UNIVERSITÉ DE GENÈVE FACULTÉ DE MÉDECINE
Section de Médecine Fondamentale Département de Pathologie Thèse préparée sous la direction des Professeurs Giulio GABBIANI et Yusuf KAPANCI
Phénotype des myofibroblastes alvéolaires dans les
différents types de fibrose pulmonaire
Thèse
Présentée à la Faculté de Médecine de l’Université de Genève
pour l’obtention du grade de Docteur en Médecine
par
Jean-Frédéric VODOZ
de
La Tour-de-Peilz (VD)
Thèse no. 10310
Genève
2003
Introduction
Parmi les cellules constituant le tissu interstitiel pulmonaire, il en existe certaines qui possèdent des propriétés contractiles, réparties en 3 types : les cellules musculaires lisses, les péricytes et les myofibroblastes, dont nous allons nous occuper dans ce travail.
Les myofibroblastes alvéolaires (MyF), initialement baptisés « cellules interstitielles contractiles » sont des éléments de type fibroblastique pourvus de microfilaments d’actine, qui jouent probablement un rôle dans la régulation de la ventilation et de la perfusion (V/Q) du poumon normal1 ; dans certaines pathologies, ces MyF expriment une isoforme d’actine qui est l’alpha-actine muscle lisse (α-SM1)2-5. De ce fait, ces cellules augmentent leurs
propriétés contractiles, qui sont en partie à l’origine de la baisse de la compliance pulmonaire lors des pathologies fibrosantes. En effet, étant disposées de façon sinusoïdale le long de la paroi des alvéoles, ces cellules réduisent le volume des alvéoles en se contractant.
Les MyF pourvus de microfilaments d’actine furent pour la première fois décrits au niveau des septa alvéolaires normaux en 1974 par Kapanci et al.1. Plus tard, dans certains processus fibrosants, Kapanci et al.3-4, Vyalov et al.5, ainsi que Mitchell et al.6, Zhang et al.7 et Low8 ont rapporté une modulation de ces MyF caractérisée par l’expression d’α-SM1. Une prolifération
septale des MyF exprimant l’α-SM1 est rapportée notamment dans la fibrose pulmonaire
idiopathique (IPF)4, la fibrose induite par la bléomycine5-8, la BOOP8 (bronchiolitis obliterans-organizing pneumonia), l’hypertension pulmonaire post-capillaire2, les poumons transplantés3 et en cas d’ARDS9. L’induction de cellules α-SM1 positives fut également
observée en dehors du poumon, entre autres dans des processus de cicatrisation10 et de réaction stromale lors de néoplasie épithéliale11.
Dans l’IPF, cette modulation paraît être effectuée par l’intermédiaire de certaines cytokines, plus particulièrement le TGF-β1 et le TNF-α4. Dans cette pathologie, le TGF-β1 semble être synthétisé au niveau de l’épithélium bronchique, alvéolaire, ainsi que dans les cellules interstitielles des septa alvéolaires. Le TNF-α serait quant à lui produit par les pneumocytes II4. Il a d’autre part été suggéré que ces cytokines jouent également un rôle dans d’autres types de fibrose pulmonaire, tels que la fibrose induite par la bléomycine12, la sarcoïdose13, la silicose14 ou au décours d’une transplantation pulmonaire3, ainsi que dans d’autres tissus, comme par exemple le cristallin15 ou la membrane synoviale16.
D’autres molécules sont susceptibles de jouer un rôle dans ce processus. Certaines sont inductrices, comme le GM-CSF, le PDGF, l’endothéline-1 (Et-1)17 ou l’angiotensine II ; d’autres ont tendance à l’inhiber, comme l’héparine, le basic fibroblastic growth factor (βFGF), l’interleukine-1β (Il-1β) ou l’interféron-γ (INF-γ)18. Il est intéressant de noter que le TGF-β1, le TNF-α et le GM-CSF peuvent être produits par les MyF eux-mêmes, impliquant des phénomènes de rétroaction positive ou négative. Il semblerait par ailleurs que le pronostic de la pneumopathie, à savoir la tendance à la progression vers une fibrose terminale, dépende du profil cytokinique de la réponse inflammatoire et immune normale.
