Université de Montréal Faculté des arts et des sciences
Cette thèse intitluée:
The Development of an Expedient Method for the Synthesis
of a Diverse Series of Cyclopropane
α
-Amino Acids.
Présentée par: Ryan P. Wurz
a été évaluée par le jury composé des personnes suivantes:
Présidente-rapporteuse: Dr. Hélène Lebel Directeur de recherche: Dr. André B. Charette Membre du jury: Dr. Richard Giasson Examinateur externe: Dr. William W. Ogilvie Représentant du doyen: Dr. Huy Ong
Sommaire
Cette thèse présente un ensemble de réalisations accomplies dans le domaine de la cyclopropanation d’oléfines par des réactifs diazo. Cette recherche a été appliquée à la synthèse d’acides aminés cyclopropanés. Ces motifs sont présents tant dans les produits naturels que synthétiques. De plus ils possèdent des propriétés biologiques intéressantes.
Une voie rapide et efficace a été developpée pour la préparation des α-nitro-α-diazocarbonyles. Ces derniers ont par la suite été utilisés dans des réactions de cyclopropanation d’oléfines catalysées par des complexes de Rh(II) ou Cu(I). Une version asymétrique a aussi été développée qui permet la synthèse des 1-nitro- cyclopropanecarbonyles avec des énantioselectivités modestes.
La réaction de cyclopropanation peut être effectuée dans l’eau, un résultat surprenant puisque les composés diazo sont reconnus pour procéder à des réactions d’insertion O-H avec des rendements élevés. Une méthodologie permettant la génération in situ de diazo acétate d’éthyle a également été developpée. Ceci représente un progrès majeur pour l’industrie car l’utilisation de composés diazo à grande échelle peut être dangeureux due à la nature explosive de ces derniers. Cette méthode permet donc la génération contrôlée dans le milieu, de petites quantités du diazo.
Une méthode de génération in situ d’ylures d’iodonium, qui sont des équivalents de diazo sans toutefois posséder leur caractère explosif, a aussi été developpée pour remplacer les α-nitro-α-diazocarbonyles. Ces ylures peuvent aussi être utilisés dans la réaction de cyclopropanation catalysée par Rh(II) au même titre que les diazos.
La réduction de nitro cyclopropanes a finalement été effectuée avec succès, malgré l’extrême sensibilité du motif cyclopropane aux nucléophiles et aux acides. Les catalyseurs de Pd qui sont souvent utilisés pour la réduction ont conduit à la destruction du motif cyclopropane, mais en utilisant de la fine poudre de zinc dans le 2-propanol, une réduction efficace a été observée, conduisant aux produits désirés avec des rendements élevés.
Le motif nitro cyclopropane sert aussi comme un synthon utile dans la synthèse organique. La synthèse de dihydropyrroles a été accomplie en traitant les 1-nitro-cyclopropanecarbonyles avec des amines primaires. Un oxydation peut donner accès à des pyrroles hautement fonctionnalisées. De plus, l’ouverture des cyclopropanes donne accès à des isoxazolines N-oxydes qui par la suite peuvent participer dans une cycloaddition 1,3-dipolaire. Cette stratégie a été envisagée pour la synthèse des β-lactames fonctionnalisées.
MOTS CLÉS: Acides aminés cyclopropanés
α-Nitro-α-diazocarbonyles
La catalyse asymétrique
La génération in situ de réactifs diazo Ylures d’iodonium
Summary
This thesis presents a collection of developments made in the field of diazo chemistry involving the cyclopropanation of olefins. The main goal of this research was to develop a new and efficient synthetic methodology for the synthesis of cyclopropane α-amino acids. This class of amino acids appears in natural and synthetic products and has been shown to have very interesting biological properties.
An expedient and efficient method was developed for the preparation of α-nitro-α-diazocarbonyls. These diazo compounds undergo efficient cyclopropanation reactions with olefins catalyzed by Rh(II) and Cu(I) complexes. Accordingly, the presence of chiral ligands on these metal complexes allow for the catalytic asymmetric preparation of 1-nitro cyclopropanecarboxylates in modest enantioselectivities.
The cyclopropanation reaction was also found to proceed in aqueous media, an interesting observation due to the synthetic efficiency of O-H insertion reactions with diazo compounds. This led to the development of an in situ method for generation of ethyl diazoacetate. This methodology permits the controlled formation of ethyl diazoacetate in the reaction mixture, representing a safe alternative for diazo-mediated cyclopropanations on a large scale.
A method involving the in situ generation of phenyliodonium ylides derived from α-nitrocarbonyls, which represent synthetic equivalents of diazos, was also developed to eliminate potential explosion hazards associated with the use of α-nitro-α-diazocarbonyls. These ylides undergo Rh(II)-catalyzed cyclopropanation reactions in a very similar manner to those observed with the corresponding diazo compounds.
The sensitive nature of 1-nitro cyclopropanecarboxylates made their reduction very challenging. Pd-based hydrogenation catalysts, which are often used to reduce nitro groups, led only to the destruction of the cyclopropane moiety. However, zinc dust in 2-propanol enabled an efficient reduction of these cyclopropanes to the desired cyclopropane amino esters in excellent yields. These compounds can then be easily transformed into the desired cyclopropane α-amino acids upon saponification of the ester.
The nitro cyclopropanecarbonyl moiety can also serve as a useful synthon in organic synthesis. Dihydropyrroles could be prepared upon treatment of 1-nitro-1-ketocyclopropanes with a variety of primary amines. Oxidation of these dihydropyrroles then allows rapid access to densely functionalized pyrroles. 1-Nitro cyclopropane carboxylates can also participate in cascade reactions. A cascade reaction involving an intramolecular rearrangement to an isoxazoline N-oxide followed by a 1,3-dipolar cycloaddition reaction with an olefin was developed. A strategy was envisioned using this approach for the synthesis of densely functionalized β-lactams.
KEY WORDS: Cyclopropane α-amino acids
α-Nitro-α-diazocarbonyls Asymmetric catalysis In situ generation of diazos Phenyliodonium ylides
Table of Contents
Page Sommaire………..…...…...………...ii Summary………..…………..………...……….……...iv Table of Contents……….………..………...vi List of Figures……….…..………....xi List of Tables………..………xiii List of Schemes………...…………...xvii List of Abbreviations...………..……..………...xix Acknowledgements……….………..xxiiCHAPTER 1 Cyclopropane α-Amino Acids…...……..……...……..1
1.1 Introduction...…………...…………..1
1.2 Naturally occurring cyclopropane α-amino acids…………...…….3
1.3 Applications of cyclopropane α-amino acids………….…...…...6
1.3.1 Applications of cyclopropane α-amino acids as low molecular weight inhibitors...6
1.3.2 Applications of cyclopropane α-amino acids in macrocycles...10
1.3.3 Applications of cyclopropane α-amino acids in peptides...13
1.4 Preparation of cyclopropane α-amino acids……...16
1.4.1 Preparation of ACC...16
1.4.2 Cyclopropanation of functionalized α,β-dehydroamino acids...18
1.4.3 Cyclopropane α-amino acid synthesis via Curtius or Hofmann rearrangements...22
1.4.4 Cyclopropanations with amino acid equivalents...26
1.4.5 Double alkylations of glycine equivalents...29
1.4.7 Modified Kulinkovich reaction...32
1.5 Conclusions...33
CHAPTER 2 The Synthesis of α-Diazocarbonyl Compounds……...…...………..35
2.1 Introduction…………...………...35
2.2 Research objectives…………...……….………...36
2.3 Preparation of α-nitro-α-diazocarbonyls…….……...…..…..……...40
2.3.1 Introduction and precedence…………...…………..……….40
2.3.2 Development of a diazo transfer methodology……...…….42
2.4 Expansion of the scope of the diazo transfer reaction...54
2.4.1 The influence of the base in the diazo transfer reaction...58
2.5 Conclusions…………...…………...………..60
CHAPTER 3 Cyclopropanation Reactions with α-Nitro-α-Diazocarbonyls...63
3.1 Introduction and precedence…………...………..……….63
3.2 Intermolecular cyclopropanation reactions…………...……..…...…66
3.2.1 Catalyst influence on the cyclopropanation diastereo-selectivities and yields...66
3.2.2 Control of diastereoselectivity in the cyclopropanation reaction...70
3.2.3 Cyclopropanation reaction mechanism...74
3.2.4 Scope of the intermolecular cyclopropanation reaction...79
3.3 Enantioselective intermolecular cyclopropanation reactions...82
3.3.1 Evaluation of chiral Rh(II) carboxylate and carboxamidate catalysts...82
3.3.2 Influence of solvents on the enantioselectivity of the cyclopropanation reaction...93
3.3.3 Influence of the diazo compound on enantiomeric excess...95
