-β-cyclodextrine (MβCD) (Sigma Aldrich®)
Acide trichloroacétique (TCA) (Sigma Aldrich)
Phalloïdine solution stock conservée à -20°C (Thermo Fisher)
3. Kit et matériel
Gel utilisé pour la technique Western Blot : Gel acrylamide, Tris Acetate 3-8% (Invitrogen®)
Plaques pour les tests d’agrégation : microplaques de 96 demi-puits à fond plat (Greiner)
Technologie de ligation de proximité : Kit Duolink (Sigma Aldrich®)
4. Anticorps
Anti-LAT : anticorps polyclonal de lapin (Upstate®) 06-807
Anti-CD36 : anticorps polyclonal de lapin (Novusbio®) NB400-144
Anti-ß3 intégrine : anticorps produit par Dr. Dominique Pidard à partir de sérum de lapin
Anti-Syk : anticorps monoclonal murin Syk Ab-1 (4D10.1) (NeoMarkers)
Anti-MRP4 : anticorps polyclonal de lapin (Abcam®) 180712 (WB); Anticorps polyclonal de lapin (Sigma Aldrich®) HPA002476 (IF)
Anti-PDE2A : anticorps polyclonal de lapin (Antibodies-online) ABIN2747086 Anti-PKA-RI : anticorps polyclonal de lapin (Cell Signaling®) 3927S
Anti-adenyl cyclase V/VI : Anticorps monoclonal murin (Santa Cruz Biotechnology®) sc-514785
Anticorps utilisés pour l’immunofluorescence :
o Anticorps secondaire anti-anticorps murin : f(ab)’2 chèvre anti-murin alexa fluor 488 (Molecular probes)
o Anticorps secondaire anti-anticorps de lapin : f(ab)’2 chèvre anti-lapin alexa fluor 488 (Molecular probes)
Anticorps utilisés pour les Western Blot :
o Anticorps secondaire anti-anticorps de lapin : anticorps de chèvre anti-IgG lapin DylightTM 800 4X PEG conjugaison (Thermo Scientific®)
o Anticorps secondaire anti-anticorps murin (Odyssey) : anticorps de chèvre anti-IgG murin dylight 680 (Sigma-Aldrich®)
II.
Méthodes
1. Préparation des plaquettes lavées
Le sang humain, prélevé à l’établissement français du sang (EFS) (convention C CPSL UNT N° 12/EFS/064), est recueilli dans des tubes contenant un anticoagulant : l’ACD (Acide citrique 4.7 mM, citrate trisodique 11.2 mM, et glucose 20 mM, concentration finale).
Le sang total (2 volumes), est dilué avec du TpL (1 volume) en présence de deux inhibiteurs, l’apyrase (concentration finale de 0,06 U/mL) et la PGE1 (concentration finale de 2.10-7 M). Après centrifugation (216 g, 11 min à 22°C), le plasma riche en plaquettes (PRP) est récupéré, dilué avec du TpL (3 vol de PRP/ 1 vol de TpL en présence d’apyrase et PGE1) puis centrifugé (1243 g, 12 minutes, à 22°C). Pour optimiser le lavage, le culot plaquettaire est de nouveau dilué avec du TpL (contenant de l’apyrase et la PGE1) puis centrifugé (1243 g, 12 minutes, à 22°C). Le culot est finalement remis en suspension dans du TpR (2 mL) et, à l’aide de l’automate Cell Coulter Z1, la concentration plaquettaire est mesurée puis ajustée soit à 1,2.109 plaquettes/mL pour l’isolement des microdomaines, soit à 3,5.108 plaquettes/mL pour les tests d'agrégation plaquettaires.
Pour terminer, 2 mM de CaCl2 sont ajoutés à la suspension plaquettaire qui est ensuite incubée 30 minutes à température ambiante.