Nous avons cherché à savoir si une modulation similaire des MyF α-SM1 positifs existait dans
d’autres types de fibrose faisant partie des pneumopathies interstitielles et tenté d’établir le rôle éventuel des cytokines, en particulier du TGF-β1. Afin de mieux analyser l’effet du
TGF-β1, nous avons également recherché l’expression de ses deux récepteurs dans les différentes conditions pathologiques.
Tab. 1. Diffuse parenchymal lung
diseases (DPLDs) consist of disorders of known causes (collagen vascular disease, environmental or drug related) as well as disorders of unknown cause. The latter include idiopathic interstitial pneumonias (IIPs), granulomatous lung disorders (e.g. sarcoïdosis), and other forms of interstitial lung disease (ILD), including lymphangioleiomyomatosis (LAM), pulmonary Langerhans’ cell histio-cytosis/histiocytosis X (HX), and eosinophilic pneumonia. The most important distinction among the idiopathic interstitial pneumonias is that between idiopathic pulmonary fibrosis and the other interstitial pneumonias (IPs), which include nonspecific interstitial pneumonia (a provisional term), desquamative interstitial pneumonia, respiratory bronchiolitis-associated interstitial lung disease, acute interstitial pneumonia, cryptogenic organizing pneumonia, and lymphocytic interstitial pneumonia.
(Tiré de: Am J Respir Crit Care Med 2002;165:276)
Rappelons que les pneumonies interstitielles idiopathiques (IIP) font partie des pneumopathies interstitielles, et sont un groupe hétérogène de maladies pulmonaires non-néoplasiques résultant d’une altération inflammatoire et fibrotique, à degrés variables, du parenchyme pulmonaire (Tab. 1). Celles-ci se divisent, selon la dernière classification19 de l’ATS/ERS* parue en juin 2001, en sept entités clinico-radio-pathologiques, à savoir la fibrose pulmonaire idiopathique (IPF), également connue sous le terme de pneumonie interstitielle courante (UIP), la pneumonie interstitielle non-spécifique (NSIP), la BOOP, également connue sous le terme de pneumonie cryptogénique organisante (COP), la pneumonie interstitielle aiguë (AIP), la pneumopathie interstitielle associée à une bronchiolite (RB-ILD), la pneumonie interstitielle désquamative (DIP) et la pneumonie interstitielle lymphoïde (LIP).
Les caractéristiques histologiques de l’IPF consistent principalement en une destruction de l’architecture du parenchyme pulmonaire surtout au niveau sous-pleural, avec une dégénérescence fibrotique en « nid d’abeille », d’aspect hétérogène.
La NSIP présente une inflammation et une fibrose de la paroi alvéolaire de degré variable, dont l’aspect histologique se démarque des autres IIP. Le tissu pulmonaire est atteint typiquement de façon uniforme, mais la distribution des lésions est souvent éparse.
La BOOP montre des foyers de fibrose intraluminale au niveau des voies aériennes distales (bronchioles, conduits alvéolaires et alvéoles), avec une architecture pulmonaire conservée. Les lésions apparaissent d’âge égal.
L’AIP est une forme de lésion alvéolaire diffuse (diffuse alveolar damage = DAD), dont l’aspect histologique ne peut être distingué de l’ARDS. Le terme d’AIP est ainsi réservé aux étiologies inconnues de DAD. La présence de membranes hyalines est pathognomonique de cette entité histopathologique.
La RB-ILD est la manifestation clinique d’une pneumopathie interstitielle associée à la lésion histopathologique de la bronchiolite habituellement trouvée chez le fumeur, et
*American Thoracic Society / European Respiratory Society
Diffuse Parenchymal Lung Disease
DPLD of known cause, e.g. drugs or association e.g. collagen vascular disease Idiopathic interstitial pneumonias Granulomatous DPLD e.g. sarcoïdosis Other forms of DPLD e.g. LAM, HX etc. Idiopathic pulmonary fibrosis
IIP other than idiopathic pulmonary fibrosis Desquamative interstitial pneumonia Acute interstitial pneumonia Nonspecific interstitial pneumonia (provisional) Respiratory bronchiolitis interstitial lung disease
Cryptogenic organising pneumonia
Lymphocytic interstitial pneumonia
caractérisée par la présence de macrophages pigmentés intraluminaux dans les bronchioles de 1er et 2ème ordre.