3.4 Enantioselective intramolecular cyclopropanation reactions...99
3.5 Conclusions…………...……....………...108
CHAPTER 4 Transition Metal-Catalyzed Cyclopropanations in Water and In Situ Generation of Ethyl Diazoacetate…...…....110
4.1 Introduction…………...…..…………...………..110
4.1.1 O-H insertion reactions with diazo compounds…………...112
4.2 Cyclopropanations in aqueous media...116
4.2.1 Cyclopropanations with α-nitro-α-diazocarbonyls in aqueous media...116
4.2.2 Cyclopropanation reactions with EDA in aqueous media...120
4.3 Enantioselective cyclopropanation reactions involving EDA in water...123
4.3.1 Introduction and precedence...123
4.3.2 Enantioselective cyclopropanation reactions involving EDA in water...126
4.4 In situ generation of EDA followed by cyclopropanation...129
4.4.1 Introduction and precedence...129
4.4.2 Development of a new in situ EDA generation cyclopropanation methodology...132
4.5 Conclusions...………...136
CHAPTER 5 Phenyliodonium Ylides as Safe Alternatives to α-Nitro-α -Diazocarbonyl Compounds...……..138
5.1 Introduction and precedence…………...………...………...138
5.1.1 Cyclopropanation reactions with phenyliodonium ylide reagents...139
5.1.2 Hypervalent iodine(III) reagents for the in situ preparation of nitrenes...140
5.2 Cyclopropanation of olefins with in situ generated phenyliodonium
ylides derived from α-nitrocarbonyls...143
5.2.1 Modification and attempted reoxidation of the iodine(III) reagent...152
5.2.2 The mechanism of the cyclopropanation reaction using phenyliodonium ylides...153
5.3 Expansion of the reaction scope: cyclopropanation with α-cyanomethyl ketones...157
5.4 Conclusions...159
CHAPTER 6 Reduction of Nitro Cyclopropanecarboxylates…….…...……162
6.1 Introduction and precedence…………...………..………...162
6.2 Development of a new reduction methodology for nitro cyclopropane- carboxylates …………...…………..………...165
6.3 Development of a “one pot” cyclopropanation reduction procedure...173
6.4 Mechanism for reduction of nitro groups using zinc(0)...175
6.5 Application of the methodology to the expedient synthesis of aryl-substituted cyclopropylamines...179
6.6 Conclusions...184
CHAPTER 7 Synthesis of Densely Substituted Dihydropyrroles and Pyrroles...186
7.1 Introduction and precedence………...………...186
7.1.1 Objectives...189
7.2 Synthesis of dihydropyrroles using doubly activated cyclopropanes...192
7.2.1 Synthesis of 4-nitro-dihydropyrroles...192
7.2.2 Synthesis of 4-cyano-dihydropyrroles...197
7.3 Oxidation of dihydropyrroles to pyrroles...201
7.4 Conclusions...201
CHAPTER 8 A Tandem Cyclopropane Rearrangement 1,3-Dipolar Cycloaddition: Pericyclic Nitrosoacetal Synthesis...203
8.1 Introduction and precedence…………...………...………..203
8.1.1 Objectives…………...………...204
8.2 Rearrangements of activated cyclopropanes to isoxazoline N-oxides and dihydrofurans...207
8.3 Development of a tandem cyclopropane rearrangement 1,3-dipolar cycloaddition process………..…...……210
8.4 Attempted reduction of the nitrosoacetal and γ-lactam formation...215
8.5 Conclusions...218 EXPERIMENTAL SECTION..……...………..……..………..219 General Notes...219 Experimental: Chapter 2...221 Experimental: Chapter 3...250 Experimental: Chapter 4...280 Experimental: Chapter 5...298 Experimental: Chapter 6...306 Experimental: Chapter 7...324 Experimental: Chapter 8...337 Appendix I…………...……….………..……….354 Appendix II…………...………...……….355 Appendix III……...…...………...……….356 Appendix IV…...……...………...……….357 Appendix V…………...…………...……….358 Appendix VI………...……….…...……….359
List of Figures
Page
Figure 1. Applications of cyclopropanes………...……….2
Figure 2. Natural products containing multiple cyclopropane subunits.…………..….3
Figure 3. Naturally occurring cyclopropane α-amino acids.…..………...………….4
Figure 4. Inhibitors of the Ethylene Forming Enzyme (EFE)………..…….7
Figure 5. Hydroxy(2-aryl) substituted cyclopropane α-amino acid based inhibitors....8
Figure 6. Cyclopropane analogues of thyronine and histidine.…..……...………8
Figure 7. 2,3-Methylenepenams as β-lactamase inhibitors.…..………...……...9
Figure 8. Refined NS3 protease inhibitor containing a 2-vinyl ACC subunit…..…...12
Figure 9. Antiopiate tetra-peptide containing a cyclopropane analogue of methionine...14
Figure 10. The induction of β- and γ-turns in tri-peptides using cyclopropane analogues of phenylalanine...………15
Figure 11. Tri-peptide based inhibitor of calpain 1 containing the cyclopropane analogue of leucine…..………...……...……...15
Figure 12. Auxiliaries based on dehydroamino acids.………...22
Figure 13. Relative reactivities of diazo compounds...…...…...38
Figure 14. Diazo compounds representing amino acid equivalents...……..….39
Figure 15. ORTEP representation of the crystal structure of methyl nitro diazoacetate...53
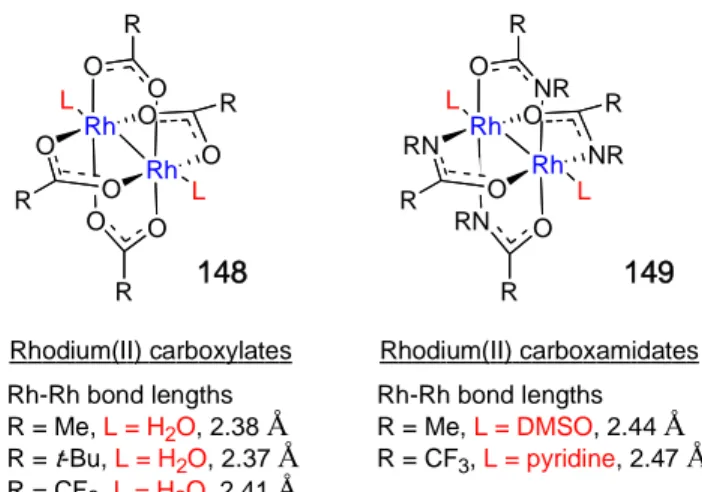
Figure 16. Rh-Rh bond lengths with various bridging and axial ligands.…………...69
Figure 17. Representative 1H NMR spectrum of ethyl-2-phenyl-1-nitrocyclopropane carboxylate (147)...…...………...…...……...72
Figure 18. ORTEP representation of the crystal structure of (E)-1-[2-(4-fluoro-phenyl)-1-nitrocyclopropyl] ethanone (151)...……...………….73
Figure 19. Proposed rhodium-carbene complexes...…....………...……...76
Figure 20. Examples of metal-carbene complexes and diazo-metal adducts...….77
Figure 21. ORTEP representation of the crystal structure of exo-1,1a,6,6a-tetrahydro-1-nitrocyclopropane[a]indene-1-carboxylate (162)...……...…….82
Figure 22. Spartan three-dimensional depiction (using MMFF 94 level) of the [Rh(S-MEAZ)2]2-carbene complex with methyl nitro diazoacetate...……….91
Figure 23. Other miscellaneous catalysts tested...………..…...92 Figure 24. ORTEP representation of the crystal structure of 10a-nitro-1a-phenyl-1,1a,2,10a-tetrahydro-8H,10H-benzo[b]cyclopropa[g][1,5]dioxonin-10-one
(181c)...103 Figure 25. Catalysts for asymmetric cyclopropanations in water involving EDA....127 Figure 26. Biologically active cyclopropylamines...179 Figure 27. Introduction of cyclopropylamines in antithrombotic therapeutics...180 Figure 28. Examples of the pyrrolomycin class of antibiotics containing a 4-nitropyrrole core...………...191 Figure 29. Biologically active tetrasubstituted pyrroles as NSAID COX-2 inhibitors...192 Figure 30. ORTEP representation of the crystal structure of 4-nitro-1,2-diphenyl-5-propyl-2,3-dihydro-1H-pyrrole (293b)...196 Figure 31. Examples of densely functionalized γ-lactams...…...205 Figure 32. ORTEP representation of the crystal structure of 2-methyl-3-nitro-4,8b-dihydro-3aH-indeno[1,2-b]furan (321)...210 Figure 33. Chem3D Pro models of a 5,5,6-nitrosoacetal (left-handed structure) and the 5,5,5-nitrosoacetal 325a (right-handed structure)...214 Figure 34. ORTEP representation of the crystal structure of 4-ethyl-7,7-diphenyl-tetrahydro-2,5,6-trioxa-5a-aza-cyclopenta[c]pentalene-1-one (325e)...215
List of Tables
Page Table 1. Potency of the HCV NS3 protease inhibitors in enzymatic (IC50) and
cell-based assays (EC50)...10
Table 2. Potency of macrocyclic HCV NS3 protease inhibitors in enzymatic (IC50) and cell-based assays (EC50)...11
Table 3. Reaction scope for the tosylhydrazone-mediated cyclopropanation...…20
Table 4. Catalytic asymmetric cyclopropanations with benzylidene diazoesters...25
Table 5. Cyclopropanation with chromium(0)-based aminocarboxycarbenes...28
Table 6. Ti(IV) mediated conversion of α-benzyloxy nitriles to cyclopropylamines.33 Table 7. A summary of various syntheses of coronamic acid...…34
Table 8. Diazo transfer reaction scope with α-nitroester substrates...47
Table 9. Diazo transfer reaction scope with α-nitroketone substrates...…..49
Table 10. Selected geometric parameters for the crystal structure of methyl nitro diazoacetate (97)...…...54
Table 11. Scope of the diazo transfer reaction with α-cyano-α-diazocarbonyl substrates...56
Table 12. Influence of the base on the yield of the diazo transfer reaction...59
Table 13. Summary of diazo transfer reaction scope with triflyl azide...…61
Table 14. Scope of the cyclopropanation reaction with various α-nitro-α-diazo compounds...…65
Table 15. The influence of the Rh(II) catalyst on the yields and diastereoselectivities of the cyclopropanation reaction...67
Table 16. The influence on diastereoselectivities of the cyclopropanation reactions by the R-group of the diazo compound...…...70
Table 17. Scope of the cyclopropanation reaction...80
Table 18. Asymmetric cyclopropanations with iodonium ylide 164 derived from Meldrum’s Acid...84
Table 19. Influence of the amine-protecting group on asymmetric induction...87