2. Isolement des radeaux lipidiques
L’isolement des microdomaines est réalisé à partir de deux trois types d’échantillons : des plaquettes au repos, des plaquettes activées par le PAR1-ap (20 µM) pendant 3 minutes ainsi que des plaquettes au repos incubées avec du MCD (5 mM) pendant 30 minutes à 37°C.
a. Séparation des fractions
Les plaquettes lavées en suspension à 1,2.109 plaquettes/mL (500 µL, soit 6.108 plaquettes) sont lysées par ajout de 500 μL de tampon de lyse 2X à 4°C. Les lysats sont vortexés puis incubés 30 minutes dans la glace, puis sont dilués dans une solution de sucrose 80% (1 mL). Cette solution est alors transférée dans un tube d’ultracentrifugation (14 mL) pour former un gradient de sucrose en ajoutant successivement 6 mL de sucrose 30% puis 6 mL de sucrose 5%. La séparation des différentes fractions est ensuite obtenue par ultracentrifugation des gradients pendant 18 heures, à 4°C, à 200 000 g (ultracentrifugeuse Optima X Beckman Coulter® ; rotor SW40 Ti.)
b. Récupération des fractions
Treize fractions sont récupérées manuellement en chambre froide (12 fractions de 1 mL et une 13ème fraction avec le volume restant) par pipetage successif en surface du gradient.
c. Précipitation des protéines
Les protéines présentes dans chaque fraction sont concentrées par précipitation à l’acide trichloroacétique (TCA). Pour ce faire, une solution de TCA 100% est ajoutée pour obtenir une concentration finale de TCA à 5%. Les échantillons sont ensuite incubés à 4°C pendant 30 minutes puis centrifugés 15 minutes, à 4°C, à 20 000g. Les surnageants sont
permettant de préparer des échantillons protéiques pour la migration électrophorétique sur gel, puis conservés à -20°C.
3. Western-blot
a. Préparation des lysats plaquettaires et migration
Dix-huit microlitres de chaque échantillon de lysats plaquettaires sont aliquotés dans des tubes eppendorf puis réduits par l’ajout de 10% de DTT (2 µL). Ces aliquotes sont chauffées dans un bain marie à sec pendant 5 minutes à 90°C puis vortexées.
Chaque échantillon (15 µL), un lysat total de plaquettes lavées (15 µL à 5.10^8 plaquettes/mL) ainsi qu’un marqueur de poids moléculaire (10 µL ; SeeBlue® Plus2 Prestained Standard), sont déposés dans les puits de gels 3-8% d’acrylamide (gels NuPAGE® Bis-Tris Gel Invitrogen®). La migration s’effectue dans du tampon de migration NuPAGE MES (MES SDS Running Buffer) sous 200V pendant 35 minutes.
b. Transfert
Les protéines, ainsi séparées, sont ensuite transférées sur une membrane de nitrocellulose en 7 minutes par électrotransfert grâce à l’appareil Invitrogen®. Afin de contrôler la qualité du transfert et visualiser le profil protéique des lysats plaquettaires, la membrane de nitrocellulose est incubée avec du rouge ponceau qui colore spécifiquement et de façon réversible les protéines en milieu acide. La membrane est ensuite lavée (3 x 5 minutes) avec du TTBS jusqu’à disparition du marquage.
c. Western-blot
La membrane est incubée une heure, à température ambiante, sous agitation douce (ou la nuit à 4°C en chambre froide), dans du lait (lait écrémé Regilait 5%) dilué dans du TTBS, afin de saturer les sites susceptibles de provoquer des fixations non spécifiques des anticorps.
Après rinçage au TTBS, la membrane est incubée une heure, sous agitation douce, avec un des anticorps primaires dilués : anti-LAT (1:500ème), anti-CD36 (1:1000ème), anti-ß3
intégrine (1:1000ème), anti-MRP4 (1:500ème), anti-Syk (1:500ème), anti-PKA-RI (1:1000ème) et anti-moesin (1:2400ème).