La DIP se caractérise par une accumulation de nombreux macrophages dans les voies aériennes les plus distales (plutôt que par une désquamation des cellules épithéliales, comme initialement décrit) et est presque invariablement associée au tabagisme. Cette entité est considérée parfois comme la phase avancée d’une RB-ILD. Elle se distingue toutefois de la RB-ILD par une atteinte diffuse et uniforme du poumon, ne touchant pas spécifiquement les bronchioles.
La LIP est souvent secondaire à une maladie auto -immune systémique et présente une infiltrat lymphoïde dense surtout au niveau des septa alvéolaires.
Matériel et méthodes
Notre étude regroupe 46 cas, dont 35 biopsies et 11 autopsies. On dénombre 10 cas d’IIP (comprenant 3 UIP, 2 DIP, 1 LIP et 4 cas de NSIP), 1 cas de fibrose induite par la bléomycine, 9 cas de BOOP, 6 cas de sarcoïdose, 4 cas de granulomatose d’autres types, 5 cas de silicose, 3 cas de fibrose non-spécifique, 5 cas d’ARDS et 3 cas témoins (Tab. 2).
Dans les cas où une biopsie pulmonaire transbronchique a été pratiquée, le diagnostic de pneumopathie interstitielle était posé sur la base de données anatomo-cliniques (contexte clinique exigé : image radiologique réticulo-nodulaire, insuffisance respiratoire restrictive et trouble de la diffusion / distribution).
Le matériel a été fixé par immersion dans une solution de formaldéhyde 10%. Il a été ensuite inclus dans de la paraffine et coupé par un microtome rotatif en séries de coupes de 5µ d’épaisseur. Ces coupes ont été colorées à l’hématoxyline-éosine (H&E) et marquées par des anticorps anti- α-SM1, desmine, cytokératine à large spectre (KWS), ainsi que TGF-β1 et ses
deux récepteurs. L’anticorps anti- α-SM1 a été préparé dans les laboratoires du Pr G. Gabbiani
du Département de Pathologie de la Faculté de Médecine de Genève. La spécificité et les caractéristiques de cet anticorps ont été décrites par Skalli et al.20 ; les caractéristiques de l’anticorps anti- TGF-β1 (R&D Systems, Minneapolis, MN, USA) sont rapportées par Perdue et Brody21. D’autre part, dans certaines pneumopathies interstitielles, une typisation des cellules lymphoïdes a été effectuée (CD3, CD20, CD23, CD43, CD68, CD79a). La membrane basale a été marquée par une immunoglobuline anti-collagène IV, et l’épithélium alvéolaire par KWS.