Table 21. Asymmetric cyclopropanations with Rh(II) azetidine-based catalysts...90 Table 22. Influence of solvent on enantioselectivity...94 Table 23. The influence of the diazo compound on the levels of asymmetric induction...95 Table 24. Copper-catalyzed cyclopropanations of styrene and methyl nitro diazoacetate...98 Table 25. Asymmetric intramolecular cyclopropanations for the synthesis of 9-membered nitrocyclopropyl lactones...102 Table 26. The influence of the Rh(II) catalyst on the yields of the intramolecular cyclopropanations...104 Table 27. Scope of the Rh(II)-catalyzed intramolecular cyclopropanation reaction...105 Table 28. Rh-catalyzed O-H insertion reactions of alcohols with EDA (101)...113 Table 29. O-H insertion reactions of alcohols with α-nitro-α-diazoester compounds. ...114 Table 30. Cyclopropanation O-H insertion competition experiments...116 Table 31. Aqueous cyclopropanation reactions involving α-nitro-α-diazocarbonyl compounds: catalyst optimization...117 Table 32. Aqueous cyclopropanations of α-nitro-α-diazocarbonyl compounds: influence of olefin equivalents on reaction yields...119 Table 33. Scope of the aqueous cyclopropanation reaction with α-nitro-α-diazocarbonyls...120 Table 34. The influence of the catalyst on cyclopropanation efficiencies of styrene with EDA (101) in aqueous media...121 Table 35. Scope and comparison of cyclopropanation reactions involving EDA (101) in aqueous media...122 Table 36. Nishiyama’s asymmetric biphasic cyclopropanation using a water-soluble Ru(II) catalyst...124 Table 37. The influence of various solvents on asymmetric cyclopropanation reactions using a β-ketoiminato Co(II) complex 204...126
Table 38. Catalytic asymmetric cyclopropanation reactions involving EDA in water...128 Table 39. In situ generation of EDA followed by cyclopropanation reported by Barrett et al...131 Table 40. In situ generation of EDA (101) followed by cyclopropanation...133 Table 41. Oxidative cyclization of carbamates using PhI(OAc)2 via in situ formation
of iminoiodinane intermediates...141 Table 42. The in situ formation of iminoiodinane intermediates using PhI=O, followed by copper-catalyzed intramolecular aziridinations or C-H insertions...142 Table 43. Cyclopropanation of styrene using an in situ generated phenyliodonium ylide derived from methyl nitroacetate: optimization of reaction conditions...145 Table 44. Cyclopropanation of styrene using an in situ generated phenyliodonium ylide: variation of the α-nitrocarbonyl substrate...147 Table 45. Comparison of the diazo-mediated cyclopropanation reaction to the in situ generated phenyliodonium ylide protocol...149 Table 46. Preparation of a series of symmetric nitro cyclopropanecarboxylates...151 Table 47. Optimization of the in situ generation of the phenyliodonium ylides with α-cyanomethyl ketones...158 Table 48. Extension of the in situ generated phenyliodonium ylide methodology to Meldrum’s Acid...160 Table 49. Reduction of nitro cyclopropanecarboxylates containing an aromatic group using zinc dust...169 Table 50. Reduction of a structurally diverse series of nitro cyclopropanecarboxylates using zinc dust...171 Table 51. Reduction of symmetric nitro cyclopropanecarboxylates using zinc dust...173 Table 52. Preparation of aromatic substituted trans-cyclopropylamines...183 Table 53. Preparation of a structurally diverse series of substituted 1-nitro-1-keto cyclopropanes...193 Table 54. Preparation of tetrasubstituted 4-nitro-dihydropyrroles by treatment of 1-nitro-1-ketocyclopropanes with primary amines...194
Table 55. Preparation of densely substituted 4-cyano-dihydropyrroles...198 Table 56. Scope of the intermolecular cyclopropanation reaction followed by a
List of Schemes
Page
Scheme 1. Synthetic strategies for the synthesis of ACC (6)...…………...…….17
Scheme 2. Strategies for the preparation of cyclopropane α-amino acids.……..…...18
Scheme 3. Cyclopropane formation via a 1,3-dipolar cycloaddition pathway.…...21
Scheme 4. Simmons-Smith approach to coronamic acid (55)....…...…23
Scheme 5. Schöllkopf’s bislactim ether method...……...………….27
Scheme 6. Synthesis of trifluoronorcoronamic acid from a chiral epoxide...…...31
Scheme 7. Possible side-reactions in the attempted diazo transfer reaction to ethyl nitroacetate...……….44
Scheme 8. The use of triflyl azide in a diazo transfer reaction for the synthesis of an α-diazoketone...44
Scheme 9. Proposed mechanism for the diazo transfer reaction...…...….50
Scheme 10. Synthesis of α-nitroesters…………...52
Scheme 11. Synthesis of α-nitroketones...……53
Scheme 12. Wolff rearrangement followed by nitroso “ene” or a hetero-Diels-Alder reaction...64
Scheme 13. Schematic representation of the catalytic cycle of a Rh(II) catalyzed cyclopropanation of an α-nitro-α-diazocarbonyl and an olefin...…….75
Scheme 14. Rationalization of diastereoselectivities using Doyle’s Model...….78
Scheme 15. Preparation of diazo compounds for intramolecular cyclopropanations...101
Scheme 16. Synthesis and intramolecular cyclopropanation of an α-nitro-α-diazoketone...107
Scheme 17. Synthesis of an α-nitro-α-diazoketone compound based on a 2-hydroxybenzyl alcohol scaffold...…………...107
Scheme 18. In situ generation of non-carbonyl stabilized diazo compounds...130
Scheme 19. Proposed intramolecular cyclopropanation reaction mechanism involving phenyliodonium ylides...154
Scheme 21. Reduction of nitrocyclopropane...……...163 Scheme 22. Pd-catalyzed reduction of methyl 2-phenyl-1-nitro cyclopropanecarboxylate (150a)...166 Scheme 23. The proposed mechanism of the zinc-mediated reduction of nitro cyclopropane carboxylates...176 Scheme 24. Mechanism of cyclopropane ring opening during reduction...177 Scheme 25. Synthesis of a cis-aryl cyclopropylamine using an in situ diazo generation protocol...181 Scheme 26. Synthesis of enantiomerically pure tranylcypromine (276)……...182 Scheme 27. Synthesis of amino cyclopropanecarboxylates: summary of preparative methods...185 Scheme 28. Total synthesis of (±)-trachelanthamidine via an intramolecular ring opening of a doubly activated cyclopropane...…...187 Scheme 29. The synthesis of γ-substituted α-amino acids from nitro cyclopropanes.... ...188 Scheme 30. Synthetic strategy for the synthesis of densely functionalized pyrroles... ...192 Scheme 31. Synthesis of a trisubstituted 4-nitro-dihydropyrrole...197 Scheme 32. Two possible mechanisms proposed for the formation of dihydropyrroles...199 Scheme 33. Mechanistic studies by Lhommet et al...200 Scheme 34. The synthesis of FR900482 congeners from doubly activated cyclopropanes and nitrones...204 Scheme 35. Envisioned retrosynthetic strategy for the construction of densely functionalized γ-lactam scaffolds...206
List of Abbreviations
[α]D optical rotation
Å angström
Ac acetyl
ACC amino cyclopropanecarboxylate anal. elementary analysis
aq. aqueous Ar aromatic group atm atmosphere Bn benzyl Boc tert-butylcarbonyl b.p. boiling point br broad o C degrees Celsius calcd calculated cat. catalytic δ chemical displacement DBU 1,8-diazabicyclo[5.4.0]undec-7-ene DCC dicyclohexylcarbodiimide DDQ 2,3-dichloro-5,6-dicyano-1,4-benzoquinone de diastereomeric excess Decomp. decomposed DMAP dimethylaminopyridine DMF dimethylformamide E entgegen ee enantiomeric excess EI electron impact equiv equivalent Et ethyl
g gram h hour
HPLC high performance liquid chromatography HRMS high resolution mass spectrometry i-Pr isopropyl
IR infrared
J coupling constant
Lit. literature M molar
MAB metastable atom bombardment
Me methyl mg milligram MHz megahertz min minute mL milliliter mm Hg millimeters of mercury mmol millimole µL microliter M.p. melting point NBS N-bromosuccinimide n-Pr n-propyl
NMR nuclear magnetic resonance ORTEP Oak Ridge Thermal Ellipsoid Plot PG protecting group
Ph phenyl
ppm parts per million
p-TsOH para-toluene sulfonic acid
pyr pyridine
ref. reference
Rf relative mobility
TBDPS tertiary butyl diphenylsilyl t-Bu tertiary butyl
temp. temperature THF tetrahydrofuran
TLC thin layer chromatography TMS trimethylsilyl
tr retention time
Acknowledgements
I would like to begin by extending my most sincere thanks to my research supervisor professor André B. Charette. I feel greatly privileged to have been accepted into his research group, an experience that allowed me to greatly expand my knowledge of synthetic organic chemistry. On a more personal note, I greatly respect his insights and knowledge of chemistry, the enthusiasm he has for the research conducted in his laboratories and his generosity and support throughout my doctoral studies. The accomplishments that I have realized can be largely attributed to his relentless support and encouragement.