Pour éliminer l’anticorps primaire non fixé, la membrane est rincée trois fois dix minutes sous agitation au TTBS. La membrane est ensuite incubée pendant 45 minutes avec des anticorps secondaires anti-anticorps de lapin ou anti-anticorps murin (suivant les anticorps primaires utilisés) couplés à une sonde fluorescente (dilués au 1:10000ème). Trois rinçages de 10 minutes sont nécessaires avant la révélation.
d. Révélation
La révélation s’effectue à l’aide de l’appareil Odyssey®, un système d’imagerie dans l’infrarouge et le proche infrarouge. La membrane est scannée et une semi-quantification des protéines est réalisée à l’aide du logiciel Image J. Les résultats sont normalisés par rapport à la somme de la fluorescence de toutes les fractions et exprimés en pourcentages.
4. Test d'agrégation plaquettaire
La suspension plaquettaire à 3,5.108 plaquettes/mL est pré-incubée avec le MβCD à différentes concentrations ou avec du TpR pendant 30 minutes à 37°C. Quarante-cinq microlitres de suspension plaquettaire sont ensuite déposés par puits dans une plaque 96 puits et mis en agitation 2 min à 37°C dans le lecteur de plaque MWG Discovery HT_R piloté par le logiciel KC4. La mesure de la densité optique (DO405) est enregistrée pendant 2 minutes afin de vérifier l'absence d'agrégation spontanée. La DO de la suspension plaquettaire mesurée au repos correspond au 0% d’agrégation. L'agoniste, le PAR1-ap (5 µM), est ensuite ajouté à la pipette multicanaux. Les mesures sont effectuées toutes les 25 secondes et la courbe d'agrégation est enregistrée pendant un temps donné, dépendant de l'expérience réalisée. La DO est inversement proportionnelle aux agrégats formés. Le 100% d’agrégation correspond alors à la DO minimale obtenue suite à l’ajout du PAR1-ap (sans MCD ni forskoline). Pour certaines séries, la forskoline est ajoutée 15 secondes avant l'agoniste.
5. Immunofluorescence
a. Préparation de la lame et incubation des anticorps
Les douze puits d’une lame Ibidi® (lame sur laquelle est disposée une chambre détachable de douze puits) sont coatés à 4°C avec du BSA 2% dilué en PBS, durant la nuit qui précède l’expérience. Après quelques rinçages aux PBS, la suspension plaquettaire est déposée dans les puits ; après quinze minutes, les plaquettes sont fixées par ajout de PFA 4% pendant 15 minutes.
Une fois fixées, les plaquettes sont lavées au PBS, perméabilisées pendant cinq minutes à température ambiante avec du Triton 0,1% puis bloquées avec du PBS/BSA 2% pendant trente minutes. L’anticorps primaire peut alors être incubé soit toute la nuit à 4°C en chambre humide, soit 2 heures à température ambiante ou 1 heure 30 à 37°C en chambre humide (les dilutions utilisées sont les suivantes : anti-MRP4 1:300ème, anti-PDE2A 1:500ème, anti-PKA-RI 1:100ème, anti-moesin 1:2400ème, anti-tubuline 1:200ème et anti-AC V/VI 1:100 ème). Les puits sont ensuite lavés et les plaquettes sont incubées 1 heure à 37°C en chambre humide avec l’anticorps secondaire anti-anticorps de lapin ou anti-anticorps murins couplé à l’Alexa fluor 488 (dilution 1:200ème dans du PBS/BSA 2%). Pour finir, après quelques lavages, la phalloïdine 546 est incubée vingt minutes à température ambiante.
b. Révélation
Après rinçage au PBS, les puits sont séchés et la chambre est détachée de la lame de verre. Une goutte de milieu de montage est alors déposée sur chaque échantillon. Une fois le milieu polymérisé, une lamelle est ensuite déposée sur les échantillons et les bords de la jonction sont vernis afin de conserver au mieux les échantillons. La révélation se fait le lendemain, au microscope à fluorescence, après une nuit à -20°C.