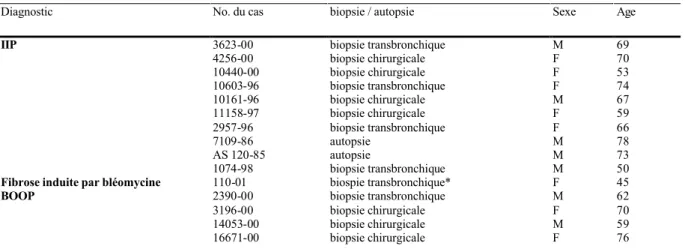
Tab. 2. Données cliniques essentielles
Diagnostic No. du cas biopsie / autopsie Sexe Age
IIP 3623-00 biopsie transbronchique M 69
4256-00 biopsie chirurgicale F 70 10440-00 biopsie chirurgicale F 53 10603-96 biopsie transbronchique F 74 10161-96 biopsie chirurgicale M 67 11158-97 biopsie chirurgicale F 59 2957-96 biopsie transbronchique F 66 7109-86 autopsie M 78 AS 120-85 autopsie M 73 1074-98 biopsie transbronchique M 50
Fibrose induite par bléomycine 110-01 biospie transbronchique* F 45
BOOP 2390-00 biopsie transbronchique M 62
3196-00 biopsie chirurgicale F 70
14053-00 biopsie chirurgicale M 59
10444-00 biopsie chirurgicale M 53
12672-98 biopsie transbronchique M 69
12892-98 biopsie transbronchique M 53
8396-00 biopsie transbronchique M 52
1911-96 biopsie transbronchique M 77
Sarcoïdose 4834-00 biopsie transbronchique F 41
2877-00 biopsie transbronchique F 59
574-00 biopsie chirurgicale M 47
6197-00 biopsie chirurgicale M 40
3626-96 biopsie chirurgicale F 33
21426-87 biopsie transbronchique F 58
Autres granulomatoses 575-00 biopsie chirurgicale M 59
7568-98 biopsie transbronchique M 74
2615-98 biopsie transbronchique M 43
9821-00 biopsie chirurgicale F 77
Silicose 9289-00 biopsie chirurgicale M 69
AS 66-99 autopsie M 79
3998-88 autopsie M 81
AS 30-87 autopsie M 67
AS 151-84 autopsie M 73
Fibrose non spécifique 2807-00 biopsie chirurgicale F 62
8141-99 biopsie chirurgicale M 34 AS 79-88 autopsie M 61 ARDS AS 16-89 autopsie M 19 AS 43-93 autopsie M 45 AS 67-90 autopsie M 60 externe autopise** externe autopsie**
Cas contrôle 7762-97 biopsie transbronchique*** F 67
pas de no. biopsie chirurgicale*** F 58
pas de no. biopsie chirurgicale**** M 63
* néoplasie mammaire traitée
**gracieusement mis à disposition par le Dr J.-C. Pache ***adénocarcinome bronchique
****carcinome épidermoïde bronchique
Résultats
Poumon normal
Dans les coupes témoins, les MyF alvéolaires ne sont pas marqués par l’α-SM1, ni par le
TGF-β1 (Fig. 1). La musculature des anneaux alvéolaires (alveolar rings), les péricytes, ainsi que la musculature des artères et des bronches sont par contre positifs pour l’α-SM1 et la
desmine. Le TGF-β1 est positif au niveau de l’épithélium bronchique, dans de rares cellules épithéliales alvéolaires disposées plus ou moins régulièrement, ainsi qu’au niveau de la musculature lisse bronchique et vasculaire.
IIP
Dans les IIP, notamment l’IPF/UIP, l’α-SM1 marque les MyF alvéolaires ; le nombre de
cellules marquées varie inversement avec l’intensité de la fibrose (Fig. 2). En effet, au fur et à mesure que le collagène se dépose au niveau des septa alvéolaires, le nombre de MyF diminue. Toutefois, les nombre de cellules marquées reste élevé dans la fibrose à la bléomycine (Fig. 3).
On assiste d’abord à un dédoublement de la membrane basale alvéolaire, puis à une stratification des fibres collagènes dans les septa et autour des bronches et des vaisseaux (Fig. 4). Dans la fibrose avancée autour de ces structures, on observe une prolifération des MyF pour former de véritables faisceaux de musculature lisse. Ces faisceaux sont positifs à l’immunomarquage par la desmine. Il se réalise ainsi l’image classique de « cirrhose musculaire du poumon » (Fig. 5). Dans les MyF alvéolaires, la desmine est absente, alors
qu’elle est présente dans la musculature péri-bronchovasculaire. Le TGF-β1 et ses récepteurs sont positifs dans les MyF, la musculature lisse et l’épithélium alvéolaire (Fig. 6 et 7).