I would also like to sincerely thank all the members of the Charette research group and Barbara Bessis whom I had the pleasure of working with throughout my doctoral studies. They have created a stimulating research environment where the mutual exchange of ideas was always welcome. I also thank them for their teaching efforts and tolerance of my inabilities to fluently speak French and for accepting me as both a colleague and a friend. I would like to especially thank Claude Legault who, not only is an exceptionally gifted chemist, but has also inspired my research over the course of my studies. He has helped me a great deal over the years and I greatly value his friendship. I would like to extend my thanks to the past members of the group including Dr. Alexandre Gagnon, Dr. Carmela Molinaro and Dr. Michel Grenon who set very high standards to follow and were also very supportive in my early training. My past and present lab mates including Jonathan Martel, Patrick Deroy, Jean-Emmanuel Bouchard, Alexandre Côté, Mark K. Janes, Wei Lin and Sébastien Nolet have my greatest respects for the creative and enjoyable atmosphere that they helped create in the laboratory.
My collaborations with the members of the various services offered by the Université de Montréal has also been a very pleasurable experience. In particular, I would like to thank Dr. Tah Phan-Viet, Sylvie Bilodeau and Robert Mayer from the nuclear magnetic resonance laboratories for their availability, good humor and expertise in repairing the delicate NMR instrumentation. Francine Bélanger-Gariépy from the X-ray diffraction laboratory, who was responsible for the solutions of all six
X-ray structures in this thesis deserves much more than a simple thank you. The employees at the electronics and machine shops, whose ingenuity and skill solved many instrumentation problems also receive my respects. I would like to extend my thanks to Huguette Dinel at the elemental analysis laboratory whose assistance was also very helpful.
I would like to acknowledge NSERC for the greatly needed financial support offered during my doctoral studies as well as the financial assistance offered by the l’Université de Montréal and the J. A. DeSève foundation.
I would like to thank my family who have supported me financially and morally and encouraged me to excel throughout my studies. My final thanks goes to my future wife, Christina, for having the strength to endure what has been a very busy and often stressful 4 1/2 years. The realization of this dream would not have been possible without her companionship, her tolerance of my grueling work schedule and her love.
CHAPTER 1
Cyclopropane
α
-Amino Acids
1.1 Introduction
Cyclopropane chemistry is amazingly diverse and continues to fascinate scientists from a broad range of backgrounds. Theoreticians, synthetic or structural chemists and biochemists with interests in natural product or medicinal chemistry have all shown great interest in this field. This cyclic three-membered carbon ring also represents a source of great wonder for the fundamental aspects of bonding and challenges the skills of organic chemists in the preparation of highly strained molecules.
On a more practical basis, the cyclopropane moiety has a significant role in medicinal chemistry. It is estimated that by the year 2010, 10% of all the pharmaceuticals on the market will contain a cyclopropane subunit.1 This compares even more favorably than the biaryl subunit, which appears in approximately 4.3% of pharmacologically active compounds.2 This virtually ubiquitous three-membered carbocycle may serve a variety of purposes in drug design including: the rigidification of molecular conformations, as in the cyclopropane analogue of tamoxifen (1),3 an effective medication for the treatment and possible prevention of breast cancer; or its introduction into peptides 2, where it rigidifies the three-dimensional conformation of the peptide acting as an excellent mimic of the bound state (Fig. 1).4
Nature has also chosen to use a cyclopropane skeleton to design a defense mechanism for certain pyrethrum flowers against insect attack.5 In the case of
1.(a) Communication S. Nguyen OMCOS 12, Toronto, ON, July 6-10, 2003. For a review on cyclopropane chemistry see: (b) de Meijere, A. Small Ring Compounds in Organic Synthesis VI Vol 207 Top. Curr. Chem. Springer: New York, 2000.
2. Communication K. Fagnou 14th QOMSBOC, Montréal, QC, December 5-7, 2003. 3. Davies, H. M. L.; Nagashima, T.; Klino III, J. L. Org. Lett. 2000, 2, 823-826. 4. Davidson, J. P.; Lubman, O.; Rose, T.; Waksman, G.; Martin, S. F. J. Am. Chem. Soc. 2002, 124, 205-215.
chrysanthemic acid (3), isolated from the petals of these plants,6 the cyclopropane is a very effective antifeedant for herbivores. It represents a broad spectrum insect repellent with low mammalian toxicity. This insecticide’s efficiency has led to commercial production of a class of biomimetic insecticides produced to a tune of over $1.5 billion (US) annually.7
Me O NMe2 Me2N H N O O H2O3PO CO2H N H O H N CO2H O OH O H Me Me Me H CO2H Me Cyclopropane analogue of tamoxifen Incorporation into peptides
trans-Chrysanthemic acid (potent insecticide)
3 2
1
Figure 1. Applications of cyclopropanes.
Multiply cyclopropanated analogues also occur in nature. Yoshida et al. isolated the potent antifungal agent FR-900848 (4) from the fermentation broth of Streptoverticillium fervens, which contains four contiguous and one isolated cyclopropane unit.8 Another related natural product isolated from Streptomyces sp.,
U-106305 (5), was found to act as an effective inhibitor of the cholesteryl ester transfer protein in the blood and can, thus, be envisioned to slow the progression of atherosclerosis (Fig. 2).9
6. Staudinger, H.; Ruzicka, L. Helv. Chim. Acta 1924, 7, 177-235.
7. Faust, R.; Knaus, G.; Siemeling, U. World Records in Chemistry (Ed. Quadbeck-Seeger, H.- J.), Wiley-VCH, Weinheim, 1999, pg. 95.
8. Yoshida, M.; Ezaki, M.; Hashimoto, M.; Yamashita, M.; Shigematsu, N.; Okuhara, M.; Kohsaka, M.; Horikoshi, K. J. Antibiotics 1990, 18, 748-754.
9. Kuo, M. S.; Zielinski, R. J.; Cialdella, J. I.; Marschke, C. K.; Dupuis, M. J.; Li, G. P.; Kloosterman, D. A.; Spilman, C. H.; Marshall, V. P.; J. Am. Chem. Soc. 1995, 117, 10629-10634.
Me HN O Me Me Me HN O O N HN OH OH O O (-)-U-106305
Cholesteryl ester transfer protein inhibitor (-)-FR-900848
Potent antifungal agent
4
5
Figure 2. Natural products containing multiple cyclopropane subunits.
The medicinal importance of these cyclopropane containing products justifies the keen interest in the development of new methods for the construction of cyclopropanes in a stereo-controlled fashion.10 Many current methods rely on toxic or highly pyrophoric reagents, thus the development of new methodologies that can avoid use of these reagents are of great interest. In the context of the development of a commercially viable methodology, the availability and toxicity of the implicated reagents must be carefully examined. Attempts to accommodate these issues in a new cyclopropanation methodology will be addressed in the following chapters.
1.2 Naturally occurring cyclopropane α-amino acids
The design and synthesis of amino acids with enhanced properties has been an ongoing subject of research in the Charette group. As a result, we became interested in the preparation of α,α-disubstituted amino acids due to their important role in the design of peptides with enhanced properties. This area of peptidomimetics has inspired keen interest in the scientific community. In conjunction with the group’s
10. For a recent review on stereo-controlled cyclopropanations see: Lebel, H.; Marcoux, J.- F.; Molinaro, C.; Charette, A. B. Chem. Rev. 2003, 103, 977-1050.
successes in the stereo-controlled synthesis of cyclopropanes, the synthesis of cyclopropane α-amino acids containing chiral quaternary centers11 represents a
particularly attractive and challenging research goal.
Many researchers have paid particular attention to the special characteristics that the cyclopropane ring confers on a peptide in which it is incorporated. The introduction of the cyclopropane unit imposes the formation of new stereocenters in addition to the creation of new stereoisomers. Therefore, a very interesting way to decrease the conformational freedom of peptides is to incorporate amino acids containing a cyclopropane subunit.
There are few known examples of naturally occurring cyclopropane α-amino acids and only one of which is abundant in nature. The parent compound, 1-amino-1-cyclopropane carboxylic acid, (ACC, (6), Fig. 3) occurs in every green plant.12 It was
discovered by two independent research groups in 1957, where it was isolated from perry pears and cider apples by Burroughs13 and extracted from cowberries by Vähätalo and Virtanen.14 This cyclopropane amino acid 6 represents the eighty-eighth natural amino acid to be discovered.
H2N CO2H HO2C NH2 H N NH2 NH H H O HO2C H HN O
ACC Coronatine Carnosadine
6 7 8
Figure 3. Naturally occurring cyclopropane α-amino acids.
11. For reviews on the catalytic enantioselective construction of molecules with quaternary carbon stereocenters see: (a) Christoffers, J.; Mann, A. Angew. Chem., Int. Ed. 2001, 40, 4591-4597. (b) Corey, E. J.; Guzman-Perez, A. Angew. Chem., Int. Ed.