6. Technique du Duolink®
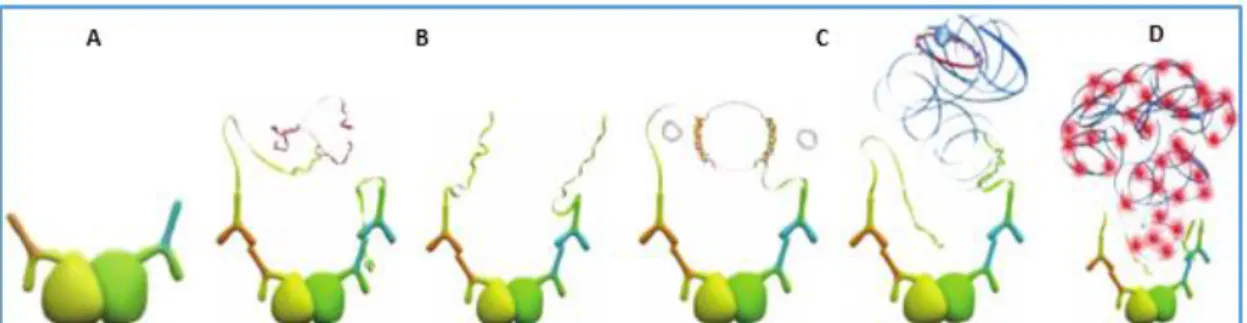
a. Principe
La technique du Duolink® permet la détection, la visualisation et la quantification d’une co-localisation entre les protéines. C’est une technique in situ basée sur une technologie de ligation de proximité. Elle utilise deux anticorps secondaires couplés chacun à un oligonucléotide (sondes PLA). Lorsque les deux protéines sont à proximités (maximum 40 nm), un signal va être généré grâce à la ligation et l’amplification des sondes PLA par l’intermédiaire des oligonucléotides (figure 11). Le signal détecté est visualisé comme un spot fluorescent.
Figure 11. Etapes successives de la technique Duolink (image issue du site de SIGMA ALDRICH)
A : incubation de deux anticorps primaires d’espèces différentes ; B : incubation des anticorps secondaires couplés à des sondes PLA d’espèces respectives aux anticorps primaires ; C : étape de ligation des sondes PLA par ajout d’une ligase ; D : étape finale d’amplification du signal par ajout d’une polymérase.
b. Méthode
Cette technique est basée sur la méthode de l’immunofluorescence. En effet, les premières étapes consistant à préparer la lame, déposer les échantillons et incuber avec les anticorps primaires, sont identiques à la technique d’immunofluorescence (voir ci-dessus). Après avoir lavé les puits au PBS sous agitation, les anticorps secondaires anticorps de lapin et anti-anticorps murins, couplés à une sonde PLA PLUS ou PLA MINUS, sont incubés une heure à 37°C en chambre humide. Les puits sont ensuite lavés avec du Buffer A (kit Duolink Sigma Aldrich®) avant d’ajouter une solution de ligation (1:5ème) contenant une ligase (ligase
Une fois la ligation effectuée et les puits lavés, ces derniers sont incubés avec une solution d’amplification (1:5ème) contenant une polymérase (1:80ème), afin d’amplifier le signal, pendant cent minutes à 37°C en chambre humide.
Pour finir, les puits sont lavés deux fois dix minutes avec du Buffer B 1X (kit Duolink Sigma Aldrich®) puis une minute avec du Buffer B 0,01X. Après une minute, les puits sont séchés et retirés de la lame de verre. Une goutte de milieu de montage (kit Duolink Sigma Aldrich®) est déposée sur chaque échantillon puis recouvert d’une lamelle. Quinze minutes plus tard, la lame est conservée à -20°C en attendant sa révélation le lendemain, au microscope à fluorescence
7. Statistiques
Les figures et les tests statistiques (Mann-whitney, test non paramétrique) sont réalisés à l’aide du logiciel GraphPad PRISM. Le seuil de significativité retenu est p < 0,05.