Lorsque la fibrose est peu marquée et l’infiltrat inflammatoire interstitiel prépondérant (UIP), il s’agit principalement d’une infiltration de lymphocytes T marqués par CD3. Quelques lymphocytes B (CD20 et CD79a positifs) se trouvent autour des bronches. Les granulocytes polymorphonucléaires sont rares. Dans cette phase de l’UIP, les MyF α-SM1 positifs
abondent. Dans la DIP, on trouve plusieurs MyF α-SM1 positifs ; dans la LIP, ils sont plus
rares, mais le nombre de cas examinés est trop restreint pour tirer des conclusions. BOOP
Dans la BOOP, seuls les moulages fibroblastiques intra-alvéolaires (corps de Masson) contiennent des cellules positives à l’α-SM1, au TGF-β1 et à ses récepteurs (Fig. 8) Les MyF
au niveau des septa alvéolaires ne sont qu’exceptionnellement marqués. Dans ces septa on n’observe que rarement une augmentation du réseau de collagène IV.
Silicose
Dans les lésions silicotiques fraîches, soit lorsqu’il s’agit d’une accumulation de macrophages avec des cristaux d’oxyde de silice accompagnée d’une réaction inflammatoire septale modérée, il n’y a pratiquement pas de prolifération fibroblastique avec expression d’α-SM1
dans la lésion. Cette observation rappelle celle de Piguet et al.25, qui, dans la silicose expérimentale chez la souris, ne constatent pas de prolifération myofibroblastique α-SM1
positive.
En revanche, lorsque les nodules silicotiques se développent avec disposition des fibres collagène en pelure d’oignon, on observe autour des nodules l’apparition de nombreux MyF
α-SM1 (Fig. 9). Les septa alvéolaires sont toujours dépourvus de MyF marqués. Les
macrophages alvéolaires, quelques MyF de même que de rares cellules épithéliales apparaissent faiblement marqués par TGF β1 et ses récepteurs.
Sarcoïdose et autres granulomatoses
Dans la sarcoïdose et les autres granulomatoses pulmonaires, les granulomes et l’interstice alvéolaire sont dépourvus de MyF α-SM1 positifs. Toutefois, lorsqu’il y a fibrose autour des
granulomes et lors de la confluence de ces derniers, les MyF péri-nodulaires expriment l’α -SM1 et le TGF-β1 (Fig. 10). Cette dernière cytokine est également présente dans certaines
cellules au sein des granulomes, correspondant cependant à des macrophages, et non à des MyF. Comme dans la silicose, les septa alvéolaires ne comportent jamais de cellules α-SM1
positives. ARDS
Dans nos cas, le début de la maladie date de plus de 15 jours et les MyF abondent. Ces cellules, avec leurs longs processus cytoplasmiques, sont localisées dans l’épaisseur des septa alvéolaires contre les capillaires congestionnés (Fig. 11 et 12). Ces observations sont compatibles avec celles de Pache et al.9, qui décrivent aux stades précoces de l’ARDS (phases
exsudative et proliférative) des cellules α-SM1 positives particulièrement abondantes dans les
septa alvéolaires. Comme pour les autres pathologies discutées précédemment, les MyF peuvent générer des forces de traction sur l’architecture alvéolaire, qui, en association avec le dépôt de collagène, contribuent au remodelage pulmonaire observé chez certains patients en phase tardive d’ARDS.
Fibrose non-spécifique
Nos 3 cas de fibrose non-spécifique (FNS) consistent en des cicatrices pulmonaires. Dans ces cicatrices, certains fibroblastes expriment l’α-SM1. Dans les septa, les MyF alvéolaires restent
négatifs pour l’α-SM1.
Discussion
Cette étude confirme que l’expression de l’α-SM1 par les MyF alvéolaires est caractéristique,
mais pas spécifique de l’IPF/UIP4,22. Comme il a été montré antérieurement23, les autres conditions où cette modification se présente sont la fibrose induite par la bléomycine, l’hypertension pulmonaire post-capillaire, les poumons transplantés et le syndrome de détresse respiratoire de l’adulte (ARDS)9. Dans cette dernière pathologie, il a été observé que les MyF contribuent, en phase chronique, au remodelage du parenchyme pulmonaire. D’après nos observations, la modulation des MyF avec expression d’α-SM1 dans l’ARDS pourrait être
due à la congestion des capillaires, qui est de règle dans cette condition. En effet, la ressemblance des septa alvéolaires entre l’hypertension pulmonaire post-capillaire et l’ARDS est frappante.
Les MyF α-SM1 positifs abondent également autour des nodules silicotiques et des
granulomes de sarcoïdose, ainsi que dans les exsudats alvéolaires organisés de la BOOP (corps de Masson). Par contre, ils sont exceptionnels dans l’interstice alvéolaire. D’après nos observations, les MyF alvéolaires sont strictement limités aux septa23, alors que dans la silicose et autour des granulomes de sarcoïdose, il s’agit de MyF de type cicatriciel. La prolifération de MyF alvéolaires contribue potentiellement à la contraction du parenchyme pulmonaire entraînant une baisse de la compliance dans ces maladies. Via l’expression d’un gène procollagène, ils participent aussi à la déposition de matrice extra-cellulaire, ce qui renforce la fibrose22.
Il est intéressant de noter que dans la silicose induite chez la souris, les nodules fibreux contiennent des macrophages produisant du TNF-α, qui inhibe l’expression du gène tropoélastine24. Selon Piguet et al.25, l’héparine protège les poumons contre la fibrose induite par la bléomycine, alors qu’elle n’a pratiquement pas d’effet en cas de silicose. On connaît l’effet inhibiteur de l’héparine sur la prolifération des MyF ; on peut donc supposer que la non-inhibition de la fibrose par l’héparine en cas de silicose est due à un phénotype différent des cellules prolifératives. Piguet pense que dans la silicose, le processus de fibrose est du à une activation des fibroblastes, et dans la fibrose induite par la bléomycine, à une activation des MyF. Cette hypothèse doit encore être confirmée, car les fibroblastes sont les cellules-mères des MyF, et qu’en cas de silicose en phase tardive (nodulaire), il y a chez l’humain de nombreux MyF autour des nodules. Il est cependant probable que l’apparition de MyF autour des nodules silicotiques chez l’être humain soit l’expression d’un processus cicatriciel, n’impliquant pas les MyF alvéolaires.
La production de la force contractile des MyF, mécanisme-clé dans les processus fibrosants, est induite par le TGF-β1, d’une part via l’expression de l’α-SM1, et d’autre part via la
formation d’éléments structuraux comme les fibres de stress, les complexes d’adhésion de type fibronexus contenant de la vinculine et des fibrilles de fibronectine. La genèse de ces éléments se déroulerait avant et indépendamment de l’expression de l’α-SM1, selon
l’hypothèse non confirmée de Vaughan et al.26 Il a déjà été montré qu’un anticorps dirigé contre l’α-SM1 limite l’évolution de la fibrose27, ce qui laisse imaginer d’éventuels débouchés
thérapeutiques dans ce sens. Le TGF-β1 est par ailleurs produit en majeure partie par les MyF eux-mêmes, ainsi que la monocyte chemo-attractant protein-1 (MCP-1), cytokine importante pour l’inflammation et la fibrose, notamment dans la fibrose induite par la bléomycine12. Nos résultats nous permettent aussi de vérifier l’hypothèse d’une sorte de « down-regulation » des MyF α-SM1 positifs lors de fibrose avancée4, phénomène déjà observé dans la
cicatrisation cutanée au stade chronique10. Il semblerait par contre que ce processus soit inverse dans la silicose, où les nodules fibreux anciens contiennent plus d’éléments α-SM1
positifs que les nodules récents, ce qui n’avait pas encore été décrit jusqu’ici.
En conclusion, il semblerait que le « pattern » cytokinique propre aux différentes pathologies interstitielles pulmonaires étudiées dans notre étude détermine l’ampleur et l’éventuelle « down-regulation » des MyF alvéolaire α-SM1, et, de ce fait, la progression ou non vers une
fibrose terminale. Le TGF-β1 semble donc être synonyme de mauvais pronostic, tout comme il a récemment été montré pour la mucoviscidose28. Cependant, l’implication des MyF dans cette maladie n’est pas mentionnée.