1998, 37, 388-401. (c) Fuji, K. Chem. Rev. 1993, 93, 2037-2066.
12. Yang, S. F.; Hoffman, N. E.; Annu. Rev. Plant Physiol. 1984, 35, 155-189. 13. Burroughs, L. F. Nature 1957, 179, 360-361.
ACC (6) plays a very important role in plants as it serves as the direct synthetic precursor to the plant hormone, ethylene. Since ethylene is a volatile gas, plants have evolved an ingenious method for ethylene fixation, storing it in the form of a simple, non-volatile amino acid. Ethylene gas can be released upon action of the Ethylene Forming Enzyme (EFE) on this amino acid 6. Ethylene is a phytohormone that initiates and regulates many aspects of plant growth including fruit ripening, wound healing, germination of seeds, senescence (wilting and other catabolisms, such as Fall colors), abscission (dropping of Fall leaves) and responses to environmental stress.15
The role of ACC (6) in plants has a great impact to human activities as well. If you consider the tremendous losses of agricultural products due to over-ripening, the development of products to control the production of ethylene could have a profound value to the global food supply. Progress made towards the understanding and development of inhibitors of the EFE with this in mind will be the topic of further discussion in Section 1.3.1 along with the structure-activity relationships of substituted ACC’s and the Ethylene Forming Enzyme (EFE).
Coronatine (7), is another naturally occurring cyclopropane α-amino acid produced by the bacteria Pseudomonas coronafacience var. atropurpurea (Fig. 3).16 Infection of host plants by these bacteria induces chlorosis on the leaves, due to production of 7. In addition, this phytotoxin also induces hypertrophy of potato cells, inhibits root elongation in corn and stimulates ethylene biosynthesis at concentrations below 1.0 µM.16 Plant defense against herbivores involves the release of volatile
substances, which act as SOS signals attracting predators that prey on herbivores. It has been shown that coronatine (7) is superior to jasmonic acid in inducing the biosynthesis and emission of volatiles.17
15. Pirrung, M. C. Acc. Chem. Res. 1999, 32, 711-718.
16. Ichihara, A.; Shiraishi, K.; Sato, H.; Sakamura, S.; Nishiyama, K.; Sakai, R.; Furusaki, A.; Matsumoto, T. J. Am. Chem. Soc. 1977, 66, 636-637.
17. Boland, W.; Hopke, J.; Donath, J.; Nüske, J.; Bublitz, F. Angew. Chem., Int. Ed.
Carnosadine (8) was isolated from the red algae, Grateloupia carmasa and its biological activities are yet unknown (Fig. 3).18 The asymmetric synthesis of carnosadine (8) and its protected analogues have been reported for use in the incorporation into peptides where it represents conformationally constrained surrogates of arginine.19
1.3 Applications of cyclopropane α-amino acids
1.3.1 Applications of cyclopropane α-amino acids as low molecular weight inhibitors
Considerable interest has been devoted to cyclopropane α-amino acids (ACC's) in recent years on account of their biological activities as low molecular weight inhibitors. The understanding and development of inhibitors related to the Ethylene Forming Enzyme (EFE) have been research interests for many groups. Yang and Ichihara found that allo-coronamic acid (9) is preferred over its other stereoisomers as a substrate for the production of 1-butene in plants.20 As a result, a variety of analogues were prepared to study their inhibition abilities of the EFE. Hydroxymethyl-ACC (10) was found to be a good inhibitor with a Ki/Km = 1.21 The cyclopropene analogue (11) of ACC (6) was found to be one of the best competitive inhibitors with a Ki/Km = 0.6, forming acetylene by plant tissue, but at a rate of only 0.2% of the rate ACC (6) is converted to ethylene (Fig. 4).22
18.(a) Wakamiya, T.; Nakamoto, H.; Shiba, T. Tetrahedron Lett. 1984, 25, 4411-4412. (b) Wakamiya, T.; Oda, Y.; Fujita, H.; Shiba, T. Tetrahedron Lett. 1986, 27, 2143-2144.
19. Burgess, K.; Ho, K.-K. Tetrahedron Lett. 1992, 33, 5677-5680.
20. Hoffman, N. E.; Yang, S. F.; Ichihara, A.; Sakamura, S. Plant Physiol. 1982, 70, 195-199.
21. Pirrung, M. C.; McGeehan, G. M.; Angew. Chem., Int. Ed. 1985, 24, 1044-1045. 22. Pirrung, M. C.; Trinks, U. P. J. Chem. Soc., Chem. Commun. 1989, 857-859.
NH3+ CO2 -NH3+ CO2 -HO NH3+ CO2
-allo-Coronamic acid Hydroxymethyl-ACC Cyclopropene analogue of ACC
9 10 11
Figure 4. Inhibitors of the Ethylene Forming Enzyme (EFE).
Since there are few known naturally occurring cyclopropane α-amino acids, specific enzymes for their degradation in mammals are rare. Amino acids that are substituted with a cyclopropane have very similar electrostatic and steric profiles. Modification of an amino acid through incorporation of the cyclopropane moiety leads to only a small increase in overall steric bulk. As a result, binding of these modified amino acids to substrates tends to resemble that of the parent amino acid. However, the presence of the cyclopropane moiety greatly decreases the rate of enzymatic degradation, hence the metabolism of the modified amino acid. This in turn could result in the inhibition of certain enzymes. This represents an area of potential interest for applications in the pharmaceutical industry.
A number of 2-substituted cyclopropane α-amino acid derivatives are efficient inhibitors of DOPA carboxylase. For instance 2-(3,4-dihydroxyphenyl) cyclopropane amino acid 12, due to its structural analogy with α-methyl DOPA (13), is a reversible, time-dependent inhibitor of DOPA carboxylase and of tyrosine amino transferase (Fig. 5).23 In related research, the four possible cyclopropane analogues of m-tyrosine were prepared, assayed and proven to be competitive inhibitors of pig liver L-aromatic amino acid decarboxylase against D-m-tyrosine. It was found that the (+)-(E)-enantiomer 14 corresponding to that of a D-amino acid was the most potent inhibitor, showing a Ki of 22 µM, 45-fold greater than that of D-m-tyrosine (Fig. 5).24
23. Suzuki, M.; Kumar, S. D.; Stammer, C. H. J. Org. Chem. 1983, 48, 4769-4771. 24. Ahmad, S.; Phillips, R. S.; Stammer, C. H. J. Med. Chem. 1992, 35, 1410-1417.
CO2 -NH3+ HO HO 12 HO HO Me CO2 -NH3+ 13 NH3+ HO 14 CO2 -α-Methyl DOPA 2-(3,4-Dihydroxyphenyl)
cyclopropane amino acid
Cyclopropane analogue of m-tyrosine
Figure 5. Hydroxy(2-aryl) substituted cyclopropane α-amino acid based inhibitors.
(Z)-2,3-Methanothyronine (15) and its dibromo derivative 16 have comparable activities with thyroxine (17), a thyroid hormone, which exhibited thyromimetic activity in basal metabolism and antigoiter tests (comparison of oxygen consumption and heart rate in normal and thyroidectomized rats), but did not have an inhibitory action on the metabolism developed by triiodothyronine (Fig. 6).25
(Z)-2,3-Methanohistidine (18) was also found to be an effective inhibitor of histidine decarboxylase when tested on rat liver (Fig. 6).26
15 16 17 CO2 -NH3+ I O I OH CO2 -NH3+ I O I OH Br Br CO2 -NH3+ I O I OH I I Thyroxine (Z)-2,3-Methanothyronine (Z)-2,3-Methanothyronine dibromo analogue CO2 -NH3+ N N H (Z)-2,3-Methanohistidine 18
Figure 6. Cyclopropane analogues of thyronine and histidine.
The cyclopropane analogues of a variety of β-lactam antibiotics have also been prepared.27 The cyclopropane derivative of penicillin G, (2,3)-β-methylenepenam
25. Pages, R. A.; Burger, A. J. Med. Chem. 1967, 10, 435-440. 26. Pages, R. A.; Burger, A. J. Med. Chem. 1966, 9, 766-768.
(19), containing an imbedded cyclopropane α-amino acid subunit, was found to possess reduced antibacterial potency when compared to its penam counterpart. Despite its poor antibacterial effectiveness, it proved to be a substrate for several β-lactamases.28 When the cyclopropane analogue was tested in several strains of
β-lactamase producing bacteria, it was found to serve as a β-β-lactamase inhibitor to protect the β-lactam antibacterials. A broad-spectrum β-lactamase inhibitor sulbactam (20) showed similar half-maximal inhibitory concentration (IC50) values
with its cyclopropane analogue 21 against some β-lactamases such as Staphylococcus aureus and Escherichia coli R1 (Fig. 7).28
O N S Me NaO2C O HN Bn N S Me RO2C O O O N S CO2R O O O (2,3)-β-Methylenepenam Cyclopropane analogue of penicillin G
R = H or Me R = H or Me
Sulbactam (Broad-spectrum
β-lactamase inhibitor) Cyclopropane analogue of sulbactam
19 20 21
Figure 7. 2,3-Methylenepenams as β-lactamase inhibitors.