Les statistiques ont été calculées pour les séries contenant aux moins cinq résultats (Curtis et al. 2015). Les résultats des agrégations sont représentés par le diagramme en boîte qui est délimitée par le minimum et le maximum d’une série, et qui permet aussi d’indiquer la médiane ainsi que les 25ème et 75ème percentiles. Dans le texte, ces résultats sont indiqués par la médiane, le maximum et le minimum.
Résultats
I.
Rôle des microdomaines dans l’activation plaquettaire
1. Rôle des microdomaines sur l’agrégation plaquettaire
Pour déterminer le rôle de ces microdomaines dans l’activation plaquettaire, des tests d’agrégations ont été réalisés en présence de MβCD. Ce dernier, en déplétant les membranes de cholestérol, désorganise les microdomaines.
Les suspensions de plaquettes lavées ont été pré-incubées avec différentes doses de MβCD allant de 0,25 mM à 5 mM pendant 30 minutes à 37°C, puis activées par un agoniste fort : le PAR1-ap à 5µM.
Pour des doses faibles de MβCD allant de 0,25 mM à 1,25 mM, aucune différence d’agrégation n’est à noter (figure 12). En revanche, à la concentration plus élevée de 2,5 mM, une diminution significative du pourcentage d’agrégation, mesuré à 200 secondes, a été observée : 45 % [25-63] versus 74% [64-81] en l’absence de MβCD (p = 0.002). De même pour la concentration de MβCD à 5 mM, l’intensité d’agrégation observée est significativement plus faible (12% [2-23] versus 74% [64-81] ; p=0.002).
Figure 12. Pourcentage d'agrégation au PAR1-ap à 5 µM en fonction de la concentration en MβCD en mM
Dose/réponse au MCD : les plaquettes lavées sont incubées 30 min à 37°C avec différentes doses de MCD puis activées au PAR1-ap (5 µM). Les résultats sont exprimés en pourcentage de l’agrégation maximale à 200 secondes (n=6). ** p = 0.002
2. Rôle des microdomaines dans la voie de l’AMPc
Après avoir montré le rôle des microdomaines dans l’activation plaquettaire, nous avons cherché à mettre en évidence une relation entre les microdomaines et la voie de signalisation de l’AMPc qui régule l’activation plaquettaire. Pour cela, un agoniste de l’AC a été utilisé : la forskoline. L’activation de l’AC augmente la synthèse de l’AMPc cytosolique et ainsi augmente l’inhibition plaquettaire.
Dans un premier temps, une dose-réponse de forskoline de 0,05 à 1 µM (incubé 15 seconde avant l’ajout de l’agoniste), en absence de MβCD, a été réalisé dans le but de choisir des concentrations n’entrainant pas une diminution significative de l’agrégation (figure 13), le but étant uniquement d’augmenter le taux basal de l’AMPc. Les concentrations de forskoline retenues pour la suite des expériences sont celles de 0,1 µM et de 0,25 µM.
Figure 13. Pourcentage d'agrégation au PAR1-ap à 5 µM en fonction de la concentration en forskoline
Les suspensions de plaquettes lavées ajustées à 3,5x108pl/mlsont pré-incubées 30 min à 37°C avec du TpR, puis pré-incubées 15 secondes avec la forskoline et immédiatement après activées au PAR1-ap (5 µM). Les résultats sont exprimés en pourcentage de l’agrégation maximale à 200 secondes (n≥2)
Dans un deuxième temps, les suspensions de plaquettes pré-incubées avec différentes doses de MβCD (allant de 0,25 mM à 5 mM), ont été incubées 15 secondes avec les concentrations de forskoline sélectionnées (0,1 µM ou 0,25 µM) avant d’être activées au PAR1-ap (5 µM). Les résultats sont exprimés en pourcentage d’inhibition de l'agrégation plaquettaire calculés par rapport à l’agrégation à 200 secondes de la suspension plaquettaire en l'absence de forskoline et de MCD.