Résumé
Le présent travail étudie l’expression d’α-actine muscle lisse (α-SM1) dans les
myofibroblastes (MyF) alvéolaires lors de différentes pneumopathies interstitielles. Les maladies choisies sont les pneumonies interstitielles idiopathiques (IIP) y compris la pneumonie interstitielle courante (UIP), la pneumonie interstitielle désquamative (DIP) et la pneumonie interstitielle lymphoïde (LIP), ainsi que la fibrose interstitielle après administration de bléomycine, la BOOP, la silicose, la sarcoïdose et l’ARDS. Quarante-six cas ont été examinés en utilisant notamment des anticorps contre l’α-actine muscle lisse (α -SM1), TGF-β1 et ses récepteurs.
Notre étude confirme que l’expression d’α-SM1 par les MyF alvéolaires est caractéristique,
mais pas spécifique des IIP. On retrouve une telle modulation dans la fibrose induite par la bléomycine, dans la phase chronique de l’ARDS et, selon d’autres publications, dans la transplantation pulmonaire et dans l’hypertension pulmonaire post-capillaire. De plus, les corps de Masson en cas de BOOP, la périphérie des nodules silicotiques, ainsi que celle des granulomes de la sarcïdose renferment des MyF α-SM1 positifs ; en revanche, les septa
alvéolaires en sont dépourvus. La modulation de MyF alvéolaires α-SM1 est induite par le
TGF-β1. Cette modulation semble jouer un rôle important dans la production de fibres collagène au niveau alvéolaire ; elle contribue par ailleurs vraisemblablement à la réduction de la compliance pulmonaire.
Remerciements
Nous tenons à exprimer notre gratitude envers le Dr J.-C. Pache pour nous avoir fourni 2 cas d’ARDS.
Références
1. Kapanci Y, Assimacopoulos A, Irlé C, Zwahlen A, Gabbiani G « Contractile interstitial cells » in pulmonary alveolar septa : a possible regulator of ventilation/perfusion ratio ? Ultrastructural, immunofluorescence and in vitro studies J Cell Biol 1974 ;60 :375-392
2. Kapanci Y, Burgan S, Pietra GG, Conne B, Gabbiani G Modulation of actin isoform expression in alveolar myofibroblasts (contractile interstitial cells) during pulmonary hypertension Am J
Pathol 1990 ; 136 :881-889
3. Kapanci Y, Kurt AM, Redard M, Gabbiani G Phenotypic modulation of alveolar myofibroblast in transplanted human lungs Mod Pathol 1997 ;10 :1134-1142
4. Kapanci Y, Desmoulière A, Pache JC, Redard M, Gabbiani G Cytoskeletal protein modulation in pulmonary alveolar myofibroblasts during idiopathic pulmonary fibrosis Am J Respir Crit Care
Med 1994 ;152 :2163-2169
5. Vyalov SL, Gabbiani G, Kapanci Y Rat alveolar myofibroblasts acquire α-smooth muscle actin expression during bleomycin-induced pulmonary fibrosis Am J Pathol 1993 ;143 :1754-1765 6. Mitchell J, Woodcock-Mitchell J, Reynolds S, Low RB, Leslie KO, Alder KB, Gabbiani G, Skalli O
α-smooth muscle actin in parenchymal cells of bleomycin-injured rats Lab Invest 1989 ;60
:643-651
7. Zhang HY, Gharaee-Kermani M, Zhang K, Kermiol S, Phan SH Lung fibroblasts α-smooth muscle actin expression and contractile phenotype in bleomycin-induced pulmonary fibrosis Am
J Pathol 1996 ;148 :527-533
8. Low RB Modulation of myofibroblast and smooth-muscle phenotypes in the lung Curr Top
Pathol 1999 ;93 :19-26
9. Pache JC, Christakos P, Gannon D, Mitchell J, Low R, Leslie K Myofibroblasts in diffuse alveolar damage of the lung Mod Pathol 1998 ;11 :1064-1070