Additionally, these cyclopropane analogues exhibit greater stability towards hydrolysis, which may explain their large differences in antibacterial activity. Once they are bound, they do not acylate the enzyme to make the attachment irreversible. Furthermore, the cyclopropane analogues are locked in an unique conformation, whereas penams are less rigid and can exist in a variety of conformations.
27. Kamiya, T.; Teraji, T.; Hashimoto, M.; Nakaguchi, O.; Oku, T. J. Am. Chem. Soc.
1975, 97, 5020-5021.
28. Keith, D. D.; Tengi, J.; Rossman, P.; Tobaro, L.; Weigele, M. Tetrahedron 1983, 39, 2445-2458.
1.3.2 Applications of cyclopropane α-amino acids in macrocycles
Researchers at Boehringer Ingelheim recently incorporated substituted cyclopropane amino acids into small peptides29 resulting in potent NS3 protease inhibitors against the hepatitis C virus (HCV). The first generation inhibitor was a tetra-peptide 22 incorporating ACC (6) as a terminal subunit. Its truncated analogue
23a was a poor inhibitor of the NS3 enzyme in vitro but upon incorporation of
2-vinyl-ACC 23b its potency again increased (Table 1).
Me O O N O R1 O N H H N O N BocHN O R1 R3 N R4 OMe O 22a R1 = R2 = H 22b R1 = X1, R2 = H 22c R1 = X1, R2 = CH=CH2 23a R1 = R2 = H, R3 = Me 23b R1 = X2, R2 = CH=CH2, R3 = (CH2)4CH3 X1 = R4 = H X2 = R4 = Ph NH CO2H R2 NH2 CO2H R2
Table 1. Potency of the HCV NS3 protease inhibitors in enzymatic (IC50) and
cell-based assays (EC50).
compound IC50 (µM)a EC50(µM)b 22a 22b 23a 23b >1000 ---1.4 >20.0 >1000 ---0.400 n.d.
a) IC50 – half maximal inhibitory concentration.
b) EC50 – mean 50% effective concentration. 22c 0.047 >5.0
29.(a) Poupart, M.- A.; Cameron, D. R.; Chabot, C.; Ghiro, E.; Goudreau, N.; Goulet, S.; Poirier, M.; Tsantrizos, Y. S. J. Org. Chem. 2001, 66, 4743-4751. (b) Rancourt, J.; Cameron, D. R.; Gorys, V.; Lamarre, D.; Poirier. M.; Thibeault, D.; Llinàs-Brunet, M. J. Med. Chem. 2004, 47, 2511-2522.
An elaborated second generation NS3 protease inhibitor included a 15-membered macrocycle with an imbedded vinyl cyclopropane amino acid subunit.30 The researchers found that upon binding of the tri-peptide 23b, the terminal carboxylic acid rotated 180° to bind in the enzyme pocket. Introduction of a macrocycle locked this optimum binding conformation.31 The vinylic cyclopropane α-amino acid subunit
was found to be 2-fold more potent that its saturated analogue (24c versus 24d, Table 2). The stereochemistry of the cyclopropane was also found to be important, with the E-isomer 200-times more potent than its Z-isomer (24c versus 25, Table 2).
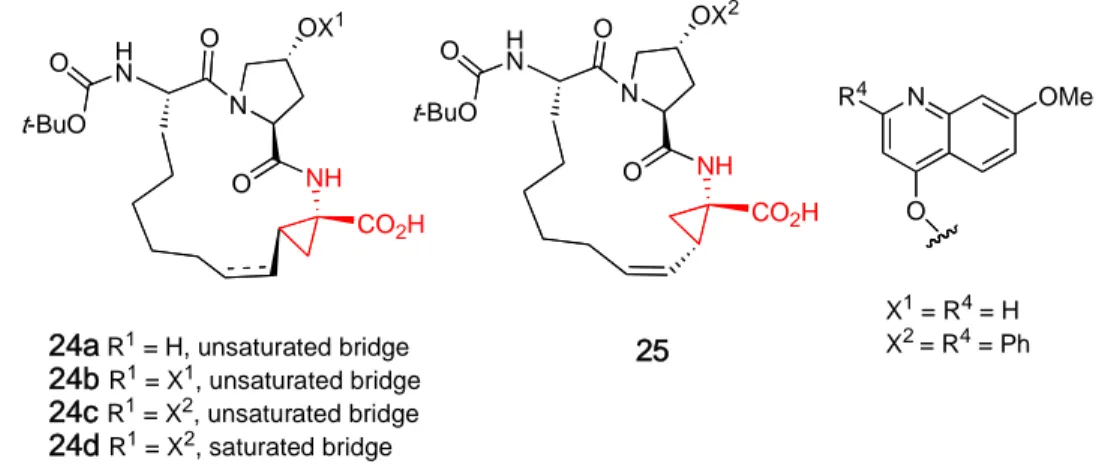
N R4 OMe O O t-BuO OX1 H N N O O NH CO2H O t-BuO OX2 H N N O O NH CO2H
24a R1 = H, unsaturated bridge
24b R1 = X1, unsaturated bridge 24c R1 = X2, unsaturated bridge 24d R1 = X2, saturated bridge X1 = R4 = H X2 = R4 = Ph 25
Table 2. Potency of macrocyclic HCV NS3 protease inhibitors in enzymatic (IC50)
and cell-based assays (EC50).
compound IC50 (µM) EC50(µM) 24a 24b 24d 25 400.0 ---0.024 1.20 0.028 0.12 2.00 ---24c 0.011 0.077
30. Tsantrizos, Y. S.; Bolger, G.; Bonneau, P.; Cameron, D. R.; Goudreau, N.; Kukolj, G.; LaPlante, S. R.; Llinàs-Brunet, M.; Nar, H.; Lamarre, D. Angew. Chem., Int. Ed. 2003, 42, 1355-1360.
31. M. Llinàs-Brunet, 14th Québec-Ontario Minisymposium on Bio-Organic and Synthetic Chemistry, Montréal, QC, December 5-7, 2003.
Additional modifications to the hydroxy-proline region of the macrocycle led to the introduction of an amino thiazole subunit, which gave a further increase in potency. The stability of the macrocycle was also increased upon replacement of the “Boc” protecting group present in macrocycles 24 and 25 with a more stable cyclopentanol derived carbamate analogue 26 (Fig. 8).32
O O O H N N O O NH CO2H 26 N S N OMe NH
Figure 8. Refined NS3 protease inhibitor containing a 2-vinyl ACC subunit.
This NS3 protease inhibitor was well tolerated at doses ranging from 5 to 2,000 mg in human clinical trials. Unspecific intestinal adverse events were observed at the highest dose (2,400 mg), probably due to local gastrointestinal irritations caused by the large quantity of drug. In small proof-of-concept trials this pharmaceutical 26 has been shown to reduce the hepatitis C RNA detectable in plasma and it represents the first of its kind in a new class of hepatitis C inhibitors. Macrocycle 26 possesses IC50
values of 4.0 and 3.0 nM for the HCV replicon 1a and 1b, respectively, and EC50
values of 1.2 nM. It has an apparent selectivity index with the addition of 50% human serum of 10,000 in Huh-7 cells, making it highly specific for the desired NS3 protease.
32. Lamarre, D.; Anderson, P. C.; Bailey, M.; Beaulieu, P.; Bolger, G.; Bonneau, P.; Bös, M.; Cameron, D. R.; Cartier, M.; Cordingley, M. G.; Faucher, A.-M.; Goudreau, N.; Kawai, S. H.; Kukolj, G.; Lagacé, L.; Laplante, S. R.; Narjes, H.; Poupart, M.-A.; Rancourt, J.; Sentjens, R. E.; St. George, R.; Simoneau, B.; Steinmann, G.; Thibeault, D.; Tsantrizos, Y. S.; Weldon, S. M.; Yong, C.- L.; Llinàs-Brunet, M. Nature, 2003, 426, 186-189.
1.3.3 Applications of cyclopropane α-amino acids in peptides
As alluded to in Section 1.2, perhaps the greatest pharmacological potential for cyclopropane α-amino acids exists in the field of conformationally constrained peptide mimetics.33 It has been observed that their incorporation into peptides influences the 3-dimensional conformation of the peptide leading to more compact structures. This compression of the peptide results in reduced rates of hydrolysis, therefore, resulting in increased bioavailability of the peptide.34 In principle, incorporation of a cyclopropane analogue could be rationally applied to almost any bioactive peptide, so the scope for this type of modification is virtually unlimited.35
Many examples of bioactive cyclopropane α-amino acid containing peptido-mimetics have already emerged. For instance, Asp-Acc-O(n-Pr) was found to be 250-300 times sweeter than sucrose,36 while replacement of phenyl alanine by its cyclopropane analogue gave tasteless analogues of aspartame (in Asp-Phe-OMe).37
The influence on conformation of adding a cyclopropane analogue into peptides is well documented in the research of Burgess et al. Peptidomimetics of the antiopiate neuropeptide (Phe-Met-Arg-Phe-NH2) were synthesized by exchanging methionine
(Met) with each of the four isomers of its cyclopropane analogue 27. All four peptidomimetics precipitated more morphine abstinence signs in morphine addicted rats, although the in vitro receptor binding studies have shown that the analogues were less strongly bound than the parent peptide (Fig. 9).38
33. For a review see: Cativiela, C.; Díaz-de-Villegas, M. D. Tetrahedron: Asymmetry
1998, 9, 3517-3599.
34.(a) Burgess, K.; Ke, C.- Y. J. Org. Chem. 1996, 61, 8627-8631. (b) Hillier, M. C.; Davidson, J. P.; Martin, S. F. J. Org. Chem. 2001, 66, 1657-1671. (c) Martin, S. F.; Dwyer, M. P.; Hartmann, B.; Knight, K. S. J. Org. Chem. 2000, 65, 1305-1318. 35. Burgess, K.; Ho. K.-K.; Moye-Sherman, D. Synlett 1994, 575-583.
36. Tsang, J. W.; Schmeid, B.; Nyfeler, R.; Goodman, M. J. Med. Chem. 1984, 27, 1663-1668.
37. Mapelli, C.; Stammer, C. H.; Lok, S.; Mierke, D. F.; Goodman, M. Int. J. Pept. Protein Res. 1988, 32, 484-487.
N H H N N H +H3N Ph O SMe O O NH2 O Ph NH NH2 +H 2N Phe-(2S,3S-cyclo-Met)-Arg-Phe-NH2 Antiopiatetetra-peptide
27
Figure 9. Antiopiate tetra-peptide containing a cyclopropane analogue of methionine.
The parent sequence (Phe-Met-Arg-Phe-NH2) was found to have the expected
random coil conformation in solution, however, introduction of cyclopropane analogues of methionine (Met) showed a bias toward defined secondary structures in solution. For example, the Phe-((2S,3S)-cyclo-Met)-Arg-Phe-NH2 analogue 27 was
found to have good correlation indicating a bias toward a γ-turn structure in solution (Fig. 9).38
In addition to the induction of three-dimensional conformational preferences in solution, both enkephalin39 and Phe-Met-Arg-Phe-NH2 peptides40 containing
cyclopropane analogues had exceptional proteolytic stability towards carboxypeptidase or leucine aminopeptidase digestion respectively. This implies that substitution with cyclopropane analogues can generally provide proteolytically stable materials.
Cativiela et al. have also reported the presence of a γ-turn induced by the introduction of a highly constrained cyclopropane analogue of phenylalanine into a tri-peptide. In solid state, one enantiomer 28 adopts a classical βΙΙ-turn while the other enantiomer 29 adopts a γ-turn centered at the cyclopropane (Fig. 10).41
39. Kimura, H.; Stammer, C. H.; Shimohigashi, Y.; Cui, R. L.; Stewart, J. Biochem. Biophys. Res. Commun. 1983, 115, 112-115.
40. Malin, D. H.; Lake, J. R.; Ho, K.-K.; Corriere, L. S.; Garber, T. M.; Waller, M.; Benson, T.; Smith, D. A.; Luu, T.-A.; Burgess, K. Peptides 1993, 14, 731-734.
N O NH O t-BuO NHi-Pr O Ph Ph N O NH O t-BuO NHi-Pr O Ph Ph
Tri-peptide which exhibits a βII-turn Tri-peptide which exhibits a γ-turn
28 29
Figure 10. The induction of β- and γ-turns in tri-peptides using cyclopropane
analogues of phenylalanine.
The influence of incorporating cyclopropane α-amino acids into tri-peptides was also studied in calpain inhibitors, a cytosolic calcium-activated neutral protease. The enzyme has been implicated in a number of pathological conditions including neurological disorders (eg. stroke), cataracts, cardiac ischemia and thrombotic platelet aggregation. A tri-peptide, already known to have good inhibition of the enzyme, was modified by the introduction of the four possible stereoisomers of leucine 30. These cyclopropane analogues were studied and found to have pronounced effects on inhibition of calpain I. The cyclopropane analogues of leucine, unfortunately, led to a 10-fold decrease in potency, but significant gains in selectivity of the peptide were observed when assayed in the presence of cathepsin B (Fig. 11).42
BnO HN N H O O O Ph N H O Ph
Tri-peptide based inhibitor of calpain I
30
Figure 11. Tri-peptide based inhibitor of calpain I containing the cyclopropane
analogue of leucine.
Incorporation of cyclopropane α-amino acids into peptides provide a means to manipulate their conformations, and in turn bioactivities, as briefly illustrated above.
42. Donkor, I. O.; Zheng, X.; Miller, D. D. Bioorg. Med. Chem. Lett. 2000, 10, 2497-2500.
Their introduction can also be greatly beneficial to the proteolytic stability of the peptide as well. However, the number of proven cases is minimal in comparison with the vast potential for this type of peptidomimetic. The main obstacle to further progress has been inaccessibility of optically pure cyclopropane α-amino acids. Consequently, efficient asymmetric syntheses of these materials are both timely and important.
1.4 Preparation of cyclopropane α-amino acids 1.4.1 Preparation of ACC
In order to gain insights into the synthesis of substituted cyclopropane α-amino acids, one has to first examine the synthesis of the simplest member of the family, namely, 1-amino-1-cyclopropanecarboxylic acid (ACC, 6). Although it is naturally occurring, it is too costly to isolate this amino acid from natural sources due to its low concentrations. There have been a number of interesting strategies for its synthesis, many of which have been applied to the synthesis of more synthetically challenging substituted cyclopropane α-amino acids.
One method of synthesis of ACC (6) includes the method of Häner and Seebach involving the nitration of a di-tert-butylhydroxyanisole (DBHA) derived cyclopropanecarboxylate enolate. This enolate was prepared upon treatment of DBHA cyclopropanecarboxylate (31) with t-BuLi.43 Reduction of the resulting nitro cyclopropane 32 was accomplished using Pd on carbon (1 atm H2) for 48 h, followed
by oxidative removal of the ester using hydrogen peroxide and formic acid (Scheme 1).
Another expedient synthesis of ACC (6) was realized upon dialkylation of isocyanide ethylester (33) with 1,2-dibromoethane, affording cyclopropane 34 in 58% yield.44 Hydrolysis and saponification of this ester 34 and treatment with HCl afforded 6 (Scheme 1) in quantitative yield. This method can also be applied to the synthesis of other cyclic amino acids.
43. Häner, R.; Seebach, D. Chimia, 1985, 39, 356-357.
An aza-Claisen rearrangement has also been used for the synthesis of ACC (6). In this approach, the readily available cyclopropylidine-ethanol (35) rearranges thermally, via a [3,3] sigmatropic rearrangement to provide the trichloroacetamide 36 in 89% yield. Oxidative cleavage of the olefin followed by refluxing in 3M HCl affords ACC (6) in 87% (Scheme 1).45
Scheme 1. Synthetic strategies for the synthesis of ACC (6).
H t-Bu O O t-Bu OMe N C OEt O OH aza-Claisen Cl3CCN, 10% NaH BrCH2CH2Br NHCOCCl3 O N C OEt NO2 O O t-Bu t-Bu OMe NH3+ CO2 -1) t-BuLi, THF 2) amyl-ONO2 1) Pd/C, H2 2) H2O2, HCO2H HCl, reflux 1) NaIO4, RuCl3 2) 3M HCl, reflux 31 32 6 33 34 35 36
NaH, DMSO, Et2O
quant.
89% 87%
63% 71%
58%
Since few naturally occurring substituted cyclopropane α-amino acids exist, their isolation from natural sources for large-scale use is impossible. Consequently, diastereoselective and enantioselective methods for their preparation become important. The stereo-controlled synthesis of chiral quaternary carbon stereocenters makes these molecules very challenging indeed. Further elements of complexity arise from the presence of the often sensitive cyclopropane moiety. To date, more than 50 synthetic methodologies have been developed to tackle this challenging problem in organic synthesis. A brief summary of these methods will follow in addition to some of the perceived benefits and drawbacks of each method.
The general strategies for the synthesis of cyclopropane α-amino acids can be placed into six main categories including: a) cyclopropanation of functionalized
dehydroamino acids, b) Curtius or Hofmann rearrangements of differentially protected cyclopropane dicarboxylic acids, c) double alkylations of glycine anion equivalents, d) cyclopropanation using amino acid equivalents, e) Strecker or allylations and f) modified Kulinkovich reactions (Scheme 2).
Scheme 2. Strategies for the preparation of cyclopropane α-amino acids.
R CH2N2 NPG2 CO2R1 R NPG2 CO2R1 R1O NPG2 O N2 RCHN2 [M] NPG2 CO2R1 N N NPG2 CO2R1 N N NPG2 CO2R1 R R R MgBr Ti(Oi-Pr)4 CO2R1 R R2 NH3+ CO2 -R NH2 R R1O CN R1O O N2 R2 [M] N CN R NHR1 CN R R X OR1 PG2N O R1NH2 NC N Ph Ph OMe OH R X R X OR1 PG2N O Cl Cl Kulinkovich Reaction Cyclopropanation of Dehydroamino acids Strecker Alkylation of Glycine Equivalents Allylation of Glycine Equivalents Cyclopropanation with Differentially Protected Carboxylic Acids Cyclopropanation with Amino Acid Equivalents
NaCN, AcOH + + + + + + + Ph Ph OR1 R R
1.4.2 Cyclopropanation of functionalized α,β-dehydroamino acids
Probably one of the most popular methods for the synthesis of cyclopropane α-amino acids involves the addition of diazo compounds or ylides to dehydroα-amino
acids. A catalytic asymmetric variation of this strategy involving Michael additions of sulfur ylides to dehydroamino acids is a good illustrative example, which was recently reported by Aggarwal et al.46 In this methodology, phenyl diazomethane is formed in situ upon treatment of the insoluble tosylhydrazone salt 37 with a phase transfer catalyst (BnEt3N+Cl-). Upon formation of phenyl diazomethane, [Rh(OAc)2]2
catalyzes the formation of a sulfur ylide derived from chiral sulphide 41. A highly enantioselective Michael addition then occurs with dehydroamino acid 38, affording the protected cyclopropane amino acid 39 in modest yields of 55% (Eq. 1). The reaction proceeds with reasonable diastereoselectivities (E:Z = 1:7) and with excellent enantiomeric excess (90%). Cleavage of the protecting groups was accomplished upon refluxing in 6N HCl, revealing the Z-cyclopropane analogue of phenylalanine (40, Eq. 1). Cyclopropanation of the di-Boc protected α-amino acrylate derivative of 38 gave slightly higher yields (72%), reduced diastereoselectivities (E:Z = 1:6) and similar enantioselectivities (92% ee).