10. Desmoulière A, Gabbiani G The role of myofibroblasts in wound healing and fibrocontractive diseases In : Clark RAF, editor. The molecular and cellular biology of wound repair. New York : Plenum Press ;1996. p. 391-423
11. Tsukada T, McNutt MA, Ross R, Gown AM HHF35, a muscle-actin-specific monoclonal antibody. II. Reactivity in normal, reactive, and neoplastic human tissues Am J Pathol
1987 ;127 :389-402
12. Phan SH Role of myofibroblast in pulmonary fibrosis Kidney Int 1996 ;54 :46-48
13. Shahar I Effects of IL-6 on alveolar fibroblast proliferation in interstitial lung diseases Clin
14. Davis G Persistant overexpression of Il-1 beta and TNF-alpha in murine silicosis J Environ
Pathol Toxicol Oncol 1998 ;17 :99-114
15. Kurosaka D Growth factors influence contractility and alpha-SMA expression in bovine lens epithelial cells Invest Ophtalmol Vis Sci 1995 ;36 :1701-1708
16. Mattey DL TGF-beta 1 and Il-4 induced alpha-SMA expression and myofibroblast-like differentiation in human synovial fibroblasts in vitro : modulation by basic fibroblast growth factor
Ann Rheum Dis 1997 ;56 :426-431
17. Shahar I, Fireman E, Toplisky M Effect of endothelin-1 on α-SM1 actin expression and alveolar
fibroblast proliferation in interstitial lung diseases Immunopharmacology 1999 ;21 :759-775 18. Zhang H, Gharaee-Kermani M, Phan SH Regulation of lung fibroblast α-smooth muscle actin
expression, contractile phenotype, and apoptosis by Il-1β J Immunol 1997 ;158 :1392-1399 19. American Thoracic Society / European Respiratory Society international multidisciplinary
consensus classification of the idiopathic interstitial pneumonias Am J Respir Crit Care Med
2002 ; 165 :277-304
20. Skalli O, Vandekerckhove J, Gabbiani G Actin isoform pattern as marker of normal or pathological smooth muscle and fibroblastic tissues Differentiation 1987 ;33 :232-238
21. Perdue TD, Brody AR Distribution of transforming growth factor-β1, fibronectin and smooth
muscle actin in asbestos-induced pulmonary fibrosis in rats J Histochem Cytochem
1994 ;42 :1061-1070
22. Ohta K, Mortenson R, Clark R, Hirose N, King T Immunohistochemical identification and characterization of smooth muscle-like cells in idiopathic pulmonary fibrosis Am J Respir Crit
Care Med 1995 ;152 :1659-1665
23. Kapanci Y, Gabbiani G Contractile cells in pulmonary alveolar tissue In : The Lung, scientific
foundations, 2nd ed. (1997) chapter 48, pp. 697-707 Edited by Crystall R B and West JD et al.
Lippincott-Raven Publ. Philadelphia
24. Mariani TJ, Arikan MC, Pierce RA Fibroblast tropoelastin and α-SM1 expression are repressed
by particulate-activated macrophage-derived TNF-α in experimental silicosis Am J Respir Cell
Mol Biol 1999 Aug ;21(2) :185-192
25. Piguet PF, Van GY, Guo J Heparin attenuates bleomycin, but non silica-induced pulmonary fibrosis in mice; possible relationship with involvement of myofibroblasts in bleomycin, and fibroblasts in silica-induced fibrosis Int J Exp Path 1996 ;77 :000-000
26. Vaughan MB, Howard EW, Tomasek JJ Transforming growth factor-β1 promotes the
morphological and functional differentiation of the myofibroblast Exp Cell Res 2000 ;257
:180-189
27. Khouw I, Von Wachem P, Plantinga J, Vujaskovic Z, Wissink M, de Leij L, van Luyn M TGF-β and bFGF affect the differentiation of proliferating porcine fibroblasts into myofibroblasts in vitro
Biomaterials 1999 ;20 :1815-1822
28. Arkwright P et al. TGF-β1 genotype accelerates lung function decline in cystic fibrosis Thorax