N CO2Et O O Ph N N -Ts Na+ BnEt3N+Cl- (20 mol%) N CO2Et Ph O O S O 6N HCl NH3+Cl -CO2H Ph sulphide (41) (20 mol%) [Rh(OAc)2]2 (1.0 mol%) 1,4-dioxane, 40 oC, 24 h + reflux, 4 h 90% ee, 55% 90% sulphide = 37 38 39 40 41 (1) E:Z 1:7
In a follow-up of this research, the scope of a related racemic methodology involving the direct cyclopropanation of protected amino acrylates 38 was explored. In this case, the absence of the chiral sulfide 41 makes the reaction mechanistically different than described in Eq. 1. The reaction scope was expanded to include other aromatic and α,β-unsaturated hydrazones 43 as diazo precursors. Table 3 summarizes
46. Aggarwal, V. K.; Alonso, E.; Fang, G.; Ferrara, M.; Hynd, G.; Porcelloni, M. Angew. Chem., Int. Ed. 2001, 40, 1433-1436.
the yields and diastereoselectivities possible for the racemic synthesis of 2-substituted cyclopropane α-amino acids.47
Table 3. Reaction scope for the tosylhydrazone-mediated cyclopropanation.
NHAc CO2Me R NHAc CO2Me Na+ R N N -Ts NHBoc CO2PNB R MeO Me F OTBS Ph Ph NHBoc CO2PNB R (c) or (d) (Z-selective) (E-selective) (c) 50 84 95:5 19:81 12:88 52 62 82 87:13 12 8:92
entry cond.a yield (%) E:Z ratio (a) or (b)
Cond. (a) BnEt3N+Cl- (5 mol%), toluene, 40 oC, 60 h. (b) Starting with RCH=NNHTs (i) LiHMDS, THF, -78 oC to rt; (ii) BnEt3N+Cl- (5 mol%), toluene, 40 oC, 60 h.
(c) ClFeTPP (1 mol%), BnEt3N+Cl- (5 mol%), toluene, 40 oC, 60 h. (d) Starting with RCH=NNHTs (i) LiHMDS, THF, -78 oC to rt; (ii) ClFeTPP (1 mol%), BnEt3N+Cl- (5 mol%), toluene, 40 oC, 60 h.
1 2 (a) 3 4 (a) (c) 72 52 95:5 5 6 (a) (c) 49 89:11 14:86 7 8 (a) (c) 14:86 9 10 (b) (d) 47 44 96:4 16:84 11 (a) (c) 76 82 66:34 13 (b) 36 72:28
a) 2.0 equiv of the dehydroamino acid was used.
43
42 44
47. Adams, L. A.; Aggarwal, V. K.; Bonnert, R. V.; Bressel, B.; Cox, R. J.; Shepherd, J.; de Vincente, J.; Walter, M.; Whittingham, W. G.; Winn, C. L. J. Org. Chem. 2003, 68, 9433-9440.
This methodology attempts to address the difficult task of selectively forming E- or Z-diastereomers by using two different sets of reaction conditions. In the case of the E-selective non-catalyzed cyclopropanation reaction conditions (a) or (b), the authors propose a 1,3-dipolar cycloaddition of the diazo compound with the dehydroamino acid followed by ring contraction and extrusion of nitrogen affording the E-diastereomer of the cyclopropane (42, Scheme 3). In contrast, the iron porphyrin catalyst leads to Z-selective cyclopropane 44 formation, presumably via an iron-carbene intermediate (Table 3). Unfortunately, this methodology is plagued by long reaction times (60 h) and the reaction yields with many aromatic or α,β-unsaturated hydrazones remain only modest (Table 3). Furthermore, issues of enantioselectivity were not addressed and the preparation of more highly functionalized cyclopropanes, such as 2,2- or 2,3-disubstituted cyclopropane α-amino acids, is not possible using this protocol.
Scheme 3. Cyclopropane formation via a 1,3-dipolar cycloaddition pathway.
Ph N N -Ts Na+ Na+ NHAc CO2PNB N N Ph H N N Ph H LUMO LUMO HOMO HOMO H NHAc CO2PNB H CO2PNB AcHN Ph N N NHAc CO2PNB Ph + - N2 37 42 + - N2 (a)
A variety of chiral auxiliaries representing dehydroamino acids have also been developed for application to related cyclopropanation reactions including: 45,48 46,49
48. Fernández, M. D.; de Frutos, M. P.; Marco, J. L.; Fernández-Alvarez, E.; Bernabé, M. Tetrahedron Lett. 1989, 30, 3101-3104.
Seebach’s dehydroalanine derivative 47,50 48 derived from (1R,2R,5R)-2-hydroxy pinan-3-one51 and 49 (Fig. 12).52 Cyclopropanation can occur with a variety of reagents including diazomethane,48,49 sulfur ylides49,51,52 and phosphorus ylides. Often cyclopropanations using these auxiliaries affords mixtures of diastereomers, which must then be separated. Cleavage of the auxiliary affords the desired amino acid, which also must be separated from the auxiliary.
N N H O O Ac Ph N O O Ph Ph Boc R O N O t-Bu COPh O N O R 45 46 47 48 N O O Ph 49 R
Figure 12. Auxiliaries based on dehydroamino acids.
1.4.3 Cyclopropane α-amino acid synthesis via Curtius or Hofmann rearrangements
Another popular method for the synthesis of cyclopropane α-amino acids involves the use of Curtius or Hofmann rearrangements for incorporation of the quaternary amine. In this strategy, a cyclopropane containing two differentially protected carboxylic acid equivalents is selectively transformed into an amine. This approach was used in the Charette laboratories for the preparation of the four possible isomers of coronamic acid employing a Simmons-Smith protocol for the highly diastereomeric preparation of 55 (Scheme 4).53
50. Chinchilla, R.; Nájera, C. Tetrahedron Lett. 1993, 34, 5799-5802.
51. Calmes, M.; Daunis, J.; Escale, F. Tetrahedron: Asymmetry 1996, 7, 395-396. 52. Chinchilla, R.; Falvello, L. R.; Galindo, N.; Nájera, C. J. Org. Chem. 2000, 65, 3034-3041.
Scheme 4. Simmons-Smith approach to coronamic acid (55). O BnO BnO OH O OBn TIPSO H Et HO TIPSO H Et Et2Zn (4.0 equiv) HO TIPSO H Et O O BnO BnO OH O OBn TIPSO H Et BocHN HO H Et +H3N H Et CH2ICl (4.0 equiv) -60 oC, 18 h 98%, 66:1 de 1) Tf2O, pyridine CH2Cl2, -20 oC 2) DMF, pyridine H2O, 120 oC 75% 83% RuCl3, NaIO4 H2O, CH3CN, CCl4 rt, 2 h 1) (PhO)2PON3 Et3N, toluene 2) t-BuOH, 120 oC 3) TBAF, AcOH, THF 1) PDC, DMF, 4 ÅMS 2) KMnO4, NaH2PO4 50 51 52 53 54 55 75% 3) TFA, CH2Cl2 84% CO2H
This method allows the preparation of the four-isomers of coronamic acid in excellent yields and excellent diastereomeric excesses, albeit a rather lengthy sequence is required. The key step of the synthesis involves use of a sugar (D-glucose) derived auxiliary in a highly diastereoselective Simmons-Smith cyclopropanation reaction yielding cyclopropane 51. Cleavage of the auxiliary followed by oxidation of the free alcohol 52 to the corresponding carboxylic acid 53 enables a Curtius rearrangement for introduction of the quaternary amine. Deprotection and oxidation of the alcohol and removal of the “Boc” protecting group affords coronamic acid (55, Scheme 4).53 The other three-isomers can be accessed using the same sugar derived auxiliary, either with protecting group inter-conversions or using the E-isomer of olefin 50.
Perhaps, the most expedient and reliable approach to date for the synthesis of substituted cyclopropane α-amino acids was reported by Davies et al.54 employing a
similar strategy. The synthesis of Z-(2R,3R)-2-phenyl-cyclopropane-1-amino acid





![Figure 18. ORTEP representation of the crystal structure of (E)-1-[2-(4-fluoro- (E)-1-[2-(4-fluoro-phenyl)-1-nitrocyclopropyl] ethanone (151)](https://thumb-eu.123doks.com/thumbv2/123doknet/7658218.238246/96.918.314.667.229.525/figure-ortep-representation-crystal-structure-fluoro-nitrocyclopropyl-ethanone.webp